
Abstract
As a research of inflammation inhibitory activity using a natural resource, the inflammation inhibitory activity by purified manassantin A from Saururus chinensis was experimented. In the result of MTT assay with manassantin A, cell viability decreased at concentration of 100 μM. LPS-treated RAW 264.7 cell group treated with 6.25–50 μM concentration of manassantin A showed approximately 4–55% NO expression compared to LPS non-treated group. Inflammation inhibitory activity and NO expression inhibition increased as RAW 264.7 cell treated with higher concentration of manassantin A. Expression inhibition of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) was also identified. An excellent prostaglandin E2 expression inhibition effect was identified with manassantin A at concentration of 6.25–25 μM. A high interleukin-1β, tumor necrosis factor-α, and IL-6 expression inhibition effect was manassantin A at concentration of 6.25–50 μM of 0–97, 6–32, and 22–66% was identified, respectively. A high interleukin-1β, tumor necrosis factor-α, and IL-6 expression inhibition effect was manassantin A at concentration of 6.25–50 μM of 0–97, 6–32, and 22–66% was identified, respectively. Expression inhibition effect was identified to be concentration dependent. Therefore, manassantin A is expected to show high inflammation inhibitory effect in RAW 264.7 induced from LPS by inhibiting iNOS and COX-2 protein expression along with cytokine expression inhibition.
Similar content being viewed by others


Introduction
Human’s living standard and eating style are changing at high and various levels due to repeated industrial and economic development, along with a medical technique development. These phenomena bring the spread of development and advance with increasing chronic diseases like a splenitis, an atopic dermatitis, and herpes zoster caused by an air pollution and environmental pollution (Lee et al. 2013). Therefore, studies about natural plants concerning antioxidants are progressed and biological materials which have anti-inflammation are researched and identified from the plants and are being tried to prevent chronic inflammation, cancer, and interaction with other disease by eating those materials (Kim 2006; Lee et al. 2010). An inflammation react is one of the defense mechanism in vital tissue against a bacterial inflammation, physical or chemical stimulation from outside. Also it is a mechanism to regenerate or recover a damaged tissue (Cho and An 2008). When an inflammation react occurs in a body, an inflammation cell like a macrophage secretes a inflammation carrier like nitric oxide (NO), prostaglandin E2 (PGE2), tumor necrosis factor-α (TNF-α), and interleukin-1β (IL-1β) (Witthoft et al. 1998). Therefore, macrophage takes charge of a major defense mechanism in immune system and produced in the bone marrow as a phagocyte which finds, and removes viruses or external matter idiosyncratically, and also secrets each cell toxic matter like H2O2, NO in order to destroy a foreign cell or tumor. Through this mechanism, macrophage secrets cytokine and enzymes like phosphatase, controls immunity phenomenon in body, and concerns an inflammation activity and hematopoietic organs (Vane 1971; Funk et al. 1991; Weisz et al. 1996; Lee et al. 2004). Recently, studies are having been processed to find natural plants having substances treating or preventing diseases (Ryu et al. 2003; Kim et al. 2013) and researched to find materials having biological activities from natural resources.
Ultra-fine grinding method is used for physicochemical transform of the raw material. Decrease the particle size changes the structure of the plant and increases the surface area. Therefore, decomposition of the powder by fermentation and enzyme is increased. Texture is improved as well as dissolution and extraction of useful compounds from non-degradable organic material. Also it has advantage of absorptance and inner body utilization ratio improvement by increasing water solubility (Yoon 2011).
In this study, manassantin A was separated and identified from ultra-fine ground Saururus chinensis Bail extract. Then LPS was treated to RAW 264.7 cell which caused inflammation and determined inhibitory effect of manassantin A on NO production quantity and expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) which is involved in inflammation. TNF-α, IL-1, and IL-1β expression inhibitory effect was determined for the development of natural material as anti-inflammatory substance.
Materials and methods
Ultra-fine grinding and extraction from Saururus chinensis
Sample used in this study was purchased at M farm at Daegu. Fresh leaves were dried and ultra-fine ground using 10 L volume of ultra-fined grinder (MKFS10-1, Koen 21 Co, Ansan, Korea) at 20 kg/h velocity and ultra-fine grinding (125 μm ISO mesh size, ASTM 140 mesh: filtered size).
