
Abstract
Herein, highly efficient sorbent with chelating and ion-exchange properties was synthesized by assembling diethylenetriamine groups on the mesoporous hybrid magnetic-chitosan core–shell nanocomposite by heterogeneous nucleation. Different analytical methods were used to investigate the physicochemical and functional characteristics of the obtained material. The structural features were superparamagnetic (MS: 30.73 emu g−1), specific surface area (~ 75.27 m2 g−1) with a dominant mesopores sizes (~ 3–9 nm), high porosity (~ 0.57 cm3 g−1) and amine content (~ 3.68 mmol g−1) and density (~ 7.36 1/nm2). Uranyl ions sorption and selectivity characteristics were exploited. Isotherm fitted by Langmuir equation: the maximal sorption capacity was reported ~ 0.75 mmolU g−1 at optimum pH0 3.5–5.0, and 298 K. Fast uptake kinetics (~ 40 min) were suited using a pseudo-second order rate equation. The sorption activation energy was 20.43 kJ mol−1. The half-sorption time (t0.5) was duplicated from 2.1 min to 4.2 min at 298 K and 323 K, respectively, indicating exothermicity. FTIR and XPS examinations demonstrated that the binding of UO22+ occurred at N- and O-functional groups via anion exchange, electrostatic attraction, chelation, and reduction mechanisms. Metal ions were recovered using acidified urea (0.25 M) with strong cycle performance and retention rate ~ 85.2% after eight sorption/desorption cycles. The sorbent was successfully evaluated for selective uranium sorption from multi-component acidic nuclear solution. Owing to such stability and reusability over multiple sorption cycles, the sorbent is promising in the field of uranium recovery. Finally, the sorption process was described using a new three-dimensional mathematical model for nonlinear quantitative description.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
With an increasing and ongoing need for energy, nuclear energy is the perfect clean energy and carbon-free answer to climate change and greenhouse gas emissions. Uranium recovery from its effluents has sparked a surge in research interest to provide a viable technique of sustaining the nuclear fuel cycle for nuclear energy production. Since uranium is necessary for the nuclear energy generation and the impending scarcity of uranium resources make this issue economically significant. Also, its adequate toxicity makes wastewater treatment a critical environmental concern. Thus, uranium separation and recovery are therefore essential for both efficient use of uranium resources and environmental conservation (Imam et al., 2018; Huang et al. 2019; Xie et al. 2019; Bai et al. 2020; Galhoum et al. 2020; Liu et al. 2020; Jun et al. 2021). For metal ion recovery the sorption technique (ion-exchange and chelating sorbents) is an acceptable, fascinating, and outstanding technology. Because of its versatility, selectivity, efficiency, rapid kinetics, and environmental friendliness (Imam et al. 2018; Liu et al. 2020). Complexing and chelating materials with multidentate N- and/or O-donor ligands, or a mix of both, which operate as basic complexing groups and their ‘‘spatial layout’’ permits to exhibit fascinating selective extraction capabilities. These sorbents have been widely researched as effective materials with specific functional groups that exhibit a selective affinity for uranyl ions (Elwakeel and Atia 2014; Galhoum et al. 2017; Rashad et al. 2021). The chelating characteristics closely follow Pearson's hard and soft acid–base theory (HSAB) (Pearson 1966). As a hard acid, uranium has a stronger affinity and greater reactivity for hard bases like amine groups than it does for carboxylic groups (Song et al. 2012; Rashad et al. 2021). Through interactions with chelating agents bearing O and N functionalities, the great range of reactive groups contained on different sorbents may explain their general attractiveness for uranyl species (Rashad et al. 2021). According to the HSAB concept, an alkaloid, 5-azacytosine (Acy) (Song et al. 2012), polyamine (Wang et al. 2011; Xu et al. 2013; Elwakeel and Atia 2014), amidoxime groups (Wang et al. 2014; Zhang et al. 2018), and aminophosphonate functionality (Imam et al. 2018; Galhoum et al. 2020; Rashad et al. 2021) belongs to multidentate N-donor ligands that have been shown noticeable selectivity toward U(VI) sorption. The amine group's acid–base characteristics result in strong ion-exchange and electrostatic attraction features (Xu et al. 2013; Elwakeel and Atia 2014; Galhoum et al. 2020): anionic metal ions species can bind to protonated amine groups (Galhoum et al. 2017; Imam et al. 2018). Grafting multifunction reactive groups improves the selectivity, sorption abilities, and pH range of sorption. There are several chelating ligands for U(VI) ion sorption, for example, catechol, polyamines (Elwakeel and Atia 2014), imino-diacetic acid and/or -phosphonic acid, phenylarsonic acid (Oshita et al. 2009), amino acids (Oshita et al. 2007; El-Bohy et al. 2017; Galhoum et al. 2017), Schiff’s base chitosan composite (Elwakeel and Atia 2014; Yang et al. 2017), and tryptophan-porous organic polymer (Bai et al. 2020). Designing new multiphase nanomaterials with high capacity and selectivity for uranium sorption is the art of creating organic–inorganic functional hybrid nanocomposite (Xie et al. 2019). Biosorbents, which are made from low-cost biological materials derived from renewable resources, offer the following advantages: high-efficiency, minimal chemical sludge, biosorbent regeneration, and metal recovery (Xu et al. 2013; Benettayeb et al. 2021).
