
Abstract
In our previous work, Asterina pectinifera was fermented with Cordyceps militaris mycelia to improve its bioactivities and was reported to have strong antioxidant activities. The aim of the current study was to investigate its anti-inflammatory effect and mechanisms of action. In this study, we observed the inhibitory effect of the extract from fermented A. pectinifera with C. militaris mycelia (FACM) on nitric oxide (NO) production and its molecular mechanism in lipopolysaccharide (LPS)-stimulated RAW264.7 cells. FACM could decrease LPS-induced NO production. Western blot analysis showed that FACM could down-regulate LPS-induced expression of inducible NO synthase without affecting cyclooxygenase-2. Moreover, FACM exhibited anti-inflammatory activity in LPS-induced RAW264.7 mouse macrophage cells through proinflammatory mediators including TNF-α and IL-6 via nuclear factor kappa B pathway. FACM inhibited LPS-induced phosphorylation of extracellular-signal-regulated kinase expression. Our results suggest that FACM may be a potential candidate for inflammation therapy by attenuating the generation of cytokines, production of NO, and generation of ROS in RAW264.7 cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The starfish species, Asterina pectinifera causes critical damage to shellfish mariculture [1]. For this reason, the Korean government has spent about 3.73 million US$ to remove starfishes from coastal areas from 2006 to 2011 [2]. Previous studies have shown that A. pectinifera extract has antimicrobial, anticancer, antimelanogenic, and anti-inflammatory activities [3,4,5,6].
Cordyceps militaris is a kind of Ascomycete, which parasitizes on insect larvae and grows gradually into a mature fruiting body [7]. The parasitic complex of fungus and caterpillar has been used for tonics and medicinal purposes for centuries in eastern Asia [8, 9]. Cordyceps extract has been reported to have various biologically active compounds such as cordycepin, cordycepic acid, adenosine, vitamins, exopolysaccharides, and enzymes [10]. Cordycepin is one of the key bioactive compounds produced by C. militaris and has exhibited antioxidant activity, anticancer activity, and anti-inflammatory properties in various cell lines [11,12,13,14,15].
Fermentation has been recognized as a useful tool for generating biological materials containing health-promoting properties. The culturing techniques are of vital importance to obtain a good yield of bioactive products because these resulting bioactive compounds are directly related to cell proliferation and metabolite biosynthesis [16,17,18].
Inflammation causes a wide range of diseases including cancer, neurological diseases, metabolic syndrome, asthma, obesity, and cardiovascular diseases [19,21,22,23]. Inflammation is a complex process regulated by inflammatory mediators and cytokines secreted from the proinflammatory cells such as macrophages [24].
Macrophages play important roles in inflammation and host defence. One of the most critical mechanisms in macrophages is the production of nitric oxide (NO). NO is produced when l-arginine converts to l-citrulline by NOS (nitric oxide synthase) in the presence of O2 and NADPH. Overproduction of NO leads to numerous inflammatory diseases, for instance, septic shock, tissue injuries, cerebral infarctions, diabetes mellitus, and neurodegenerative disorders [25,26,27].
When infection occurs, the nuclear factor kappa B (NF-κB) signaling pathway is one of the MyD88-dependent lipopolysaccharide (LPS)-induced signaling pathways that is activated after macrophages recognize the infection [28]. When the NF-κB-inhibitory kappa-B alpha (IκB-α) complex, which is located in the cytoplasm, is phosphorylated, the subunits (p65/p50) of the NF-κB are released [28]. Subsequently, NF-κB p65 enters the nucleus and induces the expression of genes which encode numerous transcript genes encode various proinflammatory cytokines including tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6) [29]. Moreover, extracellular-signal-regulated kinase (ERK), which is another downstream MyD88-dependent LPS-induced signaling pathway, plays a critical role in inflammation. When ERK is phosphorylated by (MAPK kinases) MKKs, it stimulates the transcription factor AP-1. Subsequently, AP-1 enters into the nucleus and transcript genes encode proinflammatory cytokines [24].
