
Abstract
Background:
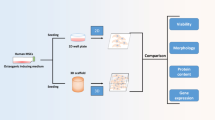
Tissue engineering is a multidisciplinary field which attracted much attention in recent years. One of the most important issue in tissue engineering is how to obtain high cell numbers and tissue regeneration while maintaining appropriate cellular characteristics in vitro for restoring damaged or dysfunctional body tissues and organs. These demands can be achieved by the use of three dimensional (3D) dynamic cultures of cells combined with cell-adhesive micro-carriers.
Method:
In this study, human mesenchymal stem cells (hMSCs) were cultured in a wave-bioreactor system for up to 100 days, after seeding on Cultisphere-S porous gelatin micro-carriers. Cell counting was performed at the time points of 7, 12, 17, 31 days and compared to those of hMSCs cultured under static condition. Higher growth and proliferation rates was achieved in wave-type dynamic culture, when cell culture continued to day 31. A scanning electron microscope (SEM) photographs, both live and dead and MTT assays were taken to confirm the survival and distribution of cells on porous gelatin micro-carrier surfaces. The results of histological stains such as hematoxylin and eosin, Masson’s trichrome, Alcian blue and Alizarin red S also showed improved proliferation and tissue regeneration of hMSCs on porous gelatin micro-carriers.
Conclusion:
The experimental results demonstrated the effect and importance of both micro-carriers and bioreactor in hMSC expansion on cell proliferation and migration as well as extracellular matrix formation on the superficial and pore surfaces of the porous gelatin micro-carriers, and then their inter-connections, leading to tissue regeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
During last decades, tissue engineering, an interdisciplinary field relating to the fabrication and implantation of engineered tissues into the body, has been considered as a cutting-edge technology in aged society. This technology has showed great promises in replacing the damaged or diseased organs/tissues by functional tissue substitutes which play roles in a variety of operations relating to structures, barrier and transport or biochemistry and bioactive molecules secretion in tissues such as bone, cartilage, skin, blood vessels, liver and pancreas [1, 2]. In tissue engineering, mesenchymal stem cells (MSCs) and their derived products have showed impressive progress with multiple clinical trials targeting on modulation of immune responses, cell therapy, drug screening and regenerations of bone, cartilage, myocardia, and diseased tissues. The better tissue regeneration it has, the higher cell number requires. However, traditional 2-dimensional (2-D) tissue culture platform can just be used when the number of cells needed is low, and it becomes impractical when doses of cells needed are above 50 million. That means that extended expansion of MSCs in 2-D plate culture does not produce sufficient numbers of cells for therapeutic and tissue engineering applications, thus leading to not only their senescence but also loss of multi-potency [3]. So Large-scale expansion of stem cells is required to generate homogeneous stem cells with biological characteristics for clinical applications.
Therefore, it is a driving force for researchers to develop 3 dimensional suspension cultures, with the use of both cell-adhesive micro-carriers and bioreactors for generating stem cells. Micro-carriers were firstly introduced in 1967 by Van Wezel [4], and have been applied successfully in culture for growing primary cells as well as anchorage-dependent cell lines for various purposes such as the production of vaccines or pharmaceuticals or cell population expansion [5]. The advantage of micro-carriers is supplying a large surface area for a given bioreactor volume, especially porous micro-carriers (e.g. 30 cm2 in 1 mL using 5 mg Cytodex 1) without having to resort to bulky equipment and tedious methodology. By providing surface for attachment, the micro-carriers induce anchorage-dependent cells to be cultivated in dynamic conditions such as stirred or wave bioreactors for a large-scale. Applying porous gelatin micro-carriers in suspension culture such as fluidized or packed bed systems, yields of cell culture are reported to be probably up to 200 million cells per milliliter. As a result, for a given quantity of (stem) cells or their products, in vitro micro-carrier cell cultures use much less space than other types of 2-D monolayer culture but generate out higher production capacity of cells. Undifferentiated MSCs were seeded onto the porous gelatin micro-carriers, expanded further, and differentiated into different cell types. Together with the demands of using micro-carriers, a variety of commercial micro-carrier types for cell expansion have been introduced with the differences in chemical composition, charge, surface coatings and porosity [6]. Micro-carriers are particles usually with diameters of 100–250 µm, usually fabricated from cellulose, glass, plastic, or polyester [7]. Recently, facilitated expansion of MSCs was obtained by micro-carriers in a suspension bioreactor culture system, that require attachment, and to provide an environment that can be easily controlled and monitored [3, 8]. Micro-carriers developed for culturing surface-adherent cell lines in suspension have been used mainly in vaccine production and research purposes [9,10,11,12,13,14,15,16].