A total of 100 mL of 50% ethanol was added to 1 g of ultra-fine ground S. chinensis extract and homogenized for 1 min at 20,000 rpm using homogenizer and then stir-extracted for 24 h. Extract was filtered using Whatman No. 1 filter paper (Whatman Inc, Piscataway, NJ, USA) and concentrated with rotary vacuum evaporator (Eyela NE, Tokyo, Japan) if necessary.
Determination of phenolic content in extracts
Sample (1 mL), and 95% ethanol (1 mL) was added to distilled water (DW) (5 mL) then 0.5 mL of 1 N-Folin-ciocalteu reagent (Junsei Chemical Co, Tokyo, Japan) was added and well mixed as color developing reagent. By leaving it for 5 min, the mixture expressed color, and then 1 mL Na2CO3 was added and OD within 1 h at 725 nm optical density (Optizen 3220UV, Mecasys, Daejeon, Korea) was determined. Then standard curve using gallic acid was prepared (Dural and Shetty 2001).
Column chromatographic conditions for purification
Compound filled was Sephadex LH-20 (Pharmacia Co, Uppsala, Sweden) in column and separated with characteristic of adsorbability. 60% EtOH and normal phase type as eluting solvent, eluted EtOH → H2O (100 → 0%) in order at flow velocity of 20 mL/min then concentrated and dried at TLC phase after identifying existence of phenolic compounds and type. MCI-gel (Supelco, Bellefonte, PA, USA) is a porous polystyrene gel, therefore used adsorbability and eluted with reverse type of H2O → MeOH (0 → 100%) as eluting solvent then identified phenolic compound separation at TLC phase.
Identification of chemical structure with anti-inflammation compound
Melting point was determined by using micro-electrothermal (Fisher-Jhons, Mexico City, Mexico) with 1 mg of sample. Halogenized alkali purification was used for infrared (IR) spectrum (Perkin-Elmer IR-1330, Perkin Elmer, Waltham, MA, USA). Purely separated sample (1 mg) and KBr (100 mg) powder were mixed and pressure was applied, making it into pressure-purified compound and then determined. In 1H and 13C-Nuclear Magnetic Resonance (NMR) spectrum (ARX-250, Burker Co, Billerica, MA, USA), pulse Fourier transform method was used by eluting pure refining 10 mg and determining solvent (CDCl3 + DMSO-D6 + D2O) at 5–20% (w/v) ratio with TMS [Tetramethylsilane; (CH3)4Si] as standard and then measured at proton magnetic resonance (PMR) 300 MHz. With solid sample (1 mg) at decompressed phase (10−4–10−6 mmHg), determined with using negative ion FAB-MS spectrum (Jeol JMS-PX 300, Tokyo, Japan) by chemical analysis method. Thioglycerol (Sigma Chemical Co, Louis, MO, USA) was used as determining solvent and emitter electric current at 22–28 eV as determining condition. Then mass spectrometric analysis was carried out with accelerative pressure of ion source at 6–7 kV.
Hyaluronidase inhibitory effect assay
There determine anti-inflammatory effect was determined by determining enzyme activity with optimal density of glucoxazoline, induced and modified from N-acetylglucosamine formed from sodium-hyaluronic acid, color expressed with p-dimethylamino benzaldehyde (DMAB) (Sigma Chemical Co) (Reissig et al. 1955). 0.1 M acetate buffer (pH 3.5) mixed with melted hyaluronidase (7900 U/mL) (Wako Pure Chemical Industries, Osaka, Japan), and sample solution (0.05, 0.1 mL) was cultured for 20 min at 37 °C then 12.5 mM CaCl2 (0.1 mL) was added and cultured for 20 min. As a substrate, melted hyaluronic acid (12 mg/mL) was added to 0.1 M acetate buffer (pH 3.5) and cultured for 40 min. 0.1 mL 0.4 N potassium tetraborate and 0.1 mL 0.4 N NaOH solution were added to reaction mixture and boiled for 3 min in water bath and then cooled completely. Cooled reactant was cultured for 20 min at 37 °C after treating with 3 mL of DMAB reagent as color former, and then inhibition activity was calculated by determining absorbance at 585 nm. Inhibition ratio (%) = (1 − absorbance of sample/absorbance of control) × 100.