In our previous study, we reported that fermentation of A. pectinifera with C. militaris mycelia (FACM) could improve its various radical scavenging activities leading to strong antioxidant activity [30]. The current study was designed to investigate the anti-inflammatory activity of FACM through the regulation of LPS-induced cellular ROS production and ERK pathway in the RAW264.7 macrophage.
Materials and methods
Materials
Dulbecco’s Modified Eagle’s medium (DMEM) culture medium and fetal bovine serum (FBS) were obtained from Life Technologies, Inc. (Carlsbad, CA, USA). LPS isolated from Escherichia coli 0111:B4 and N-acetyl-l-cysteine (NAC) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Polymyxin B (PMB) was purchased from InvivoGen (San Diego, CA, USA). TNF-α and IL-6 ELISA kits were bought from R&D Systems (Minneapolis, MN, USA). The antibodies against inducible NO synthase (iNOS), cyclooxygenase-2 (COX-2), IκB-α, phospho-p38, phospho-JNK, phospho-ERK, and β-actin were purchased from Cell Signaling Technology (Beverly, MA, USA). All other reagents were of the highest grade commercially available.
Extraction and isolation
Using the method reported in the previous study, A. pectinifera was fermented with C. militaris mycelia [30]. Briefly, A. pectinifera was cut and washed and then cooled after autoclaved at 121 °C for 15 min. A. pectinifera was inoculated with C. militaris mycelia at 10%; then, it was maintained in an incubator at 25 °C for 20 days. Next, the fermented product obtained after the fermentation process was lyophilized using a freeze dryer (Samwon industry, Seoul, Korea). The lyophilized fermented A. pectinifera was mixed with distilled water to archive tenfold dilution and boiled for 2 h. Then, the resultant extract was filtered through a Whatman No. 41 paper. Finally, the filtrate was evaporated under reduced pressure using a rotary vacuum evaporator (EYELA, Tokyo, Japan) at 50 °C, lyophilized using the freeze dryer, and stored at − 20 °C until use.
Cell culture
The murine macrophage RAW264.7 cells were maintained in DMEM supplemented with heat-inactivated 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in an incubator containing humidified atmosphere with 5% CO2. In all experiments, RAW264.7 cells were incubated in the presence or absence of different FACM concentrations 1 h prior to LPS (100 ng/mL) stimulation.
Cell viability
The effects of different FACM concentrations on RAW264.7 cell viability were evaluated using MTT assay. Briefly, RAW264.7 macrophages were pretreated with different FACM concentrations for 1 h and then exposed to LPS (100 ng/mL) for 20 h. After LPS stimulation for 20 h, 0.5 mg/mL MTT solution was added to each well and the cells were incubated for another 2 h at 37 °C in an incubator containing humidified 5% CO2 atmosphere. Then, the supernatants were aspirated and the resultant formazan crystals in each well were dissolved in 150 μL DMSO. Absorbance at 540 nm was measured using a microplate reader (SpectraMax M2/M2e, Molecular Devices, Sunnyvale, CA, USA). The optical density of MTT formazan formed in untreated cells was considered as 100% viability.
Measurement of NO production
NO production levels in the RAW264.7 cell culture supernatant after different treatments were analyzed by measuring the accumulation of nitrite (NO2 −) in the cell culture medium via reaction with Griess reagent using sodium nitrite (NaNO2) as the standard. The cell culture medium supernatant was collected and mixed with an equal amount of Griess reagent (containing 0.1% N-(1-naphthyl)-ethylenediamine, 1% sulfanilamide, and 5% phosphoric acid). Then, it was shaken lightly for 10 min at room temperature. Finally, the absorbance value at 540 nm was measured using a microplate reader (SpectraMax M2/M2e, Molecular Devices, Sunnyvale, CA, USA).