Chitosan (CS, as amino-polysaccharide) was chosen because of its chemical compatibility with the magnetite particle manufacturing technique (Galhoum et al. 2017). The synergy between the individual components to meet the requirements of ideal sorbents and enhanced characteristics (e.g., multifunctionalization, chemical stability, adsorptive property, kinetics, and being economically) (Sen et al. 2016; Benettayeb et al. 2021; Morshedy et al. 2021). This is the primary motivation for developing functionalized chitosan-based hybrid magnetic materials. Magnetic nanocomposites containing Fe3O4 nanoparticles are a unique sort of composite (Galhoum et al. 2017, 2020; Pourmortazavi et al. 2019). The magnetic solid phase extraction technology has been widely used for pollutant removal because it considerably improves specific surface area (that reduce limits caused by intraparticle diffusion), quick, easy, sensitive and incredibly effective. Besides, depleted sorbents can be recovered using an external magnetic field once the sorption process is ended (Galhoum et al. 2015a, b; Kharissova et al. 2015; Pourmortazavi et al. 2019). The thorough and in-depth characterization required for mechanistic investigations of the synthesis processes, chemical modifications, sorption and desorption mechanisms, and selectivity for uranyl ions has not yet been documented in any literature. Therefore, in this study, magnetic nanoparticles were co-generated in situ during chitosan precipitation, resulting in a magnetic-chitosan nanocomposite (MCS), which was then further modified using diethylenetriamine (TA-MCS) via continuous reaction. The physicochemical characteristics were determined by CHN, XRD, XPS, pHzpc, TEM, VSM, and FTIR. As-synthesized TA-MCS nanocomposite was tested for the UO22+ sorption from aqueous mediums as a function of the sorbent dosage, initial pH, contact time and kinetics, sorption isotherms, thermodynamic characterization, and sorbent regeneration and recycling. Furthermore, the comprehensive synthesis of magnetite and the production of magnetic nanocomposite were given in detail. Experimental and spectroscopic combined research were used to study the sorption and desorption mechanisms. Finally, uranyl ions were selectively recovered from synthetic radioactive effluents. Moreover, an innovative modeling approach has been presented to enable quantitative nonlinear representation in 3D space. As an efficient mathematical approach for evaluating sorption capabilities at certain initial pH and concentrations.
Materials and methods
Chemicals and reagents
Chitosan (DD 93%) was supplied by Acros Organics. Epichlorohydrin (CAS: 106-89-8), 1,4-dioxane (CAS: 123-91-1), diethylenetriamine, Arsenazo III, FeCl2.4H2O (≥ 99%) and FeCl3 (≥ 99.99%) were provided from Sigma-Aldrich. All reagents were used exactly as they were given without any purification. Uranyl acetate (UO2(OCOCH3)2·2H2O) (Sisco Research Laboratories Pvt. Ltd, India) was dissolved in ultrapure water to prepare a stock solution (1000 mg U L−1). The uranium content was analyzed using UV/Vis spectrophotometer (Marczenko et al. 1976).
Preparation of TA-MCS sorbent
A hybrid nanocomposite of magnetic-chitosan was prepared through one-pot hydrothermal co-precipitation of Fe(II) and Fe(III) mixture in chitosan solution with NaOH (Galhoum et al. 2015a, b; Morshedy et al. 2021; Al-Ghamdi et al. 2022). Chitosan powder (2 g) was steeped and dissolved in acetic acid solution (200 mL, 5% w/w) with ferrous chloride (FeCl2, 2.42 g), and ferric chloride (FeCl3, 3.82 g) (1:2 molar ratio, respectively). At 40–45 °C, the mixture solution was precipitated by adding NaOH (2 M) dropwise with steady stirring at pH 10.5–11. The suspension was heated for 1 h at 90 °C while stirring constantly, and the particles were magnetically collected. The freshly magnetic-chitosan nanocomposite was chemically enhanced by cross-linking with an epichlorohydrin solution (0.01 M) at 1:1 mass ratio under alkaline conditions (pH ~ 10). The suspension mixture was heated at 50 °C for 2 h with constant stirring. At the end, the cross-linked chitosan-magnetic nanocomposite was magnetically collected and washed extensively with ethanol/water to eliminate any unreacted reagent.
Diethylenetriamine was functionalized cross-linked chitosan-magnetic nanocomposite as depicted in Scheme 1 as a generic approach. The cross-linked chitosan-magnetic nanocomposite was suspended in aqueous ethanol solution (75 mL, 1:1) before adding 10 mL of epichlorohydrin; refluxing the mixture at 65–70 °C for 3 h yielded the desired results (Benettayeb et al. 2021). The product was filtered and rinsed thoroughly with ethanol and ultrapure water. Finally, the activated product and diethylenetriamine (10 mL) were added to 50 mL of DMF. The mixture was refluxed at 75–80 °C for 18 h (Galhoum et al. 2017). After the reaction, the final product was magnetically separated and washed extensively with ethanol and ultrapure water. The sorbent was finally freeze-dried.

Synthesis of TA-MCS nanocomposite (a) and revised process for triamine grafting via SN1 mechanism (b)
Characterization
(C, H, and N) was determined using an automatic analyzer (CHNS Vario EL III-elementar analyzer, Elementar, Germany). A Philips X-ray generator type PW 3710/31, Cu Kα (λ = 1.544) (Philips, Japan) was used to measure X-ray diffraction. The FTIR spectra were analyzed by a Thermo-Fisher Nicolet IS10 spectrometer (Waltham, MA, USA). The morphology and size distribution were analyzed by TEM-JEOL-2100 transmission electron microscope (JOEL, Tokyo, Japan). A Quantachrome NOVA 3200 (Boynton Beach, FL, USA) analyzer was used to measure the surface area using N2-adsorption isotherms under a degassing temperature of 40 °C for 12 h, while the pore volume was measured according to the BJH method. The magnetic characteristics were determined using a vibrating sample magnetometer (VSM; 730 T, Lakeshoper, USA). The K-Alpha spectrometer (Thermo Fisher Scientific, Waltham, USA) was used to acquire X-ray photoelectron spectroscopy (XPS) data using a monochromatic Al Kα-radiation source (200 W). The sorbent's pH zero-point of charge (pHZPC) was measured using the pH-drift method (Benettayeb et al. 2021): the sorbent was equilibrated for 24 h under agitation with a series of 0.1 M NaCl solutions with different initial pH-values (pH0); the equilibrium pH (pHeq) was recorded and the pHZPC is the pH at which ΔpH = 0. The samples preparation and measurements mostly were conducted as a research member of Prof. Ahmed A ALGhamdi group at the laboratories of King Abdulaziz University.