Cell culture for determination of anti-inflammatory effect
RAW 264.7 cell of murine macrophage cell line was purchased at Korean Cell Line Research Foundation. The cell was cultured for 72 h at 37 °C and 5% CO2 with mixed medium of 10% fetal bovine serum (HyClone, GE Healthcare Life Sciences, Chicago, IL, UT, USA), penicillin 100 U/mL, and streptomycin 100 μg/mL (HyClone, GE Healthcare Life Sciences) from Dulbecco’s modified Eagle’s medium (DMEM) (HyClone, GE Healthcare Life Sciences). The cell condition was maintained with 5% CO2 condition and subcultured with 2–3 × 106/mL cell density on cell culture dish. When experimenting, the passage condition of 80% confluency and 20 times under was kept. When experimenting, the passages condition of 80% confluency and 20 times under was kept (Cho and An 2008; Lee 2011).
Cell toxicity determination by MTT assay
Cell toxicity was determined using Carmichael et al. (1987) method. A total of 0.18 mL of RAW 264.7 cell was aliquoted at 96-well plate of 5 × 104 cells/mL. Samples (2 mL) of different concentration were treated and incubated for 24 h at 37 °C using 5% CO2 incubator. MTT solution (0.02 mL) (Sigma Chemical Co.) at concentration of 5 mg/mL was treated and incubated for 4 h and removed medium, and 0.15 mL of dimethyl sulfoxide (DMSO) was treated to each well and reacted for 30 min at room temperature. Then enzyme-linked immunosorbent assay (ELISA) reader (SPECTROstar Nano, BMG LABTECH, Ortenberg, Germany) was used and determined optical density at 540 nm. Cell toxicity determination was carried out using OD of sample solution-treated group and non-treated group. Control group was incubated at same condition treating with equal amount of distilled water. Cell survival rate (%) = (OD of sample group/OD of control group) × 100.
Nitric oxide determination
NO measurement was carried out by measuring nitric oxide (NO) amount in supernatant of the cell as nitrite and nitrate. Griess reagent (Sigma Chemical Co) was used to stabilize nitrite reduced to nitrate. At 80% confluence, 2 × 106 U of cell in 6-well plate was washed two times with phosphate-buffered saline (PBS) and cultured with non-serum culture medium for over 12 h. Then they were stimulated by adding LPS 1 μg/mL to all wells except the control group and experimented with each different concentrations after 1 h. NO production amount was determined by determining absorbance at 540 nm with the 10-min Griess reagent-reacted supernatant which is collected hourly (Ryu et al. 2003; Cho and An 2008). Inhibition ratio (%) = (1 − absorbance of sample/absorbance of control) × 100.
Western blotting analysis for inducible NO synthase (iNOS) and cyclooxygenase-2 (COX-2) activity
To determine the activity of iNOS protein, RAW 264.7 cell which is a macrophage was stabilized by culturing for 24 h in 100π tissue culture dish with suspension 2 × 104 cells/mL cell treated to each well. After removing the medium, different concentration (10, 100 μg/mL) treated medium was used to cultivate for 24 h. Then the medium removed and washed with PBS two times. It was eluted with 100 μL lysis buffer [complete mini 1 tablet added to 10 mL Radio Immuno Precipitation Assay buffer] and centrifuged (1730R, Gyrozen, Seoul, Korea) for 20 min at 12,000 rpm and 4 °C. Only supernatant was collected, transferred it into a new tube, and stored at −20 °C for further use or protein determination. Supernatant collected by centrifugation was determined with Bradford assay. A total of 20 μL of protein was separated by electrophoresis with 10% sodium sulfate polyacrylamide gel electrophoresis. Separated protein was transferred to polyvinylidene difluoride (Millipore Corp, Billerica, MA, USA) membrane using semidry transfer cell machine (Bio-Rad, Hercules, CA, USA) and incubated for 1 h with blocking buffer [5% skim milk in Tris-buffered saline Tween-20 (TBST)] at room temperature. It was washed with TBST three times every 10 min and incubated overnight with diluted primary antibody, iNOS (BD Bioscience, 1:1000, Sanjose, CA, USA), COX-2 (Cayman, 1:1000, Ann arbor, MI, USA), and GAPDH (Santa Cruz Biotechnology, Inc, 1:1000, Santa Cruz, CA, USA), at 4 °C. It was washed with TBST three times every 10 min once more and incubated for 2 h at room temperature with secondary antibodies, mouse anti-rabbit IgG HRP (Santa Cruz, 1:1000), and bovine anti-goat IgG horseradish peroxidase (HRP) (Santa Cruz, 1:1000) each diluted at 1:1000 ratio. With three times of washing, it was reacted with ECL (Millipore, Bedford, MA, USA) solution in darkroom and exposed to X-ray film. Each band was determined using Molecular Imager (Bio-Rad Laboratories, Inc) (Cho and An 2008; Cho 2011).