By using a porous gelatin micro-carriers—Cultispher-S particles in a wave-bioreactor system, the in vitro culture could be controlled for several parameters, such as temperature, pH and dissolved oxygen concentrations and rocking speed to optimize the human MSCs expansion. In a wave bioreactor system, there are two necessary elements required that are a wave bioreactor and a chamber for cell culture, the so-called cell bag. The waving agitation can keep the cells and the carriers in the culture in suspension. Besides, strict controls of culture conditions (e.g. pH, gas tensions and etc.) allows homogeneous culture systems with diverse process designs [17]. In additions, monitoring and sampling micro-carrier cultures become simpler and easier, medium amount used for the culture is probably lower than other technique in producing higher numbers of anchorage-dependent cells. These help to save cell culture costs [17], particularly when expensive fetal calf serum are required. Moreover, cells can be protected against physical and chemical stress, fewer culture vessels are required with micro-carrier cultures and in the term of contamination, the fewer number of handling steps in the micro-carrier cultures, the less risk of contamination is found. Finally, it has been considered that wave-bioreactor systems could be excellent choice for cultures of diverse cell types, including stem cells [18].
In this study, we tried to investigate the effects of micro-carrier culture in a rocking system on the expansion of hMSCs and their tissue regeneration of micro-carriers. Following that, the influence or applicable feasibility of porous gelatin micro-carrier culture in wave bioreactor system on the proliferation and differentiation of hMSCs compared to traditional culture system was evaluated. The employed gelatin micro-carrier (Cultispher-S) were commercially available and have been used for cultivation of anchorage-dependent cells and also for expansion and differentiation of many stem cells. Diverse histological evaluations such as hematoxylin and eosin (H&E), Masson’s trichrome (MT), Alizarin red S, Alcian blue and alkaline phosphatase (ALP) staining methods were performed to identify not only MSCs proliferation but also tissue regeneration from the micro-carriers.
2 Materials and methods
2.1 Materials
Minimum essential medium-α (MEM-α) and fetal bovine serum (FBS) were purchased from Gibco BRL (Thermo Fisher Scientific, Seoul, Korea), while penicillin–streptomycin was purchased from Lonza Korea (Seoul, Korea). Cell counting kit-8 (CCK-8) solution was bought from Dojindo Laboratories (Kumamoto, Japan) through Biomax Co., Ltd. (Seoul, Korea). Live and dead viability/cytotoxicity kit for mammalian cells and thiazolyl blue tetrazolium bromide (MTT; 3-(4,5-dimethylthiazol-2-yil)-2,5-diphenyltetra-zolium) were respectively supplied by Invitrogen (Thermo Fisher Scientific, Seoul, Korea) and Roche (Roche Korea Co., Seoul, Korea), whereas all staining reagents and gelatin Cultispher-S micro-carriers (the density of 1.02–1.04 g/cm3 at 25 °C, diameter of 130–380 μm, no charge) were purchased from Sigma Aldrich Co. (ST. Louis, MO, USA). Senso Lyte assay kit was obtained from AnaSpec (Fremont, CA, USA) through Bio-Medical Science Co. (Seoul, Korea). Radioimmunoprecipitation assay (RIPA) buffer, proteases and phosphatase inhibitors were purchased from Sigma Aldrich (ST. Louis, MO, USA). Primary antibodies and HRP conjugated secondary antibody were bought from Abcam (Cambridge, United Kingdom). All chemicals were employed as received.
2.2 In vitro hMSCs static culture
Isolated hMSCs (donated by professor Sungwon Kim in the Otirhinolaryngology Department, The Catholic University of Korea Seoul St. Mary’s Hospital, Seoul, Korea) were in vitro seeded at a density of 5000 cells per cm2 in 10-cm petri dish and cultured in α-MEM medium supplemented with 10% FBS, 1% penicillin–streptomycin (PS). When reaching 80% confluence, cells were trypsinized and subcultured one more time at the same cell density. Cells were fed every 2–3 day by replacing 100% of the culture medium with fresh medium (α-MEM with 10% FBS and 1% PS). The cell cultures were performed at 37 °C in a humidified atmosphere with 5% CO2. In order to prepare static in vitro culture as a control of dynamic culture, 2000 cells were seeded in tissue culture petridish and cultured for 30 days.