Batch sorption and desorption experiments
The sorption properties of synthesized sorbent were investigated: a constant UO22+ solution (V: 25 mL, C0: 0.21 mmolU L−1) was agitated and equilibrated with a given amount of sorbent (5 mg) for 180 min. at room temperature (298 ± 1 °C), 200 rpm and initial pH (pH0 4.0): except when the effect of initial pH (the pH0: 1.5–5.7), equilibration time (from 0 to 180 min. at pH0: 4.0), isotherm at various concentrations (C0: 0.093–1.253 mmolU L−1), temperature (at 298–323 ± 1 K) and sorbent dose (SD: 0.1–1.5 g/L−1) on the sorption capacities. After equilibrium and phase separation, the initial, residual UO22+ concentration at equilibrium and at different interval times (C0, Ceq, and Ct: mmolU L−1) was determined by the Arsenazo III method (using a double beam UV-11601, UV–Vis spectrophotometer (Shimadzu, Japan). The sorption capacity (qeq, mmolU g−1) was evaluated by the mass balance equation: qeq = (Co − Ceq) V/m, and the distribution coefficient (D: qeq/Ceq) in L g−1, where m (g) is the amount of sorbent and V(L) is the solution's volume. For simulating sorption kinetics and isotherms, the fundamental equations are listed in Table AM1 (see Additional Materials Section).
To investigate the selectivity for U(VI), the effect of competing cations from a synthetic simulated nuclear effluent was studied. A typical study was preformed using most common metals ions in industrial nuclear effluents, such as (REs(III): La, Ce, Eu, and Yb) and other co-ions (Co, Cd, Cs, Sr, and Ba). Equimolar multi-solutions were prepared for this study (using their nitrate and chloride salts) (Al-Ghamdi et al. 2022). The sorption studies were conducted at room temperature (298 ± 1 K), 0.5 g L−1 sorbent dose for an hour with and agitation (200 rpm). After filtration, the remaining Co, Cd, Cs, Sr, and Ba concentrations were measured by GBC atomic absorption 932AAS, manufactured by GBC Scientific Equipment Ltd. in Braeside, Victoria, Australia. Uranium and rare earth ions were determined by ICP spectrometry (ICP-OES-500, PerkinElmer, Norwalk, MA, USA). Standard experimental parameters for metal ions sorption were optimized as follow: pH: 4.05, SD: 0.2 g/L, C0: 0.74 mmolU L, T: 298 ± 1 K, and time: 60 min.
The optimization of the regeneration/recycling (for 8 successive sorption/desorption cycles) of the sorbent using acidified-urea (50 mL, 0.5 M) with a fixed sorbent weight (10 mg). The contact time was set to 30 min at 200 rpm and 298 K. At each phase, the sorption and desorption efficiencies for eight cycles were evaluated and compared to the value reached for the first cycle. The equation that describes desorption efficiency (DE = CD × V(L) × 100/qd × md) and regeneration efficiency (RE = qd × 100/qe) were systematically calculated. where qd (mmolU g−1) is the sorption capacity before first desorption, CD (mmolU L−1) is the uranium concentration in the eluate, VL is the volume of the eluent, qe (mmolU g−1) is the sorption capacity at the first cycle, and md (g) is the amount of sorbent employed in the desorption stage. The relative errors of the data were less than 6%. The pHs were adjusted using either solutions of 0.05 M/0.2 M NaOH/H2SO4 solutions, whenever low/larger pH adjustments were necessary.
Results and discussion
Basics of chitosan–magnetic nanocomposite
Magnetic-chitosan nanocomposite was synthesized by heterogeneous nucleation reaction, as Fe3O4 nanocrystals were employed as the nucleation for the chitosan film layer from the solution (Morshedy et al. 2021; Al-Ghamdi et al. 2022). A simple general procedure for the manufacture of nano-magnetic nanoparticles (Fe3O4) begins immediately after NaOH is added to the iron salts mixture, and the reaction is somewhat exothermic. The chemical reaction in question can be stated as follow (Eq. 1) (Al-Ghamdi et al. 2022):
This reaction takes place under N2(g), to prevent Fe(II) from oxidizing to Fe(III). The magnetite can be oxidized to other ferric hydroxide phases, e.g., maghemite, goethite, and some time to hematite (Pourmortazavi et al. 2019; Winsett et al. 2019).
Magnetic-chitosan nanocomposites are cross-linked with epichlorohydrin to avoid both dissolving in acidic conditions and keeping sorption capacity from declining when amine groups are added (i.e., aldehyde cross-linking is used) (Galhoum et al. 2015a, b). In fact, the cross-linking unifunctional agent is used to form covalent bonds with the C-atoms linked to the hydroxyl groups of chitosan (related to the epoxide ring rupture and a chlorine atom being released) (Galhoum et al. 2015a, b). The synthesis of magnetic polyaminated-functionalized chitosan nanocomposite is depicted in Scheme 1a. The postulated mechanism of the polyaminated product is A typical substitution nucleophilic unimolecular (SN1) reaction and favored in protic solvent such as ethanol (Wagner and Marshall 2010). The nucleophilic displacement of chloride with electron rich amine nitrogen takes place via the carbocation intermediate formation which is stabilized by ethanol solvation this will favored the product formation.