Cytokine assay
Cultured macrophage and inoculated with 1 × 106 cells/mL cell in 6-well then treated agonist (cell stimulator) in different concentration and 1 h. Treated LPS (1 μg/mL) after 1 h, determined cytokine by treating cell culture medium 1 h. Collected medium was stored at −70 °C before determination. Enzyme immuno assay kit (R&D systems Inc, Minneapolis, MN, USA) was used for determining the content of TNF-α, IL-1β, IL-6, IL-8, PGE2, etc. Each content of cytokine was converted using standard curve obtained from reaction of standard material (Anfernee et al. 2005; Iwona et al. 2006; Cho and An 2008; Nam et al. 2015).
Statistical analysis
Data were analyzed by one-way analysis of variance (one-way ANOVA) with SPSS 22.0 software followed by Duncan’s multiple range test. All values are expressed as the mean ± SD unless otherwise stated, and ρ value <0.05 was considered to indicate a statistically significant difference.
Results and discussion
Purification and identification of the anti-inflammatory compound in ultra-fine ground Saururus chinensis
Ultra-fine grind method increases the surface area of material and increases extraction yield of phenolic compound. Also increase in the solubility increases the value as medicinal food source (Yoon 2011). Therefore in this study, ultra-fine grinding method was used to increase the extraction yield of S. chinensis using ultra-fine grinding (125 μm ISO mesh size, ASTM 140 mesh: filtered size) and anti-inflammatory substance was extracted.
Obtained H2O layer, ethyl acetate layer and n-BuOH layer by solvent fractioned 200 mL of H2O/Ethyl acetate/n-BuOH with 30 g of freezing dried S. chinensis 50% ethanol extract. Solvent from three types of fractioned solvents was removed, then diluted with distilled water to certain concentration, and determined inhibition activity on hyaluronidase. As n-BuOH as target due to inhibition activity, single compound with hyaluronidase inhibition activity was obtained by purifying with Sephadex LH-20 and MCI gel CHP-20 column chromatography.
The chemical structure of active compound was C42H52O11, the molecular weight was determined 732 based on positive FAB-MS, and melting point was 80–82 °C. [[α] 25D ] was −107.6°. The 1H-NMR (CDCl3, 250 MHZ) spectrum showed a profile of 6.97–6.79 (12H, m, aromatic protons), 5.46 (2H, d, J = 5.8 Hz, H-7, 7′), 4.62 (2H, d, J = 8.2 Hz, H-7″, H-7‴), 4.08 (2H, m, H-8″, 8‴), 3.92 (6H, s, –OCH3 X 2), 3.85 (6H, s, –OCH3, X 2), 3.84 (6H, s, –OCH3, X 2), 2.29 (2H, m, H-8, 8′), 1.16 (6H, d, J = 6.1 Hz, H-9″, 9‴), 0.73 ppm (6H, d, J = 6.5 Hz, H-9, 9′), and the 13C-NMR (CDCl3, 62.9 MHz) spectrum showed a profile of 150.6 (C-4″, 4‴), 149.1 (C-3″, C-3‴), 148.7 (C-1″, 1‴), 146.3 (C-4, 4′), 136.5 (C-3, 3′), 132.7 (C-1, 1′), 119.7 (C-6″, 6‴), 118.7 (C-5″, 5‴, 2″, 2‴), 111.0 (c-6, c-6′), 110.2 (c-2, 2′), 109.8 (c-5, 5′), 84.1 (c-7″, 7‴), 83.5 (c-7, 7′), 78.4 (c-8″, 8‴), 55.6 (–OCH3, X 6), 44.3 (C-8, 8′), 17.2 (C-9″, 9‴), 14.7 ppm (C-9, 9″). This result accorded with Seo et al. (2008) report. Thus, the compound B was identified manassantin A.