Measurement of proinflammatory cytokine production
The murine macrophage RAW264.7 cells were plated in 24-well plates at a density of 4 × 105 cells per well and incubated after either LPS stimulation or 1 h pretreatment of FACM (6.25, 12.5, and 25 μg/mL) with LPS stimulation for 18 h. PMB (10 μM) was used as the positive control. Polymyxin B is an antibiotic primarily used for resistant Gram-negative infections. It has a bactericidal action against many Gram-negative bacilli. After the incubation period, the cell-free supernatants were subsequently employed in the proinflammatory cytokine level determination assays, which were performed with a mouse enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Measurement of intracellular ROS
The intracellular ROS accumulation in RAW264.7 cells was determined using the fluorescent dye DCFH-DA. Briefly, after the relevant treatments, the cells were incubated with 10 μM of DCFH-DA for 30 min at 37 °C in the dark. Then, the cells were washed twice with PBS and the intracellular levels of ROS were determined by measuring the fluorescence using a flow cytometer (FACSCalibur, Becton & Dickinson Co., Franklin Lakes, NJ, USA). NAC has been known to act as an antioxidant/free radical scavenger or reducing agent.
Western blotting
After relevant treatments with FACM and/or LPS, the cells were washed with ice-cold phosphate-buffered saline (PBS). Then, they were harvested and lysed immediately. The total protein concentration was assessed using a Bradford protein assay kit (Bio-Rad, Hercules, CA, USA). Following cell lysis, lysed cells were mixed with SDS/PAGE loading buffer and boiled for 5 min at 100 °C. Total protein (20–30 μg) was resolved by SDS/PAGE; then, separated proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane (GE Healthcare Life Sciences, Little Chalfont, Buckinghamshire, UK). Next, the PVDF membrane was washed with Tris-buffered saline Tween (TBS-T, 20 mM Tris–HCl, 150 mM NaCl, and 0.05% Tween 20). Later, nonspecific sites on the membrane were blocked by incubating the PVDF membrane with a blocking solution containing 5% skim milk in TBS-T for 1 h at room temperature. After blocking treatment, the membrane was washed with TBS-T and incubated with diluted primary antibodies at 4 °C overnight. After overnight incubation, the membrane was washed with TBS-T and incubated in a solution containing species-appropriate HRP-conjugated secondary antibodies for 1 h. Finally, the membrane was washed again with TBS-T and reacted with enhanced chemiluminescence reagent for the detection of protein bands with an image analyzer (Davinch-Western™, Youngwha Scientific Co., Seoul, Korea). Densitometric values were normalized using β-actin.
Statistical analysis
Values are presented as the mean ± standard deviation (SD) from triplicate determinations. All the data were analyzed by analysis of variance (ANOVA) followed by Dunnett’s test to determine the significance of differences using GraphPad Prism 5; p < 0.05 was considered as statistically significant.
Results and discussion
Effect of FACM on the viability of RAW264.7 cells
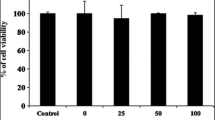
MTT assay was used to assess the cytotoxic effect of FACM on murine macrophage RAW264.7 cells. FACM showed no significant cytotoxicity on RAW264.7 cells at concentrations less than 25 μg/mL in the presence or absence of LPS after 20 h incubation [Fig. 1(A), (B)]. In contrast, higher FACM concentrations (> 25 μg/mL) showed a significant cytotoxicity on RAW264.7 cells. Hence, FACM concentrations ranging from 6.25 to 25 μg/mL were selected for use in subsequent experiments.

Effect of FACM on the viability (A, B) and NO production (C). The FACM (6.25–100 μg/mL) was tested for its cytotoxic effect on RAW264.7 cells using MTT assay. Cells were treated with only FACM for 20 h or pretreated with FACM for 1 h and then exposed to LPS for 20 h. The FACM was tested for its inhibitory effects on LPS-induced NO production using the Griess reagent. The cells were pretreated with FACM for 1 h and then exposed to LPS for 18 h. Data are expressed as the mean ± SD of three independent experiments. ### p < 0.01 versus control group, ***p < 0.001 versus LPS group
Inhibition of LPS-induced NO production by FACM in LPS-stimulated RAW264.7 cells
For the primary screening, we evaluated the effect of FACM on LPS-induced NO production. LPS markedly enhanced NO production in RAW264.7 cells. However, pretreatment with FACM could significantly suppress the LPS-mediated production of NO [Fig. 1(C)]. Compared with PMB, a representative LPS-induced TLR4 activation inhibitor, FACM displayed a similar effect on NO.