Sorbent characterization
Element analysis
The elemental analysis (CHN, wt. %) is compared during the synthesis procedure (Table AM2). When raw-chitosan (r-CS) and magnetic-chitosan's (MCS) organic fractions (related to C and N elements) are compared, the weight percentage of raw-chitosan is significantly lower (~ 52.7–53.9%): this is related to Fe3O4 nanoparticles impact. The organic fraction is ~ 46.5%, whereas Fe3O4 accounts ~ 53.5% of total weight, consistently with mass balance experiments. Surprisingly, the increase in mass of magnetite chitosan nanocomposite was about 5.35 g compared to the theoretical calculation (4 g) based on the initial chitosan-to-magnetite weight ratio (2 g/2 g, w/w). After cross-linking step, the N-content (mmol, %) decreases from 2.56 to 2.17, due to increasing epichlorohydrin binding (N-atoms thus, N-content decreased in the final products), consistently with the slightly reduce in the C and H contents. While after activation and insertion of terminal –Cl-atom via epichlorohydrin reaction (AMCS); the molar mass steadily increased; therefore, the C, H, N contents were “diluted”; and the change in the weight percent (Δwt %) were 10.8, 14.4, and 18.3%, respectively. By analogy, a similar result was observed from immobilizing Cl-atoms by the reaction of thionyl chloride (SOCl2), for subsequent immobilization of amine-bearing moieties. After chlorination the fraction of C and H elements strongly deceased by about 21.8% and 24.5%, respectively, as a result of the Cl atom's influence on the final product (El-Bohy et al. 2017). Finally, after diethylenetriamine grafting, the increased C, H, and N contents clearly demonstrate a successful synthesis process, with the N wt% increasing approximately 2.6 times from 1.98 to 5.15%.
For r-CS and TA-MCS (SBET: 4.56 and 75.27 m2 g−1, respectively), the symbolical amine density (= number of amine molecules/specific surface area of (nm2 g−1)) was around 713.74 and 7.36. The dispersion of amine density is likely owing to the inclusion of various organic reagents during the synthesis process.
pHZPC: drift titration
Figure AM1A depicts the raw-CS, MCS and TA-MCS titration profiles. The acid-based characteristics are greatly affected by the sequential chemical changes (Morshedy et al. 2021). The pHZPC of raw CS was 8.52, which corresponds to the pKa-value of 6.3–6.6 for the –NH2 groups of chitosan biopolymer (Imam et al. 2018). The pHZPC of raw-CS has been reduced to 7.29 ± 0.02 in MCS with the inclusion of the Fe3O4 core. Contrary to the DETA grafting, the pHZPC increased to 8.48 ± 0.02. This is in accordance with charge density, screening effect, and quantity of hindered Fe3O4 (Benettayeb et al. 2021). This means that these materials undergo complete protonation in acidic solutions. At pH 4.0, the noticeable increase in ∆pH is attributed to the highest protonation degree for MCS and TA-MCS; after that, protonation decreases as pH0 increases. The increase of the pHZPC after polyamine grafting was proportional with the amine groups' pKa values (3.58, 8.86 and 9.65) in DETA (Willianms 2017). Fraga et al. recorded pHZPC ~ 8.20 for DETA-functionalized graphene oxide (Fraga et al. 2019), where ∆pHZPC ~ 0.28 pH unit (the difference between them has been obtained as ~ 8.48), which may be related to the matrix effect from chitosan to graphene oxide.
Magnetic properties
The magnetic curves of MCS and TA-MCS were recorded and compared using VSM at room temperature (Figure AM1B). Obviously, both samples are superparamagnetic and characterized with no hysteresis loop or remanence and coercivity. MCS magnetic saturation (Ms) is 42.53 emu/g; however, the raw material (MCS) functionalization (DETA grafting) reduces the Ms to 30.73 emu g−1. The Ms values obtained are substantially lower than those recorded from bulk magnetite NPs (~ 80 emu/g) (Pourmortazavi et al. 2019). The polymer coating molecular weight rises as follows: MCS < TA-MCS, implying that the proportion of Fe3O4 core in the composite gradually declines, in-line with the drop in Ms values (Pourmortazavi et al. 2019; Morshedy et al. 2021). Furthermore, the Ms values correlated to the Fe3O4 content in the composites and the thickness of the organic film layer results in a decrease in magnetic characteristics. These Ms values, however, are sufficient for facile and effective magnetic separation from the aqueous phase.
TEM analysis
TEM micrographs of TA-MCS nanocomposite are shown in Fig. 1A. The Fe3O4 nanoparticles have appeared as spherical dense dots surrounded by bright areas that represent the polymeric shell, because of the electron-absorbing disparities between organic and inorganic components (Pourmortazavi et al. 2019), with a regular spherical structure, well proportioned, and apparently mono-dispersed. However, iron oxide nanoparticles have a tendency to aggregate, owing to magnetic interaction between dipoles/dipoles (Galhoum et al. 2015a, b; Stoia et al. 2016; Galhoum et al. 2020). This demonstrates that the Fe3O4 nanoparticles were successfully incorporated into the polymer matrix. The aggregation confirms the strong magnetism, as measured by magnetism experiments. The actual size of the Fe3O4 NPs was visualized using the particle size detected on the TEM images. Image software was used to get the size distribution histograms counting of at least 70 particles. The average particle size was determined to be 8.45 ± 1.4 nm, with a narrow particle size distribution using histograms counting (Fig. 1B), which matched the XRD data. Fe3O4 particles have a diameter of less than (< 20 nm), indicating that the sorbent is a nanocomposite.