Cell viability by manassantin A
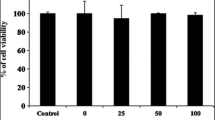
MTT assay in order to determine cell viability was executed under controlling the concentration of manassantin A at 6.25–50 μM, the cell viability was over 90% at 6.25–12.5 μM, but the cell viability tendency decrease at 20–50 μM (Fig. 1).

Cell viability of manassantin A purified from S. chinensis extracts on MTT. RAW 264.7 cells were incubated with various concentrations (6.25–50 μM) of manassantin A for 24 h. Data represent the mean ± SD with three separate experiments. One-way ANOVA was used for comparisons of multiple group means followed by Duncan’s multiple range test (significant as compared to control. ρ < 0.05)
NO producing inhibitory assay by manassantin A
NO is a type of free radical and therefore is a very unstable molecule. NO is converted to stable nitrogen oxide like NO2, N2O3, N2O4, nitrite (NO2–), or nitrate (NO3–) by oxygen or superoxide. NO is produced by NOS from l-arginine, and NOS can be classified into endothelial NOS (eNOS), which produces NO for maintaining homeostasis, neuronal NOS (nNOS) and iNOS, which is induced by inflammatory factors. An excess amount of inflammation factors such as NO and PGE2 is produced by iNOS and COX-2 during inflammation reaction processes in the body. The production of NO is important for eliminating bacteria or removing tumors, but excess amounts can cause inflammation, organ damage, gene mutation, or pathological neural damage. To determine the inhibition effect of manassantin A on NO production of RAW 264.7 cells, cells were treated with various concentrations (6.25–50 μM) of manassantin A and LPS, and the amount of NO produced was determined. As shown in Fig. 2, the LPS-treated group (control group) showed three times higher NO expression (100%) than the group without LPS non-treatment (31%). Inhibition of NO production observed weakly at 6.25–10 μM of manassantin A concentration, but the inhibition of NO production was 50–55% at 12.5–50 μM concentration than the LPS-treated group (control group). To compare with PDTC as a positive control, inhibition of NO production treated with manassantin A was lower than PDTC at a low concentration, but observed a similar effect to PDTC at 12.5 μM concentration over. Therefore, by inhibiting the expression of transcription factors that interact with NO, manassantin A is expected to show a good anti-inflammation effect in RAW 264.7 macrophages induced by LPS.

Effect of manassantin A purified from S. chinensis the production of NO in 1 μg/mL of LPS-stimulated RAW 264.7 cells. (A) manassantin A, (B) PDTC as positive control. Data represent the mean ± SD with three separate experiments. One-way ANOVA was used for comparisons of multiple group means followed by Duncan’s multiple range test (significant as compared to control. ρ < 0.05)
iNOS protein expression inhibitory assay by manassantin A
The excess amount of inflammation factors like NO, PGE2 were produced by iNOS. iNOS is known to function in a defense response against contagious pathogens including viruses with various inflammatory diseases, circulatory disorders, and cancer.
The half-life period of NO itself is very short as 6–10 s, and nitric oxide synthase (NOS) producing NO is classified into three types of enzyme (type I, type II, and type III) by a physical, and chemical icon. nNOS (type I) and eNOS (type II) are classified as constitutive NOS because they are always in sells, but iNOS (type III) is classified as iNOS because iNOS would be expressed when exposed to special stimulatives like LPS, cytokines, and cytotoxin of bacteria (Nathan and Xie 1994; Witthoft et al. 1998; Axtelle and Pribble 2001). This NOS convert L-arginine to L-citrulline at the same time product NO. This NOS is usually expressed by iNOS that plays important roles pathologically. Produced NOS reveals a cytotoxin around the tissue while an act of guanyl cyclase. Therefore, decreasing of iNOS protein levels in RAW 264.7 cells induced by LPS and treated with manassantin A might indicate an anti-inflammation effect. To identify the relationship of iNOS protein with a mechanism of NO inhibition, iNOS protein expression in the cytoplasm was quantified with a Western blot using immune blot analysis. As shown in Fig. 3, compared to the control group, the manassantin A-treated group remarkably showed iNOS inhibition of 41% at a concentration of 25 μM. Above all, the expression of iNOS was inhibited perfectly at the concentration of 25 μM. These results proved that the expression inhibition of manassantin A on iNOS was higher than that of salidroside which was reported by Won et al. (2008). Therefore, manassantin A in S. chinensis, one of medicine plants, was expected to show the anti-inflammatory effect and immune function in macrophage RAW 264.7 cells induced with LPS via inhibition of the interaction between iNOS and NO.

Effect of manassantin A purified from S. chinensis: the production of iNOS in LPS-stimulated RAW 264.7 cells. RAW 264.7 cells were treated with 25 μM concentrations of manassantin A from S. chinensis dissolved in DW for 1 h prior to the addition of LPS (1 μg/mL), and the cells were further incubated for 24 h. Control cells were incubated with vehicle alone. The concentrations of iNOS were monitored as described in the experimental procedures. Data represent the mean ± SD with three separate experiments. One-way ANOVA was used for comparisons of multiple group means followed by Duncan’s multiple range test (significant as compared to control. ρ < 0.05)
COX-2 protein expression inhibitory assay by manassantin A
COX-2 produced through activation of MEKK-1 and NFκB by pro-oxidant and pro-inflammatory stimuli plays central roles in inflammatory act. In macrophages, COX-2 protein makes the expression of pro-inflammatory cytokines’ increase such as TNF-α and IL-6 in monocytes leading to an inflammation (Naoko et al. 2005). Also COX-2 expression in monocytes is increased by pro-inflammatory agents such as IL-1β, TNF-α, phosphatidic acid, and fibroblast growth factor. COX-2 inhibition is induced by glucocorticoid, IL-4, and IL-13. Therefore, development of a selective inhibitor for COX-2 would serve as a target molecule for inflammation treatment. Figure 4 shows the effect of manassantin A treated with LPS on inflammatory factor, COX-2. Compared with the control group of 100%, manassantin A inhibited highly the expression of COX-2 about 67% at concentrations of 25 μM. Kim et al. (2012) reported Ligustrum ovalifolium extract has anti-inflammatory effect by inhibiting NO production and expression of iNOS, COX-2 protein. That would prove immune function and a high inhibitory effect of the manassantin A on expression of COX-2 in macrophage RAW 264.7 cells induced with LPS.

Effect of manassantin A purified from S. chinensis: the production of COX-2 in LPS-stimulated RAW 264.7 cells. RAW 264.7 cells were treated with 25 μM concentrations of manassantin A from S. chinensis dissolved in DW for 1 h prior to the addition of LPS (1 μg/mL), and the cells were further incubated for 24 h. Control cells were incubated with vehicle alone. The concentrations of COX-2 were monitored as described in the experimental procedures. Data represent the mean ± SD with three separate experiments. One-way ANOVA was used for comparisons of multiple group means followed by Duncan’s multiple range test (significant as compared to control. ρ < 0.05)
PGE2, TNF-α, IL-1β, and IL-6 expression inhibitory assay by manassantin A
To find the inhibitory effect of manassantin A on the expression of PGE2 and pro-inflammatory cytokines in RAW 264.7 cell by LPS, the production of TNF-α, IL-1β, and IL-6 was evaluated. As shown in Fig. 5, cells treated with manassantin A of 6.25–50 μM inhibited the PGE2 expression of 0–43% in a concentration dependent type. Compared to PDTC as using a positive control, manassantin A showed a little inhibition on the PGE2 expression at concentration of 6.25–50 μM (Fig. 5A, B). As shown in Fig. 5(C, D), TNF-α expression was inhibited weakly at 6.25–25 μM when treated with manassantin A of 6.25–50 μM concentration. But the inhibitory effect of manassantin A on TNF-α expression was observed highly as about 32% at 50 μM. The effect to inhibit TNF-α expression was more excellent than PDTC as a positive control at the concentration of 50 μM over. In the concentration of manassantin A within 6.25–50 μM, manassantin A showed the inhibition on IL-1β expression in a concentration dependent type. Compared to PDTC, the positive control, the inhibitory effect on IL-1β expression was lower than PDTC at the concentration of 6.25–12.5 μM (Fig. 5E, F). But, it was higher than PDTC at the concentration of 25 μM over. The result to treat with manassantin A of 6.25–50 μM concentrations, IL-6 expression was inhibited at 22–66% (Fig. 5G, H). Both groups treated with manassantin A and PDTC (positive control) showed analogous expressional rates when compared to control in the concentration of 6.25 μM, but the inhibitory effect of manassantin A on IL-6 expression was higher than PDTC at 25 μM over in a concentration dependent type. Thus, this result was similar to the inhibition phenomena of iNOS and COX-2 expression and therefore an effect of inflammatory inhibition in macrophage RAW 264.7 cells induced by LPS can be expected. Yun et al. (2008) reported that Artemisia princeps pampanini extracts inhibit PGE2 product at 50 μM along with cytokine like TNF-α, IL-1β, IL-6 induced by LPS. It was identified that manassantin A of S. chinensis showed higher anti-inflammatory effect than Bilnesia sarmienti hot water extract at 200 μg/mL inhibiting PGE2, TNF-α, IL-1β, IL-6 as Cheon et al. (2009) reported. Therefore, manassantin A is expected to show high inflammation inhibitory effect in RAW 264.7 induced from LPS by inhibiting iNOS and COX-2 protein expression along with cytokine (PGE2, TNF-α, IL-1β, IL-6) expression inhibition.

Expression rate of manassantin A purified from S. chinensis extracts on PGE2, TNF-α, IL-1β, and IL-6. RAW 264.7 cells incubated with various concentrations (6.25–50 μM) of manassantin A for 1 h and then treated with 1 μg/mL of LPS for 24 h. (A, C, E, G) manassantin A, (B, D, F, H) PDTC as positive control. Data represent the mean ± SD with three separate experiments. One-way ANOVA was used for comparisons of multiple group means followed by Duncan’s multiple range test (significant as compared to control. ρ < 0.05)
References
Anfernee KT, Chi-Keung W, Xiao-Ling S, Mengsu Y, Wang-Fun F (2005) Honokiol inhibits TNF-α-stimulated NF-κB activation and NF-κ B-regulated gene expression through suppression of IKK activation. Biochem Pharmcol 70:1443–1457
Axtelle T, Pribble J (2001) IC14, a CD14 specific monoclonal antibody is a potential treatment for patients with severe sepsis. J Endotoxin Res 7:310–314
Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB (1987) Evaluation of a tetrazolium based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res 47:936–942
Cheon YP, Mohammad LM, Park CH, Hong JH, Lee GD, Song JC, Kim KS (2009) Bulnesia Sarmienti aqueous extract inhibits inflammation in LPS-Stimulated RAW 264.7 cells. J Life Sci 19:479–485
Cho YJ (2011) Anti-inflammatory effect of Jatrorrhizine from Phellodendron amurense in Lipopolysaccharide-stimulated RAW 264.7 cells. J Appl Biol Chem 54:114–119
Cho YJ, An BJ (2008) Anti-inflammatory effect of extracts from Cheongmoknosang (Morus alba L.) in lipopolysaccharide-stimulated raw cells. J Korean Soc Appl Biol Chem 51:44–48
Dural B, Shetty K (2001) The stimulation of phenolics and antioxidant activity in pea (Pisum sativum) elicited by genetically transformed Anise root extract. J Food Biochem 25:361–377
Funk CD, Frunk LB, Kennedy ME, Pong AS, Fitzgerald GA (1991) Human platelet/erythroleukemia cell prostaglandin G/H synthase: cDNA cloning, expression, and gene chromosomal assignment. FASEB J 5:2304–2312
Iwona M, Barbara M, Violetta RS, Romuald M, Zbigniew S, Maciej K, Stefania GK (2006) Proinflammatory cytokine (IL-1β, IL-6, IL-12, IL-18 and TNF-α) levels in sera of patients with subacute cutaneous lupus erythematosus. Immunol Lett 102:79–82
Kim HS (2006) Effects of Saururus chinensis Baill hot-water extract intake on the lipid components and metabolic enzyme activities in hyperlipidemic rats. Korean J Exerc Nutr 10:99–106
Kim YS, Lee SJ, Hwang JW, Kim EH, Park PJ, Jeong JH (2012) Anti-inflammatory effects of extracts from Ligustrum ovalifolium H. Leaves on RAW 264.7 macrophages. J Korean Soc Food Sci Nutr 41:1205–1210
Kim HJ, Lee DJ, Ku JJ, Choi K, Park KW, Kang SH, Moon C, Lee PJ (2013) Anti-inflammatory effect of extracts from folk plants in Ulleung island. Korean J Plant Res 26:169–177
Lee E (2011) Effects of Ixeris dentata extract on the production of pro-inflammatory cytokines in the LPS stimulated rat and RAW 264.7 cells. Korean J Plant Res 24:604–612