2.3 hMSCs seeding onto gelatin micro-carriers for dynamic culture
2.3.1 Preparation of gelatin micro-carriers
Gelatin Cultispher-S MCs (0.1 g) were prepared according to the manufacturer’s instructions. In brief, dry gelatin micro-carriers were swollen and hydrated in calcium and magnesium free PBS (50 mL/g dry bead) for 2 h at room temperature, followed by autoclaving without removing PBS for sterilization the beads. After that, PBS was discarded and new PBS was added mix for washing. Prior to seeding cells, micro-carriers were washed twice with culture medium.
2.3.2 Dynamic MSC in vitro culture on micro-carrier with wave-type bioreactor
In order to cultivate cells on the MCs, hMSCs were harvested from tissue culture dishes and combined with prepared Cultispher-S micro-carriers in a 2000 mL polypropylene cell culture bag in a wave type bioreactor (Rocking Incubator, NB-207XL, N-Biotek; Bucheon, Kyunggi-Do, Korea). 1000 mL cell bag was supported from Greenpia Technology (Seoul, Korea). In details, the 0.1 g of prepared Cultispher-S beads were mixed with 1 × 107 hMSC cells in 50 mL pre-warmed α-MEM medium with 10% FBS and 1% PS. The bioreactor functions as an incubator to maintain the culture system at the temperature of 37 °C, 5% CO2, and a hypoxic condition with the oxygen tension at 5% by keeping supplying nitrogen. Besides the incubating role, in the bioreactor, wave-generating agitation is promoted. And the wave-type bioreactor system was controlled at the wave speed ranges of 5–35 rpm (Table 1) depending on stages of dynamic cell cultures. On the first day, the culture was agitated gently by wave at 15 rpm. On the second day, 50 mL of α-MEM medium with 10% FBS and 1% PS was added, and the rocking speed was increased to 30 rpm. On the third day, another 100 mL of medium was supplied into the culture bag to a final volume of 200 mL culture medium. The condition for all cultivating process was at 37 °C, 5% CO2 and 5% O2. The interval of 50% medium exchange in culture was every 2–3 day. A certain amount (3 mL) of cell suspension were taken out from the cell culture bag every other day and analyzed for cell proliferation, viability, characteristics.
2.4 Cell counting
Triplicate samples of evenly 1 mL of cell/micro-carriers suspension were collected from the culture bag at 1, 7, 12, 15, 18, 22, 26, 30, 40, 60 and 90 days and employed for evaluation of the cell expansion in micro-carrier culture in the bioreactor for 90 days. The culture lasted for up to 100 days. After settling down cell/bead complexes, 800 μL of supernatant medium from the total 1 mL of each sample was discarded, followed by the addition of 800 μL of 1 mg/mL collagenase solution and incubation at 37 °C. After 30 min of incubation, the samples were centrifuged at 1300 rpm for 5 min and then the removal of 700 μL of supernatant was performed right after. 700 μL of fresh medium was added and mixed for a proper suspension of solution to inactivate the activity of collagenase. Viable and dead cells were observed by using the test of trypan blue dye exclusion on a hemocytometer.
2.5 Scanning electron microscope
For observing cells attached on micro-carrier, the medium in 500 μL of hMSC/micro-carrier suspension was taken from the culture bag at day 100, washed with PBS and followed by fixed in 2.5% (v/v) glutaraldehyde (Merck, Darmstadt, Germany) for 24 h at room temperature in the dark. The fresh micro-carriers without cells were employed as a control. The samples were then washed and immersed in 1% OsO4 for 1 h at room temperature. After washing out the OsO4 once with PBS, samples were further incubated in fresh PBS for 30 min more. The following procedures were performed such as removing the PBS, pre-freezing the samples at − 80 °C and drying the samples in the lyophilizer in those sequence. The dried samples were coated with gold for 1 min and examined with a scanning electron microscope (SEM) TESCAN VEGA3 (Kohoutovice, Czech Republic).
2.6 Live and dead assay
Cell viability on micro-carriers was evaluated by the live and dead assay after in vitro cell culture for 20, 35, 60 and 100 days. Live and dead viability/cytotoxicity assay for mammalian cells was processed according to the protocol suggested by the vendor (Invitrogen, Carlsbad, CA, USA). 1 mL of cell/MCs suspension was obtained from the operating cell culture bag. Cell/micro-carriers were settled at the bottom of the microtube and 600 μL medium was removed. Two times of PBS washing was employed, and then the assay solutions that was composed of 1.2 μL of 2 mM ethidium homodimer-1 and 0.3 μL of 4 mM calcein AM (dead and live stains, respectively) in 600 μL PBS. Cell viability in micro-carrier culture was observed by a fluorescence microscope (Leica DMLB, Wetzlar, Germany) after 30 min incubation in 37 °C CO2 incubator.