TEM micrographs (a), histograms analysis for particle size distribution (b), selected area electron diffraction pattern (SAED) (c), and HRTEM image of crystal lattice (d)
The SAED pattern (Fig. 1C) exhibits that the majority of the particles visible in the bright-field picture are orderly spherical. Also, this pattern shows an inverted spinel structure (magnetite or hematite), supporting the two phases inferred from the XRD. With precisely scanning the patterned SAED rings, it is evident that the (311) peak of the spinel structure contributed to the initial intense wide ring (magnetite or hematite), demonstrating Fe3O4's excellent single-crystal epitaxial growth (Shahrashoub et al. 2021). Moreover, the spinel structure's (511) and (220) planes were seen. The HRTEM picture (Fig. 1D) shows that the NP ensemble is made up of a variety of single crystals that have polycrystalline particles. Additionally, the Fe3O4 planes' interplanar spacing (311) is well concordant with the lattice fringes distance of 2.48 Å (0.248 nm) (Galhoum et al. 2020; Shahrashoub et al. 2021).
XRD analysis
The XRD patterns of TA-MCS displayed in Fig. 2A show well-defined khl: (111), (220), (311), (400), (422), (511), (440), and (533) of the eight diffraction planes of the cubic spinel structure of Fe3O4. These results are in harmony with the database in PDF No. 01-076-7165 (Stoia et al. 2016). However, the potential of hematite (Fe2O3) coexisting with ferromagnetic characteristics cannot be ruled out. From XPS, the Fe 2p band on the sorbent spectrum can be deconvoluted into three signals (each with its own doublet signal) related to Fe(III) under both octahedral and tetrahedral forms, and Fe(II) (octahedral form). The resolution of the relevant satellite bands is poor. The Fe 2p3/2 signal is located at 710.6 eV; this might be delegated to the most common types of iron oxides, e.g., Fe3O4 (hematite) or g-Fe2O3 (maghemite) compounds (Kong et al. 2010; Sharma et al. 2016). The size was estimated to be ~ 9.1 nm using the highest and strongest peak matching to the (311) index (utilizing the Scherrer formula). This size is systematically (< 20 nm) reported for enhanced superparamagnetic character.

XRD of the TA-MCS nanocomposites (XRD-standard magnetite and hematite pattern is shown by the bar-type spectrum) (A), and the unit cell and atomic structure of the spinel structure (B). Textural characterization: N2-adsorption–desorption isotherms (C), and pore size analysis-PSDs (D)
Based on the above results, the structural specifics of magnetite are presented in Fig. 2B. Magnetite is made up of Fe2+Fe23+O4 (generally so-called mixed oxide “Fe3O4”) in which Fe1 and Fe2 related to (Fe2+ and Fe3+) show cation sites in the octahedron and the tetrahedron, respectively, and O indicates the oxygen anion site (Fig. 2B). Spinel magnetite's unit cell is a cube made up of 32 O2− anions and created by 8 Fe2+Fe3+2O4 molecules. The oxygen anions make up the packing of face-centered cubes (FCC), consisting of 64 tetrahedral (Fe1) and 32 octahedral (Fe2) empty spaces partly populated by Fe3+ and Fe2+ cations (Sharma et al. 2016).
Texture characteristics
The textural properties of the TA-MCS nanocomposite were evaluated using nitrogen sorption via adsorption–desorption process and the isotherms. The N2 isotherms of TA-MCS reflect type-IV isotherms which are ascribed to mesoporous materials (Fig. 2C) (Sing 1985). The specific surface area based on BET equation (SBET) was found to be 75.27 m2 g−1; however, considerable total pore volume (TPV) was attained to be 0.57 cm3 g−1 (Fig. 2C). The inferior porosity of TA-MCS was elucidated by the pore size distribution (PSDs) displaying the existence of low numbers of mesopores of an average size of 22.4 nm. However, the dominant mesopore sizes were found to be in the range 3–9 nm as depicted in Fig. 2D.
FTIR characterization
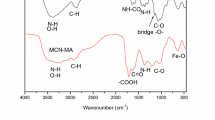
The FTIR spectra of raw TA-MCS biosorbent and consecutive compounds (after U(VI) loading and after 8th sorption–desorption cycles) are presented in Fig. 1 and Table AM3. The band at 642 and 556 cm−1 is assigned to Fe–O–Fe stretching vibration of Fe3O4 nanoparticles (Yamaura et al. 2004; Morshedy et al. 2021). These two bands are the result of a split in the ʋ1 of the bulk Fe3O4 (Fe–O) at 556 cm−1, as well as the observed band at ~ 437 cm−1, which is the blue-shift of the bulk magnetite's ʋ2 (Fe–O bond) (Yamaura et al. 2004). The FTIR spectra of TA-MCS before sorption: a strong and broad absorption band at 3424 cm−1 usually correspond to contributions from overlapped bands for –OH (and H2O traces) and –NH stretching vibrations, and O–H stretching vibration as well as polysaccharide hydrogen bonds (Galhoum et al. 2017; Imam et al. 2018; Benettayeb et al. 2021). The high-intensity peaks in 2928, 2842, and 1379 cm−1 are assigned to C–H stretching vibration (CH2 at C-6, symmetric and asymmetric) and C–H bending, respectively (Oh et al. 2005; Benettayeb et al. 2021). The peak around 2363 cm−1 might be due to stretching O=C=O in CO2. The significant peak at 1628 cm−1 can be attributed to N–H bending vibration (–NH2 and > NH amine) (Galhoum et al. 2017; Benettayeb et al. 2021). The two shoulders observed at 1068 and 1021 cm−1 are ascribed to C–O–C (in β-glucosidic linkage), C3–OH, and/or that of C–C stretching vibration (Coates 2000; Oh et al. 2005; Pourmortazavi et al. 2019). The bands at 1379 and 1068 cm−1 are assigned to primary –OH stretching and secondary –OH stretching, respectively. The bands at 892 cm−1 and 789 cm−1 represent the β-D-glucose unit (Elwakeel and Atia 2014; Galhoum et al. 2015a, b; Imam et al. 2018), and –CH2 rocking vibrations (Kuroda and Kubo 1959), respectively. The strong peak appearing at 1638 cm−1 can be attributed to N–H bending vibration (−NH2 and > NH amine) (Galhoum et al. 2015a, b; Galhoum et al. 2017). The medium/wide band in the range 800—468 cm−1 is attributed to the out-of-plane bending mode (C–C–N) (Muthuselvi et al. 2018)).
FTIR spectroscopy was also used for characterizing the interaction modes and more specifically identifying the reactive groups affected by the binding of UO22+ (Fig. 1). Basically, the two spectra were almost identical; the most noticeable characteristics of loaded-sorbent are a decrease in peak intensity and broadness, rather than band shifts when compared. In the binding mechanisms, these functional groups were included (as OH, NH2, > NH and SO42− groups). They are briefly described in the following sections:
-
(a)
The most notable changes are detected at around 3416 cm−1, the broad band (overlapped bands for –OH and –NH stretching vibrations) are little blue-shifted to 3413 cm−1 and the width and intensity have decreased, as a result of its role in metal ion chelation and sorption.
-
(b)
The bands intensity tends to strongly decrease and red-shift at 2928 cm−1 and 2842 cm−1 (assigned to –CH2 groups) moved to 2912 cm−1 and 2869 cm−1, respectively. These alterations and changes are directly related to metal ion binding that changes the –OH and –NH groups surroundings (bands superimposition that are not well-resolved).
-
(c)
The intensity of bands tends to decrease and red-shift at 1637 cm−1 to 1634 cm−1: probably due to the contribution of COO−, and –OH bending groups, demonstrating a preference for active sites with O- and N-atoms.
-
(d)
The bands at 1379 cm−1, 1065 cm−1 (attributable to N–H vibrations in secondary amines, primary and secondary –OH stretching, respectively) moved to 1382 cm−1, and 1061 cm−1—these changes are associated with UO22+–N and UO22+–O bonds of N–H and O–H vibrations.
-
(e)
The other significant change (band width and intensity) is observed at 600–1100 cm−1 affected by metal sorption. Especially, enlargement of strong peaks at 634 cm−1 (sulfate ion, SO42−), confirms the partial anion-exchange with UO2(SO4)22− on protonated amine groups involved in acidic medium (Scheme 2), and
-
(f)
Finally, the enlargement of the band at 899 cm−1 likely due to overlapped bands for UO22+ (asymmetric vibrations) and C-O str. in β-glucosidic linkage, but the pure linear vibration of UO22+ (which is characterized by the peaks at 823 cm−1 and 902 cm−1 (Song et al. 2012; Galhoum et al. 2020)) could not be observed due to overlapping with other active sites and/or the UO22+ contents is too small. However, the XPS-analysis verified it.

Tentative mechanism for UO22+ sorption and desorption using TA-MCS nanocomposite
Desorption spectrum (at the end of the eighth desorption cycle) partly recovers the sorbent and the sorbent is exceptionally stable. However, it is noteworthy that many tracer bands of metal sorption remain on the FTIR spectra of regenerated sorbents, which can be consider as “intermediary” spectra between raw and metal-loaded sorbents. The peak at 899 cm−1 (O=U=O asymmetric vibrations) begins to decline and return to its origin, indicating full uranium recovery (Fig. 3). Whereas, peaks at the 3422 cm−1, 2915–2868 cm−1, 1638 cm−1, 1428–1385 cm−1 and 1071 cm−1, or 900–400 cm−1 regions, the spectrum still hasn't been fully restored (Fig. 4).

FTIR spectra of the TA-MCS, TA-MCS + U, and after 8 desorption cycles

XPS survey spectra of TA-MCS before and after U(VI) sorption and U 4f core level
XPS spectroscopy
XPS analysis was used to determine the chemical compositions of TA-MCS before and after uranium sorption (Fig. 2). Moreover, clarify the oxidation state of metal/U species under specific acidic settings. As expected, the XPS spectra of TA-MCS showed C 1 s (~ 286.97–287.34 eV), N 1 s (~ 394.08–400.17 eV), O 1 s (~ 532.74–533.00 eV), and Fe 2p (~ 711.95–711.96 eV) signals (Galhoum et al. 2020) obtained before, and after U(VI) complexation, is characterized by a U 4f7 (~ 383.00 eV) broad-spectrum peaks and well-fitted with two components. The characteristic doublets of U 4f is identified by two peaks ~ 392.58 eV (U 4f5/2) and ~ 381.78 eV (U 4f7/2), which can well agree with match with reproduce experimental data spectra (Bai et al. 2020; Wang et al. 2021; Qiang et al. 2022), providing a direct evidence of the formation of TA-MCS-U(VI) complexes. Furthermore, two peaks at 709–710 eV (Fe 2p3/2) and 725–726 eV (Fe 2p1/2) distinguish the magnetite (Figure AM2) (Morshedy et al. 2021). Surprisingly, a new peak arises at 169.43 eV after U(VI) sorption, confirming the existence of inorganic sulfate anions (SO42−) immediately on an ion-pair (protonated amine-groups) at the sorbent's surface. This likely owing to the sorption of uranyl sulfate forms (speciation of uranyl anions from sulfate medium), confirming the anion-exchange mechanisms were involved.
Figure AM2 shows the core level spectra for key elements. The deconvoluted bands (via the assignments and relative atomic fractions for C 1s, O 2s, N 1s, S 2p, Fe 2p, and U 4f signals from HRES spectra for the TA-MCS-nanocomposite before and after uranium complexation are summarized in Table AM4. The observed variations in the relative contributions of the different bands and the shift in BEs are mainly followed by U(VI) interaction with N-containing groups (amine), and O-containing groups (-OH), as indications of reactive groups' participation in metal ion binding and/or reactive group chemical alterations.
-
(a)
C 1s signal intensities are significantly affected after U(VI)-sorption; the individual component peaks at ~ 285.56 eV shifted to 286.29 eV (C–C and C–O), while decreased at 286.23 and C–OH), and 287.49 eV (C=O, and amide (–C(=O)NH– (i.e., from raw chitosan depending on deacetylation degree)), and shifted to 286.29 eV and 290.19 eV, respectively.
-
(b)
O 1s signal shows a strongly changes after U(VI)-sorption; the intensity decreased for Fe-OH, and C–OH/C–O–C of carbohydrate ring and interchain bonding and increased for the C=O of chitosan involved in metal sorption, and due to the environment of these reactive groups. With the inclusion of magnetite (lattice O signal for magnetite Fe–O), a band at 530.7 eV (of constant intensity) develops.
-
(c)
N 1s signal, the band's relative intensity at ~ 399.12 eV (–NH2, > NH, from chitosan and diethylenetriamine moieties were involved in U(VI) sorption and reduction) is totally shifted to 399.75 eV, likely due to the N-U interaction, while that of C–N and N–H (at ~ 399.58 eV) is totally disappeared.
-
(d)
Surprisingly, a new peak arises at ~ 169.43–170.03 eV after U(VI) sorption, confirming the existence of inorganic sulfate anions (SO42−) immediately on an ion-pair (protonated amine-groups) at the sorbent's surface. This likely owing to the sorption of uranyl sulfate forms (speciation of uranyl anions from sulfate medium), confirming the anion-exchange mechanisms were involved (Scheme 2).
-
(e)
Fe 2p signal shows the main peaks of Fe3+ 2p3/2 and Fe3+ 2p1/2, which are regarded to be unique Fe3O4 spectra, are situated at ~ 710.27 and ~ 723.80 eV, respectively (Grosvenor et al. 2004; Esfandiari et al. 2020). By fitting Fe 2p double peak, the Fe2+ 2p3/2 peaks emerge at ~ 712.81 eV and Fe3+ 2p1/2 peaks appear at ~ 719.47 eV, confirming the existence of Fe2+ and Fe3+, respectively, corresponding to Fe3O4 (Esfandiari et al. 2020), and a Fe2+ 2p1/2 satellite peak appears at ~ 732.42 eV, respectively (Wang et al. 2017; Esfandiari et al. 2020). However, Fe3O4 nanoparticles, Fe2+ shakeup satellite peaks (at ~ 720.30 eV) can be observed beside Fe3+ shakeup satellite (at ~ 732.42 eV) as Fe atoms migrate to the surface and ease of identification by XPS (Wang et al. 2017; Esfandiari et al. 2020). After sorption, a slightly change is observed (Figure AM2: Fe 2p signal at 719.47 and 723.8 eV, corresponding to Fe2+-Satellite), suggesting that the Fe2+ peaks may be involved in a binding process via reduction reaction. While others peaks no impressive change is observed, because Fe atoms are located in the core of structure and sorption of UO22+ cannot affect them (Esfandiari et al. 2020).
-
(f)
High-resolution U 4f XPS spectrum can be deconvoluted into some multiple-U 4f peaks: two main U 4f7/2 and U 4f5/2 signals of U(VI) at about ~ 381.65 and ~ 392.26 eV, respectively, consistently with previously reported spectra (Wang et al. 2021; Qiang et al. 2022), while the equivalent U(IV) peaks were located at 379.84, 383.32, and 390.913 eV, respectively (Wang et al. 2021). Inspection of the satellite peaks shows the occurrence of peaks at 4 and 7 eV from the U 4f7/2. Shake-up satellites are low-intensity excitations that arise as a result of the photoelectron excitation process's shift in electrostatic potential (Gouder et al. 2018). The core level electron kinetic energy will drop as a result, and an additional/satellite peak will form on the high binding energy (BE) side of the main peak (Huang et al. 2017; Gouder et al. 2018). The multiple-U 4f peaks are well known for a tight relationship with the uranium atoms' coordination environment, besides the oxidation state. As in example, the location of U(VI): U 4f7/2 was ~ 382.3 eV in UO3, whereas in U3O8, it was ~ 381.4 eV. As a result, it is thought that the X-ray breakage of some particular bonds in uranyl molecules caused the alteration of U 4f peaks, changing the uranium's chemical environment; however, the oxidation state is not one of them (Qiang et al. 2022). Using the peak fitting method, TA-MCS reduces approximately ~ 25.2% U(VI) into U(IV) throughout the sorption process. The reducing power of the distinct functional groups ((> NH and –NH2) and –OH groups) on the sorbent's surface may have a role in U(VI) coordination onto these basic active sites.
Furthermore, following U(VI)-sorption, the intensity and atomic percent of several Fe-peaks declined noticeably and completely vanished at 716.45 eV, indicating that U(VI) was reduced to U(IV). The Eoredox (i.e., standard redox potentials) for Fe3+/Fe2+ and UO22+/UO2 is -0.771 and 0.327 V, respectively, implying that TA-MCS can reduce UO22+ thermodynamically. This is confirmed by U 4f fitting, with the 4f7/2 and 4f5/2 peaks of U(IV) peaks at 379.84, 383.32, and 390.91 eV, respectively (Fig. 2) (Wang et al. 2021). The following equation (Eq. 2) might be used to depict U(VI) reduction procedures by TA-MCS nanocomposite (Wang et al. 2021):
Metal interaction mechanism
Based on the pHs, the FTIR and XPS data show that distinct strategies were used in uranium sorption (Scheme 2):
-
(a)
Anion-exchange mechanism of anionic UO2(SO4)22− species via SO42− on the protonated amine groups,
-
(b)
Cation-exchange mechanism with cationic species (UO22+) at higher pH0, but less than pHzpc (i.e., can deprotonate active sites), with the H+ of the –OH and –NH2+/ > NH+ groups,
-
(c)
Reduction mechanism involving Fe2+-atoms on the surface, implying that U(VI) was reduced to U(IV) (XPS findings), and/or
-
(d)
Chelation and complexation mechanisms of UO22+, with different heteroatoms (O and N) on the surface of the sorbent.
The chelation and complexation interaction mechanism of U(VI) with TA-MCS, via various O- and N-atoms producing metal–ligand interactions. This was clarified with XPS findings at the molecular level by comparing the TA-MCS before and after U(VI) loading. The high-resolution C 1s spectra (Figure AM2) suggest some differences between the C 1s peak for TA-MCS before and after U(VI)sorption. Significant differences (both in intensities and peak positions) can be observed from N 1s, O 2s, signals, associated with O-, and N-containing functional groups such as the ─OH group, and > NH in chitosan molecule, and terminal –NH2 and > NH bonds in triamine moiety.
A putative mechanism for the uranyl metal ion complex may be hypothesized based on XPS and FTIR investigations (Scheme 2). Metal ions are bound via the following mechanisms: a coordinating bond of N-atom in C2-NH group and ionic bond of deprotonated O-atom of (a) primary C3-OH in chitosan molecule itself (the contribution modes (I)), another one, (b) one with the secondary (–OH) from the epichlorohydrin binder between chitosan and amine moiety (modes (II)), (c) and with > NH group of triamine (coordinating bond) (the contribution modes (III)), (d) two coordination bonds between two N-donors atoms of the triamine moiety (the contribution modes (IV and V)). Finally, contribution modes (I to V) forming a stable five-membered chelating rings. Moreover, another interaction mode between UO22+ and active sites from two chains forming two chelating rings (with five members in size). By increasing the chelate rings numbers, the formed complex’s stability improves, and the metal sorption capacity improves as a result.
The anion-exchange mechanism of uranium sorption by amine groups has been previously reported (Ritcey and Ashbrook 1984); the suspected anion-exchange reactions are illustrated as follows (Eqs. 3 and 4):
Protonated amine groups (–NH2 and > NH), formation of ion-pairs:
Overall ion-exchange reaction:
For the sorption of neutral uranium sulfate species, a different mechanism—the so-called molecular-addition mechanism (Eq. 5)—was anticipated to work as follows:
Finally, it is noteworthy that the proposed desorption process is depicted in Scheme 2: using acidified urea in desorption stage, as the acidity of this solution can facilities and breaks the contribution modes, in addition urea can coordinates and bonding with two N-donors of –NH2 in the urea CO(NH2)2 molecule, this confirm the expected mechanism (Al-Ghamdi et al. 2022).
Sorption studies
Effect of pH
The pH has a considerable influence on metal ions sorption due to many aspects linked to the right chemistry of the sorbent (degree of protonation, polymer stability and conformation), as well as the metal ion speciation of the formation of hydrolyzed species or specific complexes…. etc. (El-Bohy et al. 2017; Bai et al. 2020). Batch experiments at initial pH (1.5–5.7) were used to investigate the pH impact. Figure 3A depicts the effect of initial pH on sorption capacity. From pH 1.5 to 3.5, sorption capacity increased linearly and stabilizing above pH 3 and up to pH 5–5.5. Actually, the pH changed dramatically during the sorption stage (Fig. 5): below pH 3.5, the pH tended to decline, whereas above pH 3 and up to pH 5.7, the pH value at equilibrium was rising. The greatest pH change (by ~ 2.34) was found at initial pH 3.5, which is likely related to the existence of the triamine moiety. The equilibrium pH rises by 1.9–0.5 pH unit with sorption in the pH range 4.1–5.7, respectively, and tends to stabilize at ~ 6.05–6.18, i.e., in slightly acidic media, the TA-MCS acts as a buffer. However, uranium precipitation is affected by the initial pH and metal ion concentration, since uranyl ions begin to precipitate in a colloidal form (UO2(OH)2) when the pH is approximately 5.8 (Guibal et al. 1992).

a Influence of pH on U(VI) sorption. b Uranyl speciation (sulfuric acid solution) as a function of pH: calculated by using a Visual-Minted program. c Uptake kinetics plots for UO22+ ions sorption (Arrhenius plots of ln K2 vs. 1/T). (pH: 4.0; Co: 0.426 mmolU L−1; T: 298–318 K, SD: 0.2 g/L), and d sorption isotherms and thermodynamics of UO22+ sorption–Van’t Hoff plots of ln bL versus 1/T (pH0: 4.0, SD: 0.2 g L−1, C0: 0.093–1.253 mmolU L−1; T: 298–323 K, Time: 60 min)
Figure 5a demonstrates that raising the initial and equilibrium pH improves the sorption capacity, notably between initial pH (1.5–3.0) and pH (3.0–5.7), where the sorption capacity tends to stabilize. The maximum sorption of UO22+ ions was reported to be 0.53 mmolU g−1, at an initial pH range of 3.5–5.0. At lower pH (< 3.5), the sorbent surface charge became positive, preventing positively charged ions from approaching: H+ competed with UO22+ ions for binding sites, reducing metal ion interaction with the sorbent (Guo and Yu 2014; Bai et al. 2020). With an increase in pH, protons began to be desorbed, the protonation of hydroxyl and amine groups diminishes, allowing metal cations to bind and the UO22+ hooked up the free binding sites, hence, metal sorption increased (Galhoum et al. 2015a, b; El-Bohy et al. