
Abstract
Background:
The tumor microenvironment (TME) represents the many components occupying the space within and surrounding a tumor, including cells, signaling factors, extracellular matrix, and vasculature. Each component has the potential to assume many forms and functions which in turn contribute to the overall state of the TME, and further contribute to the progression and disposition of the tumor itself. The sum of these components can drive a tumor towards progression, keep a migratory tumor at bay, or even control chemotherapeutic response. The wide potential for interaction that the TME is an integral part of a tumor’s ecosystem, and it is imperative to include it when studying and modeling cancer in vitro. Fortunately, the development of tissue engineering and biofabrication technologies and methodologies have allowed widespread inclusion of TME-based factors into in vitro tissue-equivalent models.
Methods:
In this review, we compiled contemporary literature sources to provide an overview of the field of TME models, ranging from simple to complex.
Results:
We have identified important components of the TME, how they can be included in in vitro study, and cover examples across a range of cancer types.
Conclusion:
Our goal with this text is to provide a foundation for prospective research into the TME.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Tissue engineered models have found wide-spread adoption throughout basic research. Utilizing fundamental tissue engineering techniques such as 3D cell culture, bioprinting and biomaterial development, scientists can produce tissue equivalents with organotypic function and structure far more advanced than a traditional 2D culture. These complex 3D models can be designed to address high level queries about tissue dynamics that historically necessitated the use of animal models. Although many scientific breakthroughs were made in gold-standard animal models, they have inherent inconsistencies when compared to human physiology; 3D models can be prepared from human cells while maintaining the cell–cell and cell–matrix interactions imperative for accurate and translatable outputs. These benefits have led to a robust and wide-spread use and development of complex models, many components of which can be obtained commercially, substantially lowering the barrier to entry.
In cancer, the tumor microenvironment (TME) is the dynamic space in and around the tumor. It contains a myriad of cell types, growth and paracrine factors, and structural components, each contributing to the progression of a tumor. The combination of these interactions determines the final fate of a cancer and has a massive impact on the severity of a patient’s disease. Historically, cancer research has focused on the tumor cells in isolation, and thus, most cancer killing and controlling therapies only interact with cancer cell centered mechanisms; however, the TME’s role in tumor progression means it holds great promise as a novel vector for tumor therapy. Unfortunately, traditional animal models cannot easily replicate the human tumor-stroma dynamic as most techniques entail the use of human cancer cells injected into immune-compromised mice, without an accompanying human microenvironment, leading to slow development of new therapies. This has necessitated the development of 3D, in vitro tumor models replete with as many features of the TME as possible for identification of high value, TME-based targets and testing of experimental compounds.
Modeling the TME is an exciting and cutting-edge sub-field within cancer research that will undoubtedly lead to breakthroughs in how we identify and test novel therapeutics. Currently, there does not exist a perfect, one-size-fits-all model, but our research group and others are striving to develop models with as many replicative features as possible. In this review, we will describe the various components of the human TME, from biochemical factors to resident cell types; then, we will present contemporary examples of 3D cancer models which include features of the TME.
2 Aspects of the tumor microenvironment
The scope of the TME is vast and includes numerous cellular types, paracrine and growth factors, and cell associated physical properties (Fig. 1). Each component in the TME can contribute to proliferation, recruitment, metastasis, and treatment sensitivity of the local tumor [1]. As the tumor develops, the complex interactions between all of these disparate pieces result in a niche that can become innately protective and tumorigenic, despite being comprised of cells that may not share the same mutational signature found in the cancer itself [2]. Failure to address the synergistic relationship between the TME and tumor can result in ineffective therapies [3]. Accordingly, relevant TME factors must also be incorporated in 3D models to create a system that accurately reflects clinical cases. These complex features are often difficult or impossible to integrate into traditional 2D culture models [4, 5]. Diseases such as pancreatic ducal adenocarcinoma demonstrate how complex and impactful the TME can be on treatment, but in all cases, the TME can create an environment conducive to cancer growth [6].

The components of the tumor microenvironment. The TME is composed of many different cell types, signaling factors, extracellular matrix components and vasculature. They each interact with one another which in turn drives tumor disposition in a dynamic, multi-variate process
2.1 Components of the extracellular matrix (ECM)
The ECM surrounding a tumor acts as more than a simple scaffold for cell attachment. The composition of the matrix can have dramatic effects on cellular migration, phenotype, and genotype [7]. Protein composition of the ECM can have effects on disease progression and patient prognosis [8]. Examples of these structural proteins include collagen, laminin, and fibronectin. Different proteins are found in different densities depending on the tumor location and can account for differences in disease development and treatment effectiveness [9]. These ECM proteins contain binding sites for various growth factors which can provide stimulation and exert mechanical forces that greatly affect tumor activity [10].
Collagen is the most abundant ECM protein and is secreted and remodeled by a majority of the stromal cells in the TME, particularly cancer associated fibroblasts (CAFs). The development of an excessively collagenous ECM is often an indicator of cancer development and can allow for early detection in some tumors. Cancer cells are able utilize collagen to increase migration if they orient and move along the axis of organized collagen fibers [11]. Accordingly, increased collagen concentration correlates with increased cancer metastasis [12]. Cleavage by matrix metalloproteases (MMPs) can result in collagen fragmentation and the presence of these fragments induces integrin stimulation, resulting in increased migration and resistance to administered therapeutics [13].
Laminin is highly influential in gene expression and signaling pathways in the normal tissue microenvironment. Under normal conditions, laminin-111 and laminin-332 help mediate nitric oxide signaling, p53, HOXD10, and microRNAs in breast tissue. These traits make laminin vital for the formation of normal breast acini. When production of laminin is dysregulated, there are significant effects on these pathways and others in the microenvironment, resulting in phenotype destabilization [14]. The dense fibrotic ECM of breast cancer contains greatly upregulated laminin-332. This fibrotic layer can form a capsule that fully encompasses the tumor and aggravates surrounding normal tissue. This TME is highly conducive to enhancing epithelial-to-mesenchymal transition (EMT), leading to increased invasion [15].
Fibronectin upregulation is present in many types of cancer. In non-small cell lung cancer, the increase in this matrix glycoprotein can modulate the Akt/mTOR/p79S6K axis to induce proliferation while being associated with the downregulation of the LKB1/AMPK cell cycle checkpoint pathway [16]. The presence of fibronectin has also been shown to cause secretion of invasion-inducing MMPs [17]. Fibronectin production can be induced by p21 activated kinase-1 in pancreatic ductal adenocarcinoma, and is linked to the therapeutic resistance and low survival rates that has become synonymous with pancreatic ductal adenocarcinoma [8]. Neuopilin-1 induced fibronectin fibrils have also been linked to poorer clinical outcomes due to increased tumor growth potential [18].
2.2 Effects of physical properties of the TME
The phenotype of the cells in the TME is elastic and adaptive depending on microenvironmental conditions. In addition to chemical agents, mechanical properties of the TME are highly influential on tumor behavior. TME stiffness and perfusion are examples of physical elements that can cause alterations in cellular function and phenotype [19]. These changes have often been overlooked in favor of chemical induced signaling. However, there is a growing interest in how these factors may contribute to regulation of both normal and diseased tissue.
Stiffness is highly variable throughout the body ranging from softer tissues like fats to hard materials such as bone. Cells receive signaling through mechanotransduction mediated by integrin binding to ECM anchor points. Integrins in the cell membrane act as the main mechanical signal transduction unit in the cell, allowing physical stiffness signals to be converted into cellular signaling pathways. The Rho/ROCK pathway is an example of a stiffness regulated cell signaling pathway [20]. Similar signals can be used to help multipotent cells differentiate into the appropriate cell types [21]. The TME often contain high levels of CAFs, which can output dense ECM, or desmoplasia, to modulate the stiffness of the local area. The mechanosignaling and migration of the cells is particularly dependent on collagen and fibronectin. These dense ECMs are rich in collagen and fibronectin and are correlated with increased risk of new cancer formation, cancer cell migration, and the progression of existing cancer [22, 23].
As ECM and cellular concentrations in the local TME increases, the local vascular supply can become inadequate or cut off completely. This results in a hypoxic environment within the TME. While physiologically normal cells would likely proceed to cell death due to waste product buildup and a lack of nutrients, cancer cells can enter a state of increased metastatic potentiation to migrate to a more favorable location [24]. In cervical cancer, low tumor oxygenation was shown to correlate with increased tumor migration, more frequent lymph and vascular involvement, and significantly decreased progression free survival [25]. Hypoxic conditions have also been shown to increase the release of extracellular vesicles in tumors in an attempt to increase vascularization by tumor endothelial cells [26]. Hypoxia also induces accumulation of adenosine in the ECM, which in turn could suppress the immune reaction, leading to tumor evasion from T cell action [27]. Hypoxia can activate the HIF, PI3K, unfolded protein response, and MAPK signaling pathways among others [24, 28, 29]. Anti-angiogenic agents are sometimes used to try to deprive rapidly dividing cancer cells of essential nutrients and force them into a state of senescence or apoptosis. However, the use of these antiangiogenic creates a hypoxic environment that has been linked to the stimulation of cancer stem cells, mitigating the desired effects of the treatment [30, 31].
2.3 Chemical signaling in the tumor microenvironment
There is a vast array of chemical signals found throughout the normal microenvironment, each with their own effects and signaling pathways. Each factor family often contributes to a number of diverse functions in both the normal microenvironment and TME. Altered levels of these different factors exert dramatic effects on cancer cell behavior. Examples of these growth factor families include epidermal growth factor (EGF), transforming growth factor β (TGF-β), vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF), fibroblast growth factor (FGF), insulin-like growth factor (IGF), tumor necrosis factor (TNF), and interleukin (IL). While this is not a comprehensive list of factors present in the TME, the combined contributions of these factors on tumor survival and progression are substantial.
EGF and its multi-ligand receptor, a tyrosine kinase, are important in tumor cell proliferation, angiogenesis, metastasis, and drug resistance of cancer cells. This is mediated through the stimulation by TNF-α, stimulation of inflammatory cytokines, including the Ras and phosphatidylinositol-3 kinase signaling pathways, and CXCL5 and CXCL8 chemokines [32, 33].
The TGF superfamily of factors can be produced by cancer cells and the stromal environment in high quantities [34]. In a normal, homeostatic environment, TGF-β can induce cell cycle arrest. However, this pathway is mutated in many cancer types to increase TGF-β production, suppressing immune and other reactive cells while promoting EMT in cancer cells [35]. TGF-β can stimulate CAFs to produce and release ECM, making it a key factor in tumor fibrosis [36]. It can also have pro-tumor effects through the creation of reactive oxygen species and the downregulation of anti-oxidative pathways [37].
VEGF is a key component for induction of angiogenesis [38, 39]. This holds true for the dense TME as well [40]. The high concentration of cells in the cancer niche requires vascularization for nutrient supply and the loose endothelial cell network allows for invasion of the cancer cells through the body. Inhibition of VEGF signaling has been shown to induce apoptosis, arrest cell cycle, and prevent endothelial cell migration and tube formation [41]. However, the process of angiogenesis is essential in normal physiologic function and targeted therapy can result in highly toxic side effects. While promising, anti-VEGF therapy needs improvement [42].
Like TGF-β, PDGF can contribute to the activation of CAFs. The recruitment of these cells by PGDF helps to create the ECM dense TME niche that allows the cancer to grow and develop [43]. Downregulation of PDGF and VEGF signaling has also been shown to help prevent angiogenesis and decrease tumor-associated endothelial cell populations [41]. FGF is also a diverse multifunctional factor family that is ubiquitous in homeostatic maintenance. In the context of the TME, it promotes cancer cell survival through enhancing chemoresistance, neovascularization in the stromal space, and tumor progression [44, 45].
IGF and the ligand IGF binding protein-3 (IGFBP-3) exert transformational changes on the tumor through multiple pathways in a plethora of cancers including colorectal cancer, premenopausal breast cancer, prostate cancer, and lung cancer [46]. IGFBP-3 promotes the TGF-β mediated EMT transition leading to increased metastasis [47]. IGF also promotes EMT via up-regulation of ZEB1 [48].
Interleukins (IL) are a broad category of signaling factors involved with cellular regulation throughout the body; they often are tied to pro-inflammatory functions, such as IL-2, or anti-inflammatory functions, such as IL-10. However, IL-6 has effects reaching beyond immune capability and has been shown to induce differentiation of non-mutated stem cells into a cancer-like phenotype [49].
TNF-α is derived largely from macrophages, but can be produced by a number of other cells. It is vital for survival, apoptosis, and differentiation [50]. Evidence has shown that it TNF-α is an alternate ligand of the EGF receptor [33]. Increased serum levels of TNF-α are correlated with larger tumors and later stage progression [51].
2.4 Cell types in the TME
The stroma surrounding the tumor can be comprised of a mix of several different populations. These populations can have different origins and roles but are all part of the same support system influencing the cancer development. Major classes of cells present in the microenvironment include cancer associated fibroblasts (CAFs), tumor endothelial cells (TECs), tumor associated macrophages (TAMs), and regulatory T cells (Tregs) [3, 52].
CAFs are highly abundant in the TME, but this cell population is relatively poorly defined. On the other hand, many of the roles of CAFs have been described, as these cells play a key role in production of ECM to surround the tumor [53]. They also secrete high levels of MMPs which remodel the ECM. ECM production and modification are often conducive to proliferation, cell migration, and metastasis of tumor cells. CAFs also secrete a number of chemokines such as transforming growth factor-β (TGF-β), interleukin-6 (IL-6), vascular endothelial growth factor (VEGF), and insulin-like growth factor-I (IGF-I). The presence of these factors can combine to play a role in the EMT shift of cancer cells, leading to increased migration and metastasis, vascularization of the tissue, and suppression of the host immune environment [54]. Co-culture analysis of fibroblasts and tumor cells has been reported, often for the following objectives: tumor cell mediated activation of normal fibroblasts into CAFs, CAF-mediated induction of drug resistance in tumor cells or increased tumor cell growth and migration due to CAF-mediated cytokine release or ECM remodeling.
TECs also contribute to the migration and metastasis of tumor cells throughout the body. Endothelial cell organization in the TME differs from the typical tightly packed endothelial vascular structure found in much of the circulatory system. The chronic exposure to growth factors present in the TME results in a porous and unregulated vascular structure. These allow perfusion of nutrients into the TME, while the gaps in the vessel walls allow migrating cancer cells to exit the local environment and enter the blood stream [55]. This rapid angiogenesis greatly outpaces the normal vascular growth in the body and is related to the irregular phenotype of the TECs. This irregular population expansion often contains notable genotypic differences from typical endothelial cells [56]. One significant difference is the increased chromosomal instability and aneuploidy that is found in TECs [57]. These abnormalities also contribute to the inability of TECs to form an organized vessel with properly polarized cells [58]. Efforts to combine TECs and tumor cells often focus on either the irregular formation of blood vessels into tumor tissue or the invasion of tumor cells into or out of vascular networks.
The disruption of the natural immune environment by the TME is vital for tumor growth. Under normal circumstances, cancer cells would rapidly be targeted and destroyed by either the innate or adaptive immune system. However, TAMs and Tregs create an immunosuppressive environment that allows the cancer cells to bypass normal immune checkpoints [59,60,61]. It has been shown that the presence of TAMs can be directly related to negative clinical outcomes in diseases such as lymphoma [62]. Induced by cytokines such as IL-4, TAMs can account for up to 50% of tumor mass in some patients [63]. Aligned to an M2 macrophage phenotype, these cells create inhibit T-cell proliferation and antigen presentation. This is accomplished through the release of anti-inflammatory factors such as IL-10 and the suppression of iNOS signaling [64, 65]. In addition to suppressing natural immunity, these cells can also promote tumor growth through the secretion of stimulatory factors such as EGF, TGF-β and VEGF [65]. In this polarity, they lose their phagocytic properties, removing a vital innate immune response [63]. Tregs infiltrating the TME in high numbers further contribute to immune suppression. There is a diverse and heterogeneous population of Tregs, but the FOXP3+CD25+CD4+ immune cell population is studied for its immunosuppressive capabilities in the TME and correlation with poor clinical outcome [52, 66]. The function of Tregs under normal physiologic conditions is prevention of autoimmune response, making treatment through depletion problematic [60, 67]. When in co-culture with tumor cells, these cells are able to hinder natural antitumor response through several different pathways [68]. Tregs secrete the immunosuppressive an pro-tumor cytokines and signals including IL-10, TGF-β, and CD-35 [69,70,71]. Additionally, the suppressor Treg population can secrete perforin and granzyme B to actively destroy dendritic and effector immune cell populations [72]. Finally, they express high levels of the CTLA-4 receptor protein. This can outcompete the CD-28 receptors on the effector cells trying to bind the CD80/CD86 survival signals ligands, preventing cytotoxic T-cell differentiation and proliferation [73]. Modeling of immune and tumor cells often centers on either the beneficial effects provided by TAMs and Tregs or cytotoxic effects provided by natural killer cells and T-cells.
2.5 3D modeling of the tumor microenvironment
Three dimensional models allow for the experimental investigation of the interactions between tumor cells, endothelial cells, immune cells, and fibroblasts. Each of these cell types play a role in the development of a tumor, and they often alter or are effected by levels of ECM components, growth factors, and physical conditions to make a more favorable microenvironment. Understanding the relationship between these cell types and the TME allows an insight between fibrosis, poor vasculature, immune modulation, and the treatment issues which derive from these problems. This heterogeneity found in the TME is most easily replicable in a 3D system [74]. These microenvironment interactions are difficult to model in traditional two dimensional cell culture models due to decreased cell to cell interaction and decreased surface area for signaling that can lead to changes in growth, metabolism, and differentiation [75].
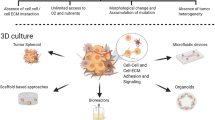
There are several methods of 3D culture utilized in the field today which can generally be divided into two categories: aggregate cultures and encapsulation cultures (Fig. 2). Aggregate cultures promote direct cell–cell adhesion through utilization of low-attachment or low-gravity environments. Common methods include spheroids fabricated in specially-made plates and utilizing rotating wall vessel bioreactors to simulate microgravity. Generally, these cultures are useful for high throughput systems needing high cellularity and their relatively simple organization can make establishment time rapid. However, they can be limited in size by lack of nutrition diffusion to cells inside the cultures depending on the size, and the highly cellular nature can give little control over non-cellular mechanical components of the tumor microenvironment. Encapsulation cultures are created by suspending cells into a hydrogel. Examples of these culture methods include ECM-based materials such as collagen, hyaluronic acid or fibrin, or using materials high in growth factors such as commercially available basement membrane extracts. Encapsulated techniques allow for complex, multi-cellular models for advanced studies due to the ability to control all aspects of the ECM, including enabling self-organization. Despite this, they typically have lower throughput than aggregation cultures and often take longer to establish due to the trend to start at lower cellular density as compared to aggregate. Complex organotypic, tissue equivalent 3D models are often called organoids which have been defined as having: 1) multiple cell types, 2) capability of replicating partial organ function, and 3) cell aggregation or grouping similar to an intact organ [76]. Typically, organoids are more complex than common 3D models and rely on some combination of both aggregation and encapsulation techniques, and often-times utilizing the fate-specification mechanisms of progenitor cells to drive the formation of organotypic structures. We have compiled models of various complexities into Table 1 however it should not be considered exhaustive as new models are developed and tested regularly.

3D culture modalities. A–C There are many types of culture formats which can be combined, tuned, and adapted to produce a final model or organoid that fits the study at hand. Generally, techniques can be separated into several types: (1) membrane-based typically used for lung, gut, and other luminal tissue models, (2) hydrogel encapsulated systems which can be adapted to all tumor types, and (3) aggregation technologies most suited to solid tumor models
2.6 Breast cancer
Breast cancer is the most diagnosed cancer in women [77]. Altered ECM remodeling leading to increased stiffness is often an early sign of breast cancer, and anti-angiogenic therapies such as bevacizumab are often prescribed to slow growth. Work to model endothelial-tumor interactions has been demonstrated utilizing microfluidic devices containing adjacent tumor and vascular organoids to mimic angiogenesis of endothelial vessels towards tumor cells and tumor cell invasion into these vessels; VEGF-2 inhibitors were shown to halt the angiogenesis and slow tumor outgrowth [78]. Another microfluidic model utilized a co-culture of breast cancer cell lines, human breast cancer associated endothelial cells, and fibroblasts in fibrin gel to demonstrate intact endothelial lumens formed under flow; these cells demonstrated increased chemoresistance when in co-culture as opposed to individual groups [79]. The use of flow was also demonstrated utilizing aggregate cultures of tumor cells, endothelial cells, and normal lung fibroblasts to monitor angiogenic sprouting towards co-culture [80]. Histological analysis showed organized lumen formation and higher cellular division in co-cultures as compared to mono or dual cultures, and flow cytometry analysis demonstrated the potential to track target molecules through the cultures. The remodeling of ECM by fibroblasts has also been analyzed, with the action of DDR2 shown to both increase ECM production and collagen fiber organization leading to increased tumor cell migration in a 3D collagen I tumor cell and fibroblast co-cultures, but not in similar Matrigel™ controls [81]. Another study analyzed the role of Hic-5 in fibroblast ECM deposition, determining fibroblasts that were deficient in the expression of Hic-5 caused the formation of more disorganized fibronectin fibers in 3D ECM deposited matrices, leading to decreased tumor migration by failure to directionally migrate along these disorganized fibers [82]. Finally, modeling of breast tumors using tumor, endothelial and stromal fibroblasts in a collagen-hyaluronic acid ECM microenvironment mimicked the formation of disorganized blood vessel-like structures and increased collagen deposition as found in native breast cancer [83].
2.7 Colon cancer
Colon cancer displays predictable invasive activity, with the majority of progressive disease metastasis to the liver. Determining methods to analyze, predict and disrupt this invasiveness may aid in development of therapies to halt or inhibit progression of the disease. We have shown that co-culture of colorectal cancer cells, human hepatocytes and mesenchymal stem cells (MSCs) in a rotating wall vessel bioreactor containing HA-dextran beads demonstrated increased tumor growth and chemotherapeutic resistance compared to aggregates omitting mesenchymal stem cells [84]. We further extended this model, using colonic smooth muscle cells to create a stromal cell assembled, collagen I based TME organoids [85]. The colon cancer organoids demonstrated stromal cell-organized ECM that in turn reduced WNT signaling, in tumor cells, resulting in reduced expression of EMT markers decreased proliferation, and reduced chemotherapeutic sensitivity compared to cancer cells cultured in bare collagen I hydrogels. Another co-culture model of tumor cells and fibroblasts was developed in collagen I constructs placed into microfluidic devices; these tumor cells demonstrated increased growth and increased chemotherapeutic resistance in co-culture conditions [86]. Additionally, the fibroblasts demonstrated an activated phenotype as confirmed by increased alpha-smooth muscle actin (SMA) and tumor cell migration in co-culture conditions.
2.8 Liver cancer
Liver cancer can begin in a manner similar to the development of other liver diseases including fibrosis and cirrhosis, with inflammation due to the activation of hepatic stellate cells (HSCs) and immune infiltration [87]. Research efforts to understand the mechanisms of HSC activation can improve our understanding of the beginning stages of liver cancer development and lead to potential methods to reverse it. Recently, analysis of stellate cells surrounding a tumor core in a microfluidic device was performed to understand co-culture effects on activation and drug resistance; the stellate cells became activated with a change in morphology and an increase in alpha-SMA while the tumor cells showed increased expression of EMT markers vimentin and TGF-β1 [88]. Another group analyzed co-cultures of hepatocellular carcinoma (HCC) cells and stellate cells and observed an increase in collagen I expression in HSCs and altered EMT markers in tumor cells [89]. Additionally, HCCs demonstrated increased drug resistance and enhanced cell motility in co-culture conditions. A separate group used a similar model to analyze HCC cells and stellate cells and observed co-culture of the two groups caused stellate cells to increase alpha-SMA and collagen I expression, and by blocking the synthesis of collagen I they could significantly alter the chemotherapeutic resistance of HCCs in the co-cultured group. They also reported an increase of migration through the upregulation of MMP9 in tumor cells in co-culture [90].
2.9 Lung cancer
The lungs are a highly complex network of stromal and epithelial tissues which are in direct contact with the outside environment. Environmental factors such as smoking or inhalation of irritants can lead to inflammation and fibrosis, with the highly vascularized environment providing sustenance for both primary and metastatic disease. A recent study demonstrated that co-culture of podoplanin+ fibroblasts dramatically increased the growth and establishment of organoid cultures in both lung tumor cell lines and patient tumor cells compared to either monoculture or podoplanin− fibroblasts [91]. A similar result was observed in lung squamous carcinoma cells and CAFs; in particular tumor cells would form acinar structures and become more invasive with decreased SOX2 expression in the presence of CAFs, suggesting that fibroblasts can dampen stem cell-like signaling [92]. In a separate study, CAFs and non-small cell lung cancer (NSCLC) cells were placed into connected wells containing basement membrane extract microfluidic devices to analyze GRP78, a regulatory protein associated with increased protein expression, and its role in tumor invasion [93]. It was shown that overexpression of GRP78 mediated by CAFs caused increased migration, and blocking GRP78 prevented this invasion. Recently, an organoid consisting of fibroblasts, endothelial, epithelial and lymphatic cells was developed to model metastatic colonization of the lung [94]. Pretreatment of these organoids with tumor exosomes increased tumor cell colonization, and angiogenesis of tumor infiltrated constructs was observed and reversible by anti-VEGF treatment. Experiments utilizing patient derived cells demonstrated treated cultures reflected clinically observed patient sensitivities to treatments.
2.10 Pancreatic cancer
Pancreatic cancer displays a poor 5-year prognosis, with few effective treatment options available. One of the difficulties in treating the cancer is related to poor chemotherapeutic penetration due to ECM remodeling and activation in pancreatic stellate cells (PSCs) to form distinct, tumor supportive subpopulations [95]. Identification of two distinct CAF subpopulations in pancreatic tumor-fibroblast co-cultures was performed in both murine and patient derived cell populations embedded in Matrigel™, with one CAF population remaining nearby the tumor cells expressing increased alpha-SMA and a more distant CAF population secreting IL-6 and other inflammatory molecules [96]. A similar study utilized tumor-stromal Matrigel™ cultures to identify TGF-β and IL-1 were secreted by tumor cells to promote differentiation of PSCs into multiple subtypes, with TGF-β downregulating IL1R1 expression to form myofibroblasts and IL-1 activating JAK-STAT signaling to form inflammatory CAFs [97]. A different study analyzing the relationship between CAFs and pancreatic tumor cels on a Matrigel™ scaffold for chemoresistance determined fibroblasts induced increased treatment resistance to oxaliplatin and benzoporphyrin derivative mediated photodynamic treatment, and this resistance could be overcome by adding metformin to the treatment regimen [98]. Utilizing a variety of primary tumor cell sources including resections, ascites and rapid biopsies, Matrigel™ organoids were developed utilizing patient derived tumor cells, CAFs and sometimes matched T-cells; the system was shown to maintain viability and would reorganize into physiologically relevant tumor organization, gaining chemoresistance to gemcitabine as compared to any condition alone [99]. One study created aggregations of tumor cells and PSCs to demonstrate a correlation between the increased collagen I deposition and drug resistance by displaying a decrease in drug penetration in co-cultured conditions [100]. Another study comprehensively analyzed the differences in 2D and 3D normal and cancer fibroblast cultured with tumor cells in rotating vessel aggregates cultures [101]. Expression of several genes such as multiple collagens, MMPs, versican and periostin were found to be highest in CAF co-culture with tumor cells, while there was also a significant increase in ECM deposition in the CAF co-cultures. Finally, a study using collagen encapsulated cells in a microfluidic device analyzed the relationship between pancreatic tumor cells and hypovascularity [102]. The study determined pancreatic cells invaded nearby endothelial lumen structures and would ablate the cells through ALK-7 mediated pathways, leaving behind tumor cells invaded into the, now empty, luminal space.
2.11 Prostate cancer
Prostate cancer is one of most diagnosed cancers in men, with age highly correlated with diagnosis [77]. Despite excellent screening techniques, advanced prostate cancer remains a large cause of death due to metastasis to distant organs. Co-culture of bone marrow-derived stromal cells and prostate cancer cells has been shown to increase expression of ECM remodeling genes and chemo-attractive cytokines in stromal cells, suggesting the tumor cells were influencing the stromal cells to create a more favorable microenvironment for further growth and migration in rotating vessel derived aggregate cultures [103]. Microtissues of prostate cancer cells would be combined with CAFs in both collagen I and Matrigel™ cultures to analyze their co-relationship in the increased migration of tumor cells; efforts to prevent this determined that FAK inhibitors blocked tumor growth and migration [104]. Another group determined the co-culture of patient derived tumor cells with stromal cells in a Transwell-oriented organoid extended growth and maintenance of elevated AMACR expression, a noted prostate tumor biomarker, when compared to monoculture of patient derived tumor cells alone [105].
2.12 Other cancer types
Several studies have been performed analyzing the tumor microenvironment for other tumor origins. An early co-culture model of salivary gland adenoid cystic carcinoma cells and cancer associated fibroblasts placed into adjacent channels of a microfluidic device demonstrated an increased tumor cell migration, which could be blocked by using an MMP inhibitor [106]. Efforts to model the tumor microenvironment in brain metastases using aggregate culture has demonstrated that CAFs from brain metastases in breast tissue displayed elevated CXCL12 and CXCL16 chemokines as compared to primary tumor fibroblasts, and these fibroblasts promoted higher cancer migration [107]. Treatment with a CXCR4 or CXCL16 antibody reduced this tumor cell migration. Another group modeled bladder cancer cell invasion of nearby stromal and endothelial tissue in a multi-layered, tissue engineered construct resembling the healthy bladder, and demonstrate co-cultured tumor cells displayed increased chemoresistance [108]. Modeling of ovarian cancer-stromal interactions demonstrated the role of SNAI2 in activation of stromal fibroblasts, which in turn increased tumor cell growth [109]. Analysis of patient outcomes demonstrated high SNAI2 expression was linked to poorer outcomes.
2.13 Tumor microenvironment and immune cell modeling
Promising work has been performed recently to combine immune cells and tumor cells into tumor-immune hybrid systems to analyze the immune response in helping treatment. The effects of the chemical and mechanical microenvironment on immune cell activation in detecting tumor cells has been a topic of considerable focus [110]. Early work has focused on creating interactions between tumor cells and immune cells derived from cell lines, including natural killer cells [111] and T-cells [112,113,114], often through the creation of targetable proteins on the tumor cells through genetic engineering. More recently, we and other groups have worked to include primary patient immune and tumor cells into single constructs to study patient matched interactions. Models consisting of combinations of patient T-cells [94, 99, 115,116,117,118,119,120,121], natural killer cells [94, 115, 117], and B-cell populations [94, 117, 119, 122] have been recorded with preservation of each cell population. In particular, there has been significant effort focused on demonstrating interactions of patient matched cytotoxic T-cells and tumor cells in order to display evidence of direct T-cell mediated killing in vitro using anti-PD-1 [117, 119,120,121], PD-L1 [117], CTL4 [120], and MICA/B and NKG2A [115] therapies.
On the other hand, other models have focused on the anti-inflammatory effects of macrophages. M2 tumor associated macrophages (TAMs) have been studied in tumor and immune co-culture microfluidic devices for their effects on inflammation [123] and tumor cell migration [124]. A recent study with murine tumor organoids displayed rapid progression of the tumor cells into highly metastatic disease due to the increased presence of TAMs, including causing an inverse relationship of cytotoxic T-cell presence and activity [125].
3 Conclusions and future directions
Modeling the TME, using bioengineered model systems is a field still in its infancy. As more advanced tissue engineering techniques are developed with the potential for incorporating higher orders of tissue complexity, such as intact vasculature, immune fractions, and physiologically structured ECM, the predictive potential of TME bearing models may approach that of in vivo systems. Currently, most research focuses on a singular aspect of the TME in isolation, to study a specific interaction rather than the dynamics of the entire space. Models incorporating many components of the TME may elucidate new mechanisms behind cancer control leading to novel therapeutics which target the support system of a cancer rather than the cancer itself. These strategies could lead to a new landscape of cancer therapies which more directly target cancer cells and their stroma while sparing the body’s healthy tissues. Additionally, effort is being directed at methods to upscale the use of 3-D models due to its relatively low throughput. Traditional cell culture has a much higher throughput capacity than current organoid technology. However, advances in the area of bioprinting are making large scale organoid studies increasingly feasible. Different methods of bioprinting include inkjet, laser-based, and extrusion printing. Each one allows at least partial automation of the creation of the TME. With this increased throughput, it is likely that organoid culture will become increasingly prevalent in areas such as precision medicine and pharmaceutical testing, improving our ability to treat patients and develop new therapeutics.
References
Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125:5591–6.
Emon B, Bauer J, Jain Y, Jung B, Saif T. Biophysics of tumor microenvironment and cancer metastasis—a mini review. Comput Struct Biotechnol J. 2018;16:279–87.
Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–22.
Herrera-Perez RM, Voytik-Harbin SL, Sarkaria JN, Pollok KE, Fishel ML, Rickus JL. Presence of stromal cells in a bioengineered tumor microenvironment alters glioblastoma migration and response to STAT3 inhibition. PLoS One. 2018;13:e0194183.
Puca L, Bareja R, Prandi D, Shaw R, Benelli M, Karthaus WR, et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat Commun. 2018;9:2404.
Neesse A, Bauer CA, Öhlund D, Lauth M, Buchholz M, Michl P, et al. Stromal biology and therapy in pancreatic cancer: ready for clinical translation? Gut. 2019;68:159–71.
Belgodere JA, King CT, Bursavich JB, Burow ME, Martin EC, Jung JP. Engineering breast cancer microenvironments and 3D bioprinting. Front Bioeng Biotechnol. 2018;6:66.
Jagadeeshan S, Krishnamoorthy YR, Singhal M, Subramanian A, Mavuluri J, Lakshmi A, et al. Transcriptional regulation of fibronectin by p21-activated kinase-1 modulates pancreatic tumorigenesis. Oncogene. 2015;34:455–64.
Amrutkar M, Gladhaug IP. Pancreatic cancer chemoresistance to gemcitabine. Cancers (Basel). 2017;9:E157.
Walker C, Mojares E, Del Rio Hernandez A. Role of extracellular matrix in development and cancer progression. Int J Mol Sci. 2018;19:E3028.
Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4:38.
Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, et al. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11.
Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol. 2010;22:697–706.
Furuta S, Ren G, Mao JH, Bissell MJ. Laminin signals initiate the reciprocal loop that informs breast-specific gene expression and homeostasis by activating NO, p53 and microRNAs. Elife. 2018;7:e26148.
Kim BG, An HJ, Kang S, Choi YP, Gao MQ, Park H, et al. Laminin-332-rich tumor microenvironment for tumor invasion in the interface zone of breast cancer. Am J Pathol. 2011;178:373–81.
Han S, Khuri FR, Roman J. Fibronectin stimulates non-small cell lung carcinoma cell growth through activation of Akt/mammalian target of rapamycin/S6 kinase and inactivation of LKB1/AMP-activated protein kinase signal pathways. Cancer Res. 2006;66:315–23.
Deng Z, Cheng Z, Xiang X, Yan J, Zhuang X, Liu C, et al. Tumor cell cross talk with tumor-associated leukocytes leads to induction of tumor exosomal fibronectin and promotes tumor progression. Am J Pathol. 2012;180:390–8.
Yaqoob U, Cao S, Shergill U, Jagavelu K, Geng Z, Yin M, et al. Neuropilin-1 stimulates tumor growth by increasing fibronectin fibril assembly in the tumor microenvironment. Cancer Res. 2012;72:4047–59.
Shivashankar GV. Mechanosignaling to the cell nucleus and gene regulation. Annu Rev Biophys. 2011;40:361–78.
Peng Y, Chen Z, Chen Y, Li S, Jiang Y, Yang H, et al. ROCK isoforms differentially modulate cancer cell motility by mechanosensing the substrate stiffness. Acta Biomater. 2019;88:86–101.
Handorf AM, Zhou Y, Halanski MA, Li WJ. Tissue stiffness dictates development, homeostasis, and disease progression. Organogenesis. 2015;11:1–15.
Schedin P, Keely PJ. Mammary gland ECM remodeling, stiffness, and mechanosignaling in normal development and tumor progression. Cold Spring Harb Perspect Biol. 2011;3:a003228.
Chin L, Xia Y, Discher DE, Janmey PA. Mechanotransduction in cancer. Curr Opin Chem Eng. 2016;11:77–84.
Nagelkerke A, Bussink J, Mujcic H, Wouters BG, Lehmann S, Sweep FC, et al. Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res. 2013;15:R2.
Hockel M, Schlenger K, Aral B, Mitze M, Schaffer U, Vaupel P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996;56:4509–15.
Eguchi T, Sogawa C, Okusha Y, Uchibe K, Iinuma R, Ono K, et al. Organoids with cancer stem cell-like properties secrete exosomes and HSP90 in a 3D nanoenvironment. PLoS One. 2018;13:e0191109.
Sitkovsky MV, Kjaergaard J, Lukashev D, Ohta A. Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin Cancer Res. 2008;14:5947–52.
Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl). 2015;3:83–92.
Sang N, Stiehl DP, Bohensky J, Leshchinsky I, Srinivas V, Caro J. MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J Biol Chem. 2003;278:14013–9.
Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci U S A. 2012;109:2784–9.
Hubert CG, Rivera M, Spangler LC, Wu Q, Mack SC, Prager BC, et al. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016;76:2465–77.
Huang P, Xu X, Wang L, Zhu B, Wang X, Xia J. The role of EGF-EGFR signalling pathway in hepatocellular carcinoma inflammatory microenvironment. J Cell Mol Med. 2014;18:218–30.
Sasaki T, Hiroki K, Yamashita Y. The role of epidermal growth factor receptor in cancer metastasis and microenvironment. Biomed Res Int. 2013;2013:546318.
Saji H, Nakamura H, Awut I, Kawasaki N, Hagiwara M, Ogata A, et al. Significance of expression of TGF-beta in pulmonary metastasis in non-small cell lung cancer tissues. Ann Thorac Cardiovasc Surg. 2003;9:295–300.
Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–8.
Hawinkels LJ, Ten Dijke P. Exploring anti-TGF-beta therapies in cancer and fibrosis. Growth Factors. 2011;29:140–52.
Krstić J, Trivanović D, Mojsilović S, Santibanez JF. Transforming growth factor-beta and oxidative stress interplay: implications in tumorigenesis and cancer progression. Oxid Med Cell Longev. 2015;2015:654594.
Soker S, Fidder H, Neufeld G, Klagsbrun M. Characterization of novel vascular endothelial growth factor (VEGF) receptors on tumor cells that bind VEGF165 via its exon 7-encoded domain. J Biol Chem. 1996;271:5761–7.
Klagsbrun M, Soker S. VEGF/VPF: the angiogenesis factor found? Curr Biol. 1993;3:699–702.
Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23:1011–27.
Timke C, Zieher H, Roth A, Hauser K, Lipson KE, Weber KJ, et al. Combination of vascular endothelial growth factor receptor/platelet-derived growth factor receptor inhibition markedly improves radiation tumor therapy. Clin Cancer Res. 2008;14:2210–9.
Eskens FA, Verweij J. The clinical toxicity profile of vascular endothelial growth factor (VEGF) and vascular endothelial growth factor receptor (VEGFR) targeting angiogenesis inhibitors; a review. Eur J Cancer. 2006;42:3127–39.
Anderberg C, Li H, Fredriksson L, Andrae J, Betsholtz C, Li X, et al. Paracrine signaling by platelet-derived growth factor-CC promotes tumor growth by recruitment of cancer-associated fibroblasts. Cancer Res. 2009;69:369–78.
Korc M, Friesel RE. The role of fibroblast growth factors in tumor growth. Curr Cancer Drug Targets. 2009;9:639–51.
Murakami M, Simons M. Fibroblast growth factor regulation of neovascularization. Curr Opin Hematol. 2008;15:215–20.
Renehan AG, Zwahlen M, Minder C, O'Dwyer ST, Shalet SM, Egger M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: systematic review and meta-regression analysis. Lancet. 2004;363:1346–53.
Natsuizaka M, Ohashi S, Wong GS, Ahmadi A, Kalman RA, Budo D, et al. Insulin-like growth factor-binding protein-3 promotes transforming growth factor-{beta}1-mediated epithelial-to-mesenchymal transition and motility in transformed human esophageal cells. Carcinogenesis. 2010;31:1344–53.
Graham TR, Zhau HE, Odero-Marah VA, Osunkoya AO, Kimbro KS, Tighiouart M, et al. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008;68:2479–88.
Welner RS, Amabile G, Bararia D, Czibere A, Yang H, Zhang H, et al. Treatment of chronic myelogenous leukemia by blocking cytokine alterations found in normal stem and progenitor cells. Cancer Cell. 2015;27:671–81.
Parameswaran N, Patial S. Tumor necrosis factor-α signaling in macrophages. Crit Rev Eukaryot Gene Expr. 2010;20:87–103.
Sheen-Chen SM, Chen WJ, Eng HL, Chou FF. Serum concentration of tumor necrosis factor in patients with breast cancer. Breast Cancer Res Treat. 1997;43:211–5.
Chaudhary B, Elkord E. Regulatory T cells in the tumor microenvironment and cancer progression: role and therapeutic targeting. Vaccines (Basel). 2016;4:E28.
Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol. 2019;12:86.
Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 2016;30:1002–19.
Dudley AC. Tumor endothelial cells. Cold Spring Harb Perspect Med. 2012;2:a006536.
Hida K, Maishi N, Annan DA, Hida Y. Contribution of tumor endothelial cells in cancer progression. Int J Mol Sci. 2018;19:E1272.
Lin PP, Gires O, Wang DD, Li L, Wang H. Comprehensive in situ co-detection of aneuploid circulating endothelial and tumor cells. Sci Rep. 2017;7:9789.
Hida K, Hida Y, Amin DN, Flint AF, Panigrahy D, Morton CC, et al. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 2004;64:8249–55.
Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006;25:315–22.
Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127:759–67.
Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019;40:310–27.
Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med. 2010;362:875–85.
Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019;79:4557–66.
Almatroodi SA, McDonald CF, Darby IA, Pouniotis DS. Characterization of M1/M2 tumour-associated macrophages (TAMs) and Th1/Th2 cytokine profiles in patients with NSCLC. Cancer Microenviron. 2016;9:1–11.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55.
Horwitz DA, Zheng SG, Wang J, Gray JD. Critical role of IL-2 and TGF-beta in generation, function and stabilization of Foxp3+CD4+ Treg. Eur J Immunol. 2008;38:912–5.
Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109–18.
Wang YA, Li XL, Mo YZ, Fan CM, Tang L, Xiong F, et al. Effects of tumor metabolic microenvironment on regulatory T cells. Mol Cancer. 2018;17:168.
Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–32.
Khazaie K, von Boehmer H. The impact of CD4+CD25+ Treg on tumor specific CD8+ T cell cytotoxicity and cancer. Semin Cancer Biol. 2006;16:124–36.
Sato T, Terai M, Tamura Y, Alexeev V, Mastrangelo MJ, Selvan SR. Interleukin 10 in the tumor microenvironment: a target for anticancer immunotherapy. Immunol Res. 2011;51:170–82.
Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. 2007;27:635–46.
Walker LS. Treg and CTLA-4: two intertwining pathways to immune tolerance. J Autoimmun. 2013;45:49–57.
Kim HN, Habbit NL, Su CY, Choi N, Ahn EH, Lipke EA, et al. Microphysiological systems as enabling tools for modeling complexity in the tumor microenvironment and accelerating cancer drug development. Adv Funct Mater. 2019;29:1807553.
Asghar W, El Assal R, Shafiee H, Pitteri S, Paulmurugan R, Demirci U. Engineering cancer microenvironments for in vitro 3-D tumor models. Mater Today (Kidlington). 2015;18:539–53.
Lancaster MA, Knoblich JA. Organogenesis in a dish: modeling development and disease using organoid technologies. Science. 2014;345:1247125.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34.
Shirure VS, Bi Y, Curtis MB, Lezia A, Goedegebuure MM, Goedegebuure SP, et al. Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids. Lab Chip. 2018;18:3687–702.
Pradhan S, Smith AM, Garson CJ, Hassani I, Seeto WJ, Pant K, et al. A microvascularized tumor-mimetic platform for assessing anti-cancer drug efficacy. Sci Rep. 2018;8:3171.
Nashimoto Y, Okada R, Hanada S, Arima Y, Nishiyama K, Miura T, et al. Vascularized cancer on a chip: the effect of perfusion on growth and drug delivery of tumor spheroid. Biomaterials. 2019;229:119547.
Corsa CA, Brenot A, Grither WR, Van Hove S, Loza AJ, Zhang K, et al. The action of discoidin domain receptor 2 in basal tumor cells and stromal cancer-associated fibroblasts is critical for breast cancer metastasis. Cell Rep. 2016;15:2510–23.
Goreczny GJ, Ouderkirk-Pecone JL, Olson EC, Krendel M, Turner CE. Hic-5 remodeling of the stromal matrix promotes breast tumor progression. Oncogene. 2017;36:2693–703.
Mazio C, Casale C, Imparato G, Urciuolo F, Netti PA. Recapitulating spatiotemporal tumor heterogeneity in vitro through engineered breast cancer microtissues. Acta Biomater. 2018;73:236–49.
Devarasetty M, Wang E, Soker S, Skardal A. Mesenchymal stem cells support growth and organization of host-liver colorectal-tumor organoids and possibly resistance to chemotherapy. Biofabrication. 2017;9:021002.
Devarasetty M, Skardal A, Cowdrick K, Marini F, Soker S. Bioengineered submucosal organoids for in vitro modeling of colorectal cancer. Tissue Eng Part A. 2017;23:1026–41.
Jeong SY, Lee JH, Shin Y, Chung S, Kuh HJ. Co-culture of tumor spheroids and fibroblasts in a collagen matrix-incorporated microfluidic chip mimics reciprocal activation in solid tumor microenvironment. PLoS One. 2016;11:e0159013.
Zhang CY, Yuan WG, He P, Lei JH, Wang CX. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J Gastroenterol. 2016;22:10512–22.
Chen Y, Sun W, Kang L, Wang Y, Zhang M, Zhang H, et al. Microfluidic co-culture of liver tumor spheroids with stellate cells for the investigation of drug resistance and intercellular interactions. Analyst. 2019;144:4233–40.
Khawar IA, Park JK, Jung ES, Lee MA, Chang S, Kuh HJ. Three dimensional mixed-cell spheroids mimic stroma-mediated chemoresistance and invasive migration in hepatocellular carcinoma. Neoplasia. 2018;20:800–12.
Song Y, Kim SH, Kim KM, Choi EK, Kim J, Seo HR. Activated hepatic stellate cells play pivotal roles in hepatocellular carcinoma cell chemoresistance and migration in multicellular tumor spheroids. Sci Rep. 2016;6:36750.
Nakamura H, Sugano M, Miyashita T, Hashimoto H, Ochiai A, Suzuki K, Tsuboi M, et al. Organoid culture containing cancer cells and stromal cells reveals that podoplanin-positive cancer-associated fibroblasts enhance proliferation of lung cancer cells. Lung Cancer. 2019;134:100–107.
Chen S, Giannakou A, Wyman S, Gruzas J, Golas J, Zhong W, et al. Cancer-associated fibroblasts suppress SOX2-induced dysplasia in a lung squamous cancer coculture. Proc Natl Acad Sci U S A. 2018;115:E11671–80.
Yu T, Guo Z, Fan H, Song J, Liu Y, Gao Z, Wang Q. Cancer-associated fibroblasts promote non-small cell lung cancer cell invasion by upregulation of glucose-regulated protein 78 (GRP78) expression in an integrated bionic microfluidic device. Oncotarget. 2016;7:25593–603.
Ramamoorthy P, Thomas SM, Kaushik G, Subramaniam D, Chastain KM, Dhar A, et al. Metastatic tumor-in-a-dish, a novel multicellular organoid to study lung colonization and predict therapeutic response. Cancer Res. 2019;79:1681–95.
Weniger M, Honselmann KC, Liss AS. The extracellular matrix and pancreatic cancer: a complex relationship. Cancers (Basel). 2018;10:E316.
Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214:579–96.
Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9:282–301.
Broekgaarden M, Anbil S, Bulin AL, Obaid G, Mai Z, Baglo Y, et al. Modulation of redox metabolism negates cancer-associated fibroblasts-induced treatment resistance in a heterotypic 3D culture platform of pancreatic cancer. Biomaterials. 2019;222:119421.
Tsai S, McOlash L, Palen K, Johnson B, Duris C, Yang Q, et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer. 2018;18:335.
Ware MJ, Keshishian V, Law JJ, Ho JC, Favela CA, Rees P, et al. Generation of an in vitro 3D PDAC stroma rich spheroid model. Biomaterials. 2016;108:129–42.
Brancato V, Comunanza V, Imparato G, Corà D, Urciuolo F, Noghero A, et al. Bioengineered tumoral microtissues recapitulate desmoplastic reaction of pancreatic cancer. Acta Biomater. 2017;49:152–66.
Nguyen DT, Lee E, Alimperti S, Norgard RJ, Wong A, Lee JJ, et al. A biomimetic pancreatic cancer on-chip reveals endothelial ablation via ALK7 signaling. Sci Adv. 2019;5:eaav6789.
Sung SY, Hsieh CL, Law A, Zhau HE, Pathak S, Multani AS, et al. Coevolution of prostate cancer and bone stroma in three-dimensional coculture: implications for cancer growth and metastasis. Cancer Res. 2008;68:9996–10003.
Akerfelt M, Bayramoglu N, Robinson S, Toriseva M, Schukov HP, Härmä V, et al. Automated tracking of tumor-stroma morphology in microtissues identifies functional targets within the tumor microenvironment for therapeutic intervention. Oncotarget. 2015;6:30035–56.
Richards Z, McCray T, Marsili J, Zenner ML, Manlucu JT, Garcia J, et al. Prostate stroma increases the viability and maintains the branching phenotype of human prostate organoids. iScience. 2019;12:304–17.
Liu T, Lin B, Qin J. Carcinoma-associated fibroblasts promoted tumor spheroid invasion on a microfluidic 3D co-culture device. Lab Chip. 2010;10:1671–7.
Chung B, Esmaeili AA, Gopalakrishna-Pillai S, Murad JP, Andersen ES, Kumar Reddy N, et al. Human brain metastatic stroma attracts breast cancer cells via chemokines CXCL16 and CXCL12. NPJ Breast Cancer. 2017;3:6.
Ringuette Goulet C, Bernard G, Chabaud S, Couture A, Langlois A, Neveu B, et al. Tissue-engineered human 3D model of bladder cancer for invasion study and drug discovery. Biomaterials. 2017;145:233–41.
Yang Z, Yang X, Xu S, Jin P, Li X, Wei X, et al. Reprogramming of stromal fibroblasts by SNAI2 contributes to tumor desmoplasia and ovarian cancer progression. Mol Cancer. 2017;16:163.
Kim JK, Shin YJ, Ha LJ, Kim DH, Kim DH. Unraveling the mechanobiology of the immune system. Adv Healthc Mater. 2019;8:e1801332.
Christakou AE, Ohlin M, Önfelt B, Wiklund M. Ultrasonic three-dimensional on-chip cell culture for dynamic studies of tumor immune surveillance by natural killer cells. Lab Chip. 2015;15:3222–31.
Pavesi A, Tan AT, Koh S, Chia A, Colombo M, Antonecchia E, et al. A 3D microfluidic model for preclinical evaluation of TCR-engineered T cells against solid tumors. JCI Insight. 2017;2:89762.
Schnalzger TE, et al. 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J. 2019;38:e100928.
Varesano S, Zocchi MR, Poggi A. Zoledronate triggers Vδ2 T cells to destroy and kill spheroids of colon carcinoma: quantitative image analysis of three-dimensional cultures. Front Immunol. 2018;9:998.
Courau T, Bonnereau J, Chicoteau J, Bottois H, Remark R, Assante Miranda L, et al. Cocultures of human colorectal tumor spheroids with immune cells reveal the therapeutic potential of MICA/B and NKG2A targeting for cancer treatment. J Immunother Cancer. 2019;7:74.
Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, et al. Generation of tumor-reactive t cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell. 2018;174:1586–98.e12.
Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, et al. Organoid modeling of the tumor immune microenvironment. Cell. 2018;175:1972–88.e16.
Finnberg NK, Gokare P, Lev A, Grivennikov SI, MacFarlane AW 4th, Campbell KS, et al. Application of 3D tumoroid systems to define immune and cytotoxic therapeutic responses based on tumoroid and tissue slice culture molecular signatures. Oncotarget. 2017;8:66747–57.
Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, et al. Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov. 2018;8:196–215.
Aref AR, Campisi M, Ivanova E, Portell A, Larios D, Piel BP, et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip. 2018;18:3129–43.
Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 2018;8:216–33.
Ou Z, Wang Y, Liu L, Li L, Yeh S, Qi L, et al. Tumor microenvironment B cells increase bladder cancer metastasis via modulation of the IL-8/androgen receptor (AR)/MMPs signals. Oncotarget. 2015;6:26065–78.
Bai J, Adriani G, Dang TM, Tu TY, Penny HX, Wong SC, et al. Contact-dependent carcinoma aggregate dispersion by M2a macrophages via ICAM-1 and β2 integrin interactions. Oncotarget. 2015;6:25295–307.
Li R, Hebert JD, Lee TA, Xing H, Boussommier-Calleja A, Hynes RO, et al. Macrophage-secreted TNFα and TGFβ1 influence migration speed and persistence of cancer cells in 3D tissue culture via independent pathways. Cancer Res. 2017;77:279–90.
Filippini D, Agosto S, Delfino P, Simbolo M, Piro G, Rusev B, et al. Immunoevolution of mouse pancreatic organoid isografts from preinvasive to metastatic disease. Sci Rep. 2019;9:12286.
Acknowledgement
This research was supported in part by the National Institutes of Health (R33CA202822).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts to disclose.
Ethical statement
There are no animal experiments carried out for this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Devarasetty, M., Forsythe, S.D., Shelkey, E. et al. In Vitro Modeling of the Tumor Microenvironment in Tumor Organoids. Tissue Eng Regen Med 17, 759–771 (2020). https://doi.org/10.1007/s13770-020-00258-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13770-020-00258-4