2.7 Metabolic activity by MTT staining
Cell viability on Cultispher-S micro-carriers was detected by MTT assay. 400 μL of cultured cell/bead suspension sample was mixed with 40 μL of 5 mg/mL MTT solution and incubated for 45 min at 37 °C. MTT is cleaved by an enzyme in the respiration chain in the mitochondria in viable cells when cells are metabolically active, generating MTT formazan, a dark blue compound, visible by optical microscopy (CKX41; Olympus-Korea, Seoul, Korea). The samples were run in triplicates.
2.8 Hematoxylin and eosin stain
500 μL of hMSC/micro-carrier suspension was substrated from cell culture bag and immersed in 10% neutral-buffered formalin (Sigma) for 30 min, and then washed with PBS for 3 times. The samples were stained with H&E solution by soaking in 1 mL of Mayor’s hematoxylin stain solution for 10 min, washing for 1 min in 1 mL of PBS, and then staining with the eosin stain solution for 5 min. The stained samples were visualized with a light microscopy (CKX41; Olympus-Korea, Seoul, Korea).
2.9 Masson’s trichrome stain
After washing 500 μL hMSC/micro-carriers suspension twice with PBS, cells were fixed in 10% neutral buffered formalin for 30 min at room temperature in the dark. Subsequent to fixation, cells/micro-carrier samples were washed three times with Ca2+ and Mg2+ free PBS and stained with Bouin’s fixative solution for 10 min, followed by a 2-time washing with PBS. Following that, Weigert’s Hematoxilin mixture for 5 min, Biebrich scarlet-acid fuchsin solution for 5 min, phosphotungstic/phosphomolybdic acid solution for 5 min and aniline blue solution for 3 min were respectively used to stain the samples. The intervals of stains were washing the samples with PBS twice. Prior to observing under the microscope, the stained sample was immersed in 1% acetic acid for 2 min followed by soaked in distilled water and then observed under a light microscope (CKX41; Olympus-Korea, Seoul, Korea).
2.10 Alcian blue stain
For chondrogenic differentiation detection, the medium in 500 μL of hMSC/MC suspension was taken from culture bag, and then hMSC/MC complexes were washed twice in PBS and followed by fixed in neutral buffered formalin (10%) for 30 min at room temperature in the dark. After fixed, hMSC/MC sample was washed three times with Ca2+ and Mg2+ free PBS and 1% (w/v) Alcian blue solution was added and incubated at room temperature for 1 h. The samples were then washed with PBS once before stained with Nucleus fast red for 5 min. Prior to observing under the microscope, the stained sample was washed twice in PBS and then observed under a light microscope (CKX41; Olympus-Korea, Seoul, Korea).
2.11 Alkaline phosphatase (ALP) stain
The osteoblastic tissue regeneration of hMSCs was assessed by ALP stain. Samples of micro-carriers containing hMSCs were washed with PBS and fixed in 10% neutral-buffered formalin (Sigma) for 1 min. After fixed, the formalin was aspirated and cells were washed using PBS. Following the washing step, the cells were incubated in 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (BCIP/NBT) substrate solution in dark at room temperature for 10 min. The BCIP/NBT substrate solution was prepared as the instruction of the manufacturer by dissolving one BCIP/NBT tablet (SigmaFast™ BCIP-NBT; Sigma-Aldrich, St. Louis, MO, USA) in 10 mL distilled water. The substrate solution was then removed and cells/beads sample were washed with washing buffer (0.05% Tween 20—including PBS). After discarding the washing buffer, the obtained samples observed under an optical microscope (CKX41; Olympus-Korea, Seoul, Korea).
2.12 Alizarin red S stain
For evaluation of tissue regeneration of hMSCs in the micro-carrier culture, Alizarin red S staining was employed. The medium in 500 μL of hMSC/micro-carrier suspension was removed and cell/micro-carrier complexes were washed twice in PBS and followed by fixed in neutral buffered formalin (10%) for 30 min at room temperature in the dark. After fixed, cell/micro-carrier complex sample was washed 3 times with Ca2+ and Mg2+ free PBS. 2% (w/v) Alizarin red S solution was added and incubated at room temperature for 45 min. The staining solution was then aspirated and the samples were washed with PBS four times with PBS. The stained sample was then observed under a light microscope (CKX41; Olympus-Korea, Seoul, Korea).