Inhibitory effects of FACM on LPS-induced production of proinflammatory cytokines
Proinflammatory cytokines are mainly produced by activated macrophage cells, and they are involved in the up-regulation of inflammatory reactions [31]. To further investigate the anti-inflammatory effect of FACM, the levels of TNF-α and IL-6 in the culture medium of RAW264.7 cells were measured by ELISA. FACM could significantly reduce the levels of LPS-mediated TNF-α and IL-6 production in a dose-dependent manner [Fig. 2(A), (B)], and the effectiveness was approximate to the positive control PMB at 10 μg/mL. Proinflammatory cytokines such as TNF-α and IL-6 are mainly released from macrophages when they are activated by LPS or other bacterial components in an event of infection [31]. Our findings implied that FACM could participate in the anti-inflammatory response, possibly through the reduction of IL-6 and TNF-α expression.

Inhibitory effect of FACM on LPS-induced production of proinflammatory cytokine IL-6 (A) and TNF-α (B). The release of the proinflammatory cytokine IL-6 into the supernatants from RAW264.7 cells incubated with FACM (6.25, 12.5, and 25 μg/mL) and PMB (10 μg/mL) with or without LPS (100 ng/mL) for 18 h was examined by the ELISA method. Data are expressed as the mean ± SD of three independent experiments. ### p < 0.01 versus control group, **p < 0.01, ***p < 0.001 versus LPS group
Measurement of intracellular ROS
Pathogenesis of many diseases is associated with increased ROS accumulation including inflammatory diseases [32]. The imbalance between the free radical production and removal in the cellular system leads to cellular damage by oxidizing macromolecules such as proteins, lipids, and DNA in the cell. Exposing the cells to LPS leads to an increased production of cellular ROS [33], and excess ROS levels are associated with the expression of various inflammations.
The LPS-induced ROS generation in RAW264.7 cells in the presence or absence of FACM was detected using the fluorescent indicator DCFH-DA. As shown in Fig. 3, RAW264.7 cells exposed to LPS (100 ng/mL) showed a significant increase in the intracellular ROS level (83.22 ± 2.26%). In contrast, NAC (positive control, 20 mM) reduced the cellular ROS production (12.14 ± 2.16%). The addition of FACM (6.25–25 μg/mL) could attenuate the LPS-induced ROS generation in RAW264.7 cells in a dose-dependent manner.

Inhibitory effect of FACM on LPS-induced ROS production. RAW264.7 cells were pretreated with FACM for 1 h and incubated with LPS (100 ng/mL) for 18 h. The medium was then replaced with fresh medium containing DCFH-DA (10 μM). After incubation for 30 min, the level of intracellular ROS was determined using a flow cytometer. (A) Control, (B) LPS (100 ng/mL), (C) LPS (100 ng/mL) + FACM (6.25 μg/mL), (D) LPS (100 ng/mL) + FACM (12.5 μg/mL), (E) LPS (100 ng/mL) + FACM (25 μg/mL), and (F) LPS (100 ng/mL) + NAC (20 mM). Values are expressed as the mean ± SD of three independent experiments
Inhibitory effect of FACM on LPS-induced iNOS expression
To investigate whether the inhibitory effect of FACM on NO production is mediated by the expression level of related inflammatory mediators, the effect of FACM on LPS-induced iNOS expression level in RAW 264.7 cells was investigated by Western blotting. RAW264.7 cells were pretreated with various concentrations (6.25, 12.5, and 25 μg/mL) of FACM for 1 h, followed by LPS (100 ng/mL) stimulation for 18 h. The iNOS protein level was markedly upregulated by the LPS treatment. As shown in Fig. 4, FACM could attenuate the high iNOS expression levels upregulated by LPS induction. However, FACM had no effect on the COX-2 expression levels in RAW264.7 cells (data not shown). COX-2 is associated with the conversion of arachidonic acid to prostaglandin E2 [34]. COX-2 is believed to be responsible for the production of proinflammatory prostaglandins in various models of inflammation [35]. As reported in this study, the inhibition of NO production through the down regulation of iNOS protein expression level while there is no effect on COX-2 protein expression level has been previously reported [36] and it is possible due to the fact that FACM could possibly contain more than a single biologically active compound that could exert effects on different molecules to different extents in the LPS-induced inflammatory signaling pathway, leading to its final anti-inflammatory property.