Lee ES, Ju HK, Moon TC, Lee E, Jahng Y, Lee SH, Son JK, Baek SH, Chang HW (2004) Inhibition of nitric oxide and tumor necrosis factor-α production by propenone compound through blockade of nuclear factor (NF)-kB activation in cultured murine macrophages. Biol Pharm Bull 27:617–620
Lee JJ, Kim DH, Lim JJ, Kim DG, Kim GS, Min WG, Lee HJ, Chang HH, Kim S (2010) Antibacterial effect of Saururus chinensis Baill ethanol extract against Salmonella typhimurium infection in RAW 264.7 macrophage. Korean J Vet Public Health 34(4):285–291
Lee JH, Choe YH, Park YJ, Zhang XW, Kim BS (2013) Antimicrobial and antiviral activity of Saururus chinensis extract by n-Hexane. Korean J Vet Serv 36:87–93
Nam KS, Jang SA, Sohn EH, Bak JP, Sohn ES, Koo HJ, Yoon WJ, Kwon JE, Jeong YJ, Meng X, Han HS, Kang SC (2015) Comparative study of Litsea japonica Leaf and fruit extract on the anti-inflammatory effects. Korean J Plant Res 28:145–152
Naoko K, Satsuki K, Shinichi W (2005) IL-17 supress TNF-α-induced CCL 27 production through induction of COX-2 in human ketatinocytes. J Allergy Clin Immunol 116:1144–1150
Nathan C, Xie QW (1994) Regulation of biosynthesis of nitric oxide. J Biol Chem 269:13725–13728
Reissig JL, Strominger JL, Leloir LF (1955) A modified colorimetric method for the estimation of N-acetylamino sugars. J Biol Chem 217:959–966
Ryu JH, Ahn H, Kim JY, Kim YK (2003) Inhibitory activity of plant extracts on nitric oxide synthesis in LPS-activated macrophage. Phytother Res 17:485–489
Seo CS, Lee YK, Kim YJ, Jung JS, Hahng Y, Chang HW, Song DK, Son JK (2008) Protective effect of lignans against sepsis from the roots of Saururus chinensis. Biol Pharm Bull 31:523–526
Vane JA (1971) Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 23:232–235
Weisz A, Cicatiello L, Esumi H (1996) Regulation of the mouse inducible-type nitric oxide synthase gene promotor by interferon-gamma, bacterial lipopolysaccharide and N G-monomethyl-l-arginine. Biochem J 316:209–215
Witthoft T, Eckmann L, Kim JM, Kagnoff MF (1998) Enteroinvasive bacteria directly activate expression of iNOS and NO production in human colon epithelial cell. Am J Physiol 275:G564–G571
Won SJ, Park HJ, Lee KT (2008) Inhibition of LPS induced iNOS, COX-2 and cytokines expression by salidroside through the NF-κB inactivation in RAW 264.7 cells. Korean J Pharmacogn 39:110–117
Yoon WB (2011) Effects of particle size and high pressure process on the extraction yield of oil compounds from soybean powder using hexane and supercritical fluid. Food Eng Prog 15:203–208
Yun JY, Choi SY, Park PJ, Chung HG, Shin HM, Suk KH, Lim BO (2008) Extracts of Artemisia princeps pampanini inhibit Lipopolysaccharide-induced nitric oxide, cyclooxygenase-2, prostaglandin E2, and tumor necrosis factor-α production from murine macrophage RAW 264.7 cells. Korean J Med Crop Sci 16:326–331
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lee, EH., Cho, JH., Kim, DH. et al. Anti-inflammatory activity of manassantin A from ultra-fine ground Saururus chinensis in lipopolysaccharide-stimulated RAW 264.7 cells. Appl Biol Chem 60, 63–71 (2017). https://doi.org/10.1007/s13765-016-0249-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13765-016-0249-5