2.13 Statistical analysis
Data are reported as mean ± standard deviation. Statistical significance was assessed with one-way ANOVA of the SPSS 14.0 program (ver. 14.0, SPSS Inc., Chicago, IL, USA). The values were considered as significantly different when p < 0.05.
3 Results
3.1 Cell proliferation rate of dynamic culture
hMSC was cultured for 100 days with a 0.1 g Cultispher-S micro-carrier in a 5% O2, 5% CO2, and 36 °C hypoxia condition using a rocking incubator equipped with a stirrer inside. In the closed inner chamber of the rocking incubator, the cell bag can be attached to the oscillating device of up and down reciprocating vibration type, the stirring speed can be controlled, and the composition of the inside of the chamber can be set by supplying CO2 and N2 gas.
The cell counting was applied to evaluate the effect of dynamic culture on cell proliferation using a bioreactor versus static in vitro culture in a general incubator within 30 days. In static in vitro cultures, 2000 hMSCs seeded in 10 cm diameter tissue culture petridish were measured at the time points of 7, 12, 17 and 30 days and then proliferated to 885.3 ± 35% proliferated for 30 days compared to those of the first day (Fig. 1A).

In vitro proliferation of hMSC cells. A Comparison of static and dynamic culture in 30 days. In static in vitro culture system, cells grew on 10 cm tissue culture petri dish surface at 37 °C, 5% CO2, 20% O2. In dynamic in vitro culture system, cells proliferated on micro-carriers in rocking bioreactor for 60 days at the conditions of 5% CO2, 5% O2 and 37 °C. B Dynamic in vitro culture in 100 days
On the other hands, the number of cells in dynamic in vitro cultures increased to 1506 ± 38% after 30 days (Fig. 1A), indicating clearly that much higher cell proliferation even under hypoxia condition. Then, in vitro dynamic culture was continued up to 100 days (Fig. 1B). The highest proliferation rate in dynamic culture was observed to be 2644 ± 178% at day 40. After that, cell growth rate started to fall down gradually and reached 874 ± 209% at day 90, probably cell migration into the pore inside, thus leading to difficulty in measuring total live cells. All results were normalized based on initial seeding cell concentration.
3.2 Cell behaviors on gelatin micro-carrier
SEM photographs were taken to confirm the growth of hMSCs attached to Cultispher-S in dynamic culture condition. On the surface and cross-section of Cultispher-S, it was confirmed that pores with a diameter of 70 ± 30 μm were irregularly distributed with scattered smaller pores on both surface and cross sections (Fig. 2A: low magnification, B: surface and C: cross section). After 60 days of in vitro bioreactor culture, the cells were found to cover the whole surface of the porous particles (Fig. 2D) and the cells and extracellular matrix distributed in the particles were identified (Fig. 2E, F).

SEM images of micro-carrier (A–C) and hMSC/micro-carrier after dynamic in vitro cell cultured for 60 days (E–H) [surface (A, B, D) and X-section (C, E, F)], [(A; × 500), (B, D, F; × 1000) (C; × 3000), (F; × 5000)]. The red arrows indicated the proliferated cells on the pore surface
Live and dead assay and MTT stain confirmed that the cells in culture were adhered to and proliferated on the surface of the micro-carriers, and as the days of culture increased, significant aggregation of the particles with cells was observed. MTT statin (Fig. 3A–D) and a Live and Dead assay (Fig. 3E–H) showed cell viability and tissue formation on whole surfaces as observed at the time intervals of 20, 35, 60 and 100 days.

hMSC viabilities on micro-carrier were analyzed with MTT stain (A–D) and a Live and Dead assay (E–H) dynamic-in vitro cultured for 20, 35, 60 and 100 days (× 200). (Color figure online)
3.3 Tissue regeneration: histological observation during dynamic culture of hMSC
In order to identify formation of the extracellular matrix from the hMSCs and observe the tissue regeneration and differentiation of the cells in dynamic culture using the micro-carriers, diverse staining of H&E (Fig. 4A, F, K and P), MT (Fig. 4B, G, L and Q), Alcian blue (Fig. 4C, H, M and R), ALP (Fig. 4D, I, N and S), and alizarin red S (Fig. 4E, J, O and T) histological staining were performed at the time points of 20, 35, 60 and 100 days, respectively. From the results of the overall staining, the micro-carriers were observed to agglomerate as the incubation period increased, possibly from day 35, and the formations of extracellular matrix around the particle were clearly confirmed. H&E and MT staining confirmed the presence of pink and blue collagen around the particles with overlapping of thick extracellular matrix. Alcian blue and green stains the proteoglycan components of the extracellular matrix associated with chondrocytes. Alcian blue staining of the cultured particles confirmed glycosaminoglycan formed broadly and they were stranded between the particles tissue cultured for 60 and 100 days (Fig. 4M and R).