Inhibitory effects of FACM on LPS-induced iNOS expression. Expression levels of iNOS protein in RAW264.7 cells incubated with the FACM (6.25, 12.5, and 25 μg/mL) or PMB (10 μg/mL) in the presence/absence of LPS (100 ng/mL) for 18 h were examined by Western blot analysis
Inhibitory effect of the FACM on LPS-induced activation of MAPKs
The MAPKs, including ERK1/2, JNK1/2, and p38 MAPK, signaling pathway has a key role in regulating the production and secretion of inflammatory mediators in LPS-stimulated macrophages [37]. Previous studies have reported that LPS-induced activation of MAPKs up-regulates the synthesis of proinflammatory cytokines (TNF-a, IL-6, and IL-1β) and iNOS expression level [33]. The MAPKs could be activated by LPS, a key stimulator of the inflammation response in macrophages as well as many cell types [36]. To determine whether MAPKs are affected by FACM, phosphorylation levels of ERK1/2, JNK1/2, and p38 MAPK were analyzed using Western blotting. As shown in Fig. 5, the expression of phosphorylated ERK1/2, JNK1/2, and p38 MAPK protein levels were increased by the LPS treatments, whereas these increased phosphorylation levels of MAPK proteins induced by LPS were attenuated by PMB pretreatment (positive control, 10 μg/mL). However, our results showed that FACM could only attenuate the increased phosphorylation level of ERK1/2 induced by LPS but not the p38 and JNK protein levels.

Inhibitory effect of FACM on LPS-induced activation of MAPKs. Effects of FACM on LPS-induced activation of MAPKs, including ERK, p38, and JNK, in RAW264.7 were examined after cell treatments with different FACM concentrations (6.25, 12.5, and 25 μg/mL) in the presence or absence of LPS (100 ng/mL) for 30 min using Western blot analysis
Effect of FACM on IκB-α degradation
Because IκB-α degradation is thought to be the key step in NF-κB activation, we investigated the effect of FACM on IκB-α degradation. NF-κB is inactivated in the cytosol by binding with IκB-α. Induced phosphorylation level of IκB-α leads to its subsequent ubiquitination and degradation via the proteasome pathway leading to translocate freed NF-κB into the nucleus [28]. To determine whether FACM could possibly affect NF-κB activation, the degradation of IκB-α was assessed. RAW264.7 cells were pretreated with different FACM concentrations for 1 h before exposure to LPS for 30 min, and the IκB-α protein levels were examined using Western blotting analysis. As shown in Fig. 6, stimulation with LPS for 30 min induced the degradation of the IκB-α protein. Pretreating the cells with FACM could protect the LPS-induced IκB-α degradation and PMB (as a positive control) also rescued IκB-α from its LPS-induced degradation. These findings indicate that FACM has an effect on IκB-α/NF-κB activation pathway. These results indicate that FACM inhibits NO production and iNOS expression through the suppression of NF-κB activity mediated by blocking of IκB-α degradation. Our current study may contribute to the knowledge of understanding the FACM’s anti-inflammatory property by recognizing its ability to down-regulate NO production through iNOS signaling pathways and suppression of cellular ROS production.

Effect of FACM on IκB-α degradation RAW264.7 macrophages were pretreated with FACM (6.25, 12.5, and 25 μg/mL) and PMB (10 μg/mL) for 1 h and then stimulated with LPS (100 ng/mL) for 15 min. Total cellular proteins were prepared and Western blotted for IκB-α
References
Kang KH, Kim JM. The predation of trumpet shell, Charonia sp., on eight different marine invertebrate species. Aquacult Res. 35: 1202–1206 (2004).
National Federat. Fish. Cooperate, Report of National Federation of Fisheries Cooperatives about starfish removal project, Seoul (2011).
Seo JK. Conformation and biological activity of Mastoparan B and its analogs: Isolation and characterization of the biological substances from inshore hagfish (Eptatretus burgeri) skin and starfish (Asterina pectinifera), Thesis, Pukyong National University (1997).
Jo WS, Choi YJ, Kim HJ, Nam BH, Lee GA, Seo SY, Lee SW, Jeong MH. Methanolic extract of Asterina pectinifera inhibits LPS-induced inflammatory mediators in murine macrophage. Toxicol Res. 26: 37–46 (2010).
Jeong MH, Yang KM, Kim JM, Nam BH, Kim GY, Lee SW, Seo SY, Jo WS. Inhibitory effects of Asterina pectinifera extracts on melanin biosynthesis through tyrosinase activity. Int J Mol Med. 31: 205–212 (2013).
Nam KS, Shon YH. Chemopreventive effects of polysaccharides extract from Asterina pectinifera on HT-29 human colon adenocarcinoma cells. BMB Rep. 42: 277–280 (2009).
Yang CH, Kao YH, Huang KS, Wang CY, Lin LW. Cordyceps militaris and mycelial fermentation induced apoptosis and autophagy of human glioblastoma cells. Cell Death Dis. 3: e431 (2012).
Stone R. Last stand for the body snatcher of the himalayas? Science 322: 1182 (2008).
Stone R. Improbable partners aim to bring biotechnology to a himalayan kingdom. Science 327: 940–941 (2010).
Tuli HS, Sharma AK, Sandhu SS. Optimization of fermentation conditions for cordycepin production using Cordyceps militaris 3936. JBCS 1: 35–47 (2014).
Yoshikaw N, Nakamura K, Yamaguchi Y, Kagota S, Shinozuka K, Kunitomo M. Antitumour activity of cordycepin in mice. Clin Exp Pharmacol Physiol. 31: S51–S53 (2004).
Chen Y, Chen YC, Lin YT, Huang SH, Wang SM. Cordycepin induces apoptosis of CGTH W-2 thyroid carcinoma cells through the calcium-calpain-caspase 7-PARP pathway. J Agric Food Chem. 58: 11645–11652 (2010).
Wehbe-Janek H, Shi Q, Kearney CM. Cordycepin/hydroxyurea synergy allows low dosage efficacy of cordycepin in MOLT-4 leukemia cells. Anticancer Res. 27: 3143–3146 (2007).
Xu HL, Zhang LJ, Shi H, Zhu X, He X. Effects of cordycepin on Hep G2 and EA. hy926 cells: potential antiproliferative, antimetastatic and anti-angiogenic effects on hepatocellular carcinoma. Oncol Lett. 7: 1556–62 (2014).
Won SY, Park EH. Anti-inflammatory and related pharmacological activities of cultured mycelia and fruiting bodies of Cordyceps militaris. J Ethnopharmacol. 96: 555–561 (2005).
Tang YJ, Zhong JJ. Fed-batch fermentation of Ganoderma lucidum for hyperproduction of polysaccharide and ganoderic acids. Enzyme Microb Tech. 31: 20–8 (2002).
Park JP, Kim SW, Hwang HJ, Yun JW. Optimization of submerged culture conditions for the mycelial growth and exo-biopolymer production by Cordyceps militaris. Lett App Microb. 33: 76–81 (2001).
Fang QH, Zhong JJ. Submerged fermentation of higher fungus Ganoderma lucidum for production of valuable bioactive metabolites—ganoderic acid and polysaccharide. Biochem Eng J. 10: 61–5 (2002).
Baniyash M, Sade-Feldman M, Kanterman J. Chronic inflammation and cancer: suppressing the suppressors. Cancer Immunol Immunother. 63: 11–20 (2014).
Ferreira ST, Clarke JR, Bomfim TR, De Felice FG. Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer’s disease. Alzheimers Dement. 10: S76–S83 (2014).
Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract. 105: 141–150 (2014).
Golia E, Limongelli G, Natale F, Fimiani F, Maddaloni V, Pariggiano I, Bianchi R, Crisci M, D’Acierno L, Giordano R, Palma GD, Conte M, Golino P, Russo MG, Calabrò R, Calabrò P. Inflammation and cardiovascular disease: from pathogenesis to therapeutic target. Curr Atheroscler Rep. 16: 1–7 (2014).
Li H, Yoon JH, Won HJ, Ji HS, Yuk HJ, Park KH, Park HY, Jeong TS. Isotrifoliol inhibits pro-inflammatory mediators by suppression of TLR/NF-κB and TLR/MAPK signaling in LPS-induced RAW264.7 cells. Int Immunopharmacol. 45: 110–119 (2017).
Abarikwu SO. Kolaviron, a natural flavonoid from the seeds of Garcinia kola, reduces LPS-induced inflammation in macrophages by combined inhibition of IL-6 secretion, and inflammatory transcription factors, ERK1/2, NF-kappaB, p38, Akt, p-c-JUN and JNK. Biochim Biophys Acta. 1840: 2373–2381 (2014).
Park PJ, Je JY, Kim SK. Free radical scavenging activities of differently deacetylated chitosans using an ESR spectrometer. Carbohydr Polym. 55: 17–22 (2004).
Chung HT, Pae HO, Choi BM, Billiar TR, Kim YM. Nitric oxide as a bioregulator of apoptosis. Biochem Biophys Res Commun. 282: 1075–1079 (2001).
Xie QW, Cho HJ, Calaycay J, Mumford RA, Swiderek KM, Lee TD. Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science 256: 225–228 (1992).
Asehnoune K, Strassheim D, Mitra S, Kim JY, Abraham E. Involvement of PKC alpha/beta in TLR4 and TLR2 dependent activation of NF-κB. Cell Signal. 17: 385–394 (2005).
Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 17: 1–14 (2005).
Kim YS, Kim EK, Natarajan SB, Hwang JW, Kim SE, Jeon NJ, Lee JW, Jeong JH, Kim HJ, Park PJ. Radical scavenging activities of Asterina pectinifera fermented with Cordyceps militaris mycelia. Food Sci Biotechnol. 25(S): 97–101 (2016).
Duque GA, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Frontiers in Immunol. 5(491): 1–12 (2014).
Hancock JT, Desikan R, Neill SJ. Role of reactive oxygen species in cell signaling pathways. Biochem Soc Trans. 29: 345–350 (2001).
Kao SJ, Lei HC, Kuo CT, Chang MS, Chen BC, Chang YC, Chiu WT, Lin CH. Lipoteichoic acid induces nuclear factor-kappa B activation and nitric oxide synthase expression via phosphatidylinositol 3-kinase, Akt, and p38 MAPK in RAW 264.7 macrophages. Immunol. 115: 366–374 (2005).
Murakami A, Ohigashi H. Targeting NOX, INOS and COX-2 in inflammatory cells: chemoprevention using food phytochemicals. Int J Cancer. 121: 2357–2363 (2007).
Subbaramaiah K, Dannenberg AJ. Cyclooxygenase 2: a molecular target for cancer prevention and treatment. Trends Pharmacol Sci. 24: 96–102 (2003).
Jung CH, Jung H, Shin YC, Park JH, Jun CY, Kim HM, Yim HS, Shin MG, Bae HS, Kim SH, Ko SG. Eleutherococcus senticosus extract attenuates LPS-induced iNOS expression through the inhibition of Akt and JNK pathways in murine macrophage. J Ethnopharmacol. 113: 183–187 (2007).
Coskun M, Olsen J, Seidelin JB, Nielsen OH. MAP kinases in inflammatory bowel disease. Clinica Chimica Acta. 412: 513–520 (2011).
Acknowledgements
This research was a part of the project, the Development and Industrialization of Organism Defense Regulatory Materials from Extracts of Starfish Fermented with Cordyceps militaris, funded by the Ministry of Oceans and Fisheries, Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Kim, YS., Shin, WB., Dong, X. et al. Anti-inflammatory effect of the extract from fermented Asterina pectinifera with Cordyceps militaris mycelia in LPS-induced RAW264.7 macrophages. Food Sci Biotechnol 26, 1633–1640 (2017). https://doi.org/10.1007/s10068-017-0233-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10068-017-0233-9