Histological stain was introduced to observe hMSC differentiation on gelatin micro-carrier according to dynamic culture condition, where hematoxylin and eosin (H&E) (A, F, K, P; × 200), MT (B, G, L, Q; × 200), Alcian blue (C, H, M, R; × 200), ALP (D, I, N, S; × 200) and Alizarin red S (E, J, O, T; × 80) stain. The yellow arrows indicate the results of Alizarin red S staining, which indicates mineralization of the regenerated tissues by Ca++ ions. The scale bar represents 50 μm. (Color figure online)
Alkaline phosphatase and alizarin red S staining confirmed the possibility of osteogenic differentiation of hMSCs according to dynamic culture. ALP stain is a stem cell imaging product and is a cell-permeable fluorescent substrate for alkaline phosphatase, diffusing out over the course of a couple of hours. It was observed in this stain process that the staining became more intense over time at 20 days, 35 days, 60 days and 100 days (Fig. 4D, I, N and S, respectively). Alizarin Red S, an anthraquinone derivative, has been used to identify calcium in regenerated tissue sections of the micro-carriers. Even the reaction is not strictly specific for calcium formation due to possible interference by magnesium, barium, strontium, manganese, and iron, calcium ions form an Alizarin Red S-calcium complex in a chelation process.
4 Discussion
Diverse micro-carriers such as gelatin, polystyrene, collagen and dextran with properties of composition, morphologies and sizes of ranging from 100 to 300 μm have been reported for dynamic cultures of stem cells. The porosity and adhesive surfaces of porous micro-carriers supported adhesion sites for stem cells in dynamic suspension cultures. Initial adhesion of cells on the micro-carrier surface are extremely important for promotion of cell proliferations and subsequent differentiations into specific cell lineages such as mesenchymal stem cells [19], hematopoietic and endothelial cells [20] hepatocyte [21] as well as osteogenic [22] and chondrogenic arthritis [23, 24]. Normally micro-carriers were employed for prevention of cell aggregation of cells and induction of cell growth to high densities, and then the high density cells were collected by enzymatic dissociation for further therapeutic applications. However, tissue regeneration of dynamic micro-carriers have not reported yet using rocking bioreactor over long time.
In this study, however, effects of dynamic signals by wave type bioreactors on mesenchymal stem cell behaviors on porous gelatin micro-carriers have been studied for mass culture of mesenchymal stem cells and subsequent dynamic tissue regeneration over long time in vitro culture by controlling the wave speeds at the ranges of 5–35 rpm. While the porous gelatin micro-carrier with a higher surface area is expected to induce higher initial cell adhesion on and then migration into the pores of the micro-particles, forming tissue regeneration of micro-spheroids. The wave signals of the bioreactors are also expected to speed up the potential of tissue regeneration of the micro-carriers under hypoxia conditions.
The significantly higher rates of cell proliferation and higher degree of secretion of extracellular matrix of gelatin micro-carrier by the bioreactor’s wave signals were clearly observed in long-term dynamic in vitro culture of mesenchymal stem cells with cell counting and viability kits and histological observations over those of the control 2D static in vitro cell culture. Dynamic in vitro culture system was favorable for large-scale culture of cells, because dynamic culture was actively carried out in all of the 31-day cultures using 2D tissue culture dish and micro-carriers. The cells attached well to both the superficial surface and the porous surface of the gelatin micro-carrier during in vitro cell culture, and then secreted the extracellular matrix. Furthermore, the cells seemed to be migrated and proliferated into the pores over dynamic in vitro cultures for 100 days as observed by SEM results, which is transformation of micro-carriers to tissue regeneration over long term culture. This shows that micro-carriers with cells were inter-connected with extracellular matrix between micro-carriers.
Interestingly, the number of cells decreased after 40 days in dynamic cell culture, even though characteristics of the mesenchymal stem cells were observed after 20 days or more in vitro dynamic culturing. As the period of dynamic cell culture increased, the gelatin micro-carriers with discrete cells gradually clumped with the nearby micro-carriers, and agglomerates consisting of 2–5 micro-carriers were observed from the 20th day. After the aggregate was formed, large amounts of extracellular matrix including collagens by the MT assay were produced, and bone marrow differentiation of stem cells was observed around some aggregates. These results suggest that the number of cells counting seems to be limited due to the limited number on the surface of micro-carriers, as well as difficulty in counting total number of live cells deep in pore channels. After a certain period of time, the number of cells does not increase any more, and the shape of regenerated thick tissues is formed through the aggregates, resulting in inter-connection of extracellular matrix produced on each gelatin micro-carrier.
These results and dynamic cell techniques can be used to culture large amount of stem cells before differentiation, and further, to obtain differentiated cells such as bone and cartilage as examples. Subsequent specific studies on the dynamic culture conditions and detailed study of cell characterizations may be needed for further research. As for the culture conditions, studies on the proliferation and characteristics of the cells according to the biological and physical culture conditions may be carried out such as the components of the culture medium, the continuous supply of new micro-carrier, and the stirring speed. Mass production of diverse cells like mesenchymal stems, keratinocytes and chondrocytes [25–27] is expected to be employed as sources for tissue engineering of bone, cartilage, blood vessels and skins by using diverse recent technologies such as 3D bioprinting [28, 29], microchips, hydrogels and others [30–32].
References
Polo-Corrales L, Latorre-Esteves M, Ramirez-Vick JE. Scaffold design for bone regeneration. J Nanosci Nanotechnol. 2014;14:15–56.
Berthiaume F, Maguire TJ, Yarmush ML. Tissue engineering and regenerative medicine: history, progress, and challenges. Annu Rev Chem Biomol Eng. 2011;2:403–30.
Sun LY, Hsieh DK, Syu WS, Li YS, Chiu HT, Chiou TW. Cell proliferation of human bone marrow mesenchymal stem cells on biodegradable microcarriers enhances in vitro differentiation potential. Cell Prolif. 2010;43:445–6.
Sun LY, Lin SZ, Li YS, Harn HJ, Chiou TW. Functional cells cultured on microcarriers for use in regenerative medicine research. Cell Transplant. 2011;20:49–62.
van Wezel AL. Growth of cell-strains and primary cells on micro-carriers in homogeneous culture. Nature. 1967;216:64–5.
Nilsson K. Microcarrier cell culture. Biotechnol Genet Eng Rev. 1988;6:403–39.
Nie Y, Bergendahl V, Hei DJ, Jones JM, Palecek SP. Scalable culture and cryopreservation of human embryonic stem cells on microcarriers. Biotechnol Prog. 2009;25:20–31.
Frauenschuh S, Reichmann E, Ibold Y, Goetz PM, Sittinger M, Ringe J. Microcarrier-based cultivation system for expansion of primary mesenchymal stem cells. Biotechnol Prog. 2007;23:187–93.
Singh H, Mok P, Balakrishnan T, Rahmat SN, Zweigerdt R. Up-scaling single cell-inoculated suspension culture of human embryonic stem cells. Stem Cell Res. 2010;4:165–79.
Jain E, Kumar A. Upstream processes in antibody production: evaluation of critical parameters. Biotechnol Adv. 2008;26:46–72.
Gallo-Ramírez LE, Nikolay A, Genzel Y, Reichl U. Bioreactor concepts for cell culture-based viral vaccine production. Expert Rev Vaccines. 2015;14:1181–95.
Liu JY, Hafner J, Dragieva G, Burg G. Bioreactor microcarrier cell culture system (Bio-MCCS) for large-scale production of autologous melanocytes. Cell Transplant. 2004;13:809–16.
Abeille F, Mittler F, Obeid P, Huet M, Kermarrec F, Dolega ME, et al. Continuous microcarrier-based cell culture in a benchtop microfluidic bioreactor. Lab Chip. 2014;14:3510–8.
Wu CY, Stoecklein D, Kommajosula A, Lin J, Owsley K, Ganapathysubramanian B, et al. Shaped 3D microcarriers for adherent cell culture and alaysis. Microsyst Nanoeng. 2018;4:21.
Merten OW. Advances in cell culture: anchorage dependence. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140040.
Mrakovcic M, Absenger M, Riedl R, Smole C, Roblegg E, Fröhlich LF, et al. Assessment of long-term effects of nanoparticles in a microcarrier cell culture system. PLoS One. 2013;8:e56791.
Liu JY, Hafner J, Dragieva G, Burg G. A novel bioreactor microcarrier cell culture system for high yields of proliferating autologous human keratinocytes. Cell Transplant. 2006;15:435–43.
Zhang Y, Wang X, Pong M, Chen L, Ye Z. Application of bioreactor in stem cell culture. J Biomed Sci Eng. 2017;10:485–99.
Rafiq QA, Coopman K, Nienow AW, Hewitt CJ. Systematic microcarrier screening and agitated culture conditions improves human mesenchymal stem cell yield in bioreactors. Biotechnol J. 2016;11:473–86.
Lam AT, Chen AK, Li J, Birch WR, Reuveny S, Oh SK. Conjoint propagation and differentiation of human embryonic stem cells to cardiomyocytes in a defined microcarrier spinner culture. Stem Cell Res Ther. 2014;5:110.
Park Y, Chen Y, Ordovas L, Verfaillie CM. Hepatic differentiation of human embryonic stem cells on microcarriers. J Biotechnol. 2014;174:39–48.
Shekaran A, Sim E, Tan KY, Chan JK, Choolani M, Reuveny S, et al. Enhanced in vitro osteogenic differentiation of human fetal MSCs attached to 3D microcarriers versus harvested from 2D monolayers. BMC Biotechnol. 2015;15:102.
Lin YM, Lim JF, Lee J, Choolani M, Chan JK, Reuveny S, et al. Expansion in microcarrier-spinner cultures improves the chondrogenic potential of human early mesenchymal stromal cells. Cytotherapy. 2016;18:740–53.
Morille M, Toupet K, Montero-Menei CN, Jorgensen C, Noël D. PLGA-based Microcarriers induce mesenchymal stem cell chondrogenesis and stimulate cartilage repair in osteoarthritis. Biomaterials. 2016;88:60–9.
Kakkar A, Sorout A, Tiwari M, Shrivastava P, Meena P, Saraswat SK, et al. Current status of stem cell treatment for type I diabetes mellitus. Tissue Eng Regen Med. 2018;15:699–709.
Oliva J, Ochiai K, Florentino A, Bardag-Gorce F, Wood A, Niihara Y. Feeder cells free rabbit oral mucosa epithelial cell sheet engineering. Tissue Eng Regen Med. 2018;15:321–32.
Iyer K, Chen Z, Ganapa T, Wu BM, Tawil B, Linsley CS. Keratinocyte migration in a three-dimensional in vitro wound healing model co-cultured with fibroblasts. Tissue Eng Regen Med. 2018;15:721–33.
Jung CS, Kim BK, Lee J, Min BH, Park SH. Development of printable natural cartilage matrix bioink for 3D printing of irregular tissue shape. Tissue Eng Regen Med. 2018;15:155–62.
Das D, Pham TTH, Lee S, Noh I. Fabrication of alginate-based stimuli-responsive, non-cytotoxic, terpolymric semi-IPN hydrogel as a carrier for controlled release of bovine albumin serum and 5-amino salicylic acid. Mater Sci Eng C Mater Biol Appl. 2019;98:42–53.
Das D, Pham TTH, Noh I. Characterizations of hyaluronate-based terpolymeric hydrogel synthesized via free radical polymerization mechanism for biomedical applications. Colloids Surf B Biointerfaces. 2018;170:64–75.
Janarthanan G, Noh I. Click chemistry-based injectable hydrogels and bioprinting inks for tissue engineering applications. Tissue Eng Regen Med. 2018;15:531–46.
Das D, Cho H, Kim N, Pham TTH, Kim IG, Chung EJ, et al. A terpolymeric hydrogel of hyaluronate-hydroxyethyl acrylate-gelatin methacryloyl with tunable properties as biomaterial. Carbohydr Polym. 2019;207:628–39.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) Grant (2015R1A2A1A10054592). Human mesenchymal stem cells has been donated by Professor Sungwon Kim in the Catholic University of Korea St Mary’s Hospital, Seoul, Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statement
There are no animal experiments carried out in this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nguyen, L., Bang, S. & Noh, I. Tissue Regeneration of Human Mesenchymal Stem Cells on Porous Gelatin Micro-Carriers by Long-Term Dynamic In Vitro Culture. Tissue Eng Regen Med 16, 19–28 (2019). https://doi.org/10.1007/s13770-018-00174-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13770-018-00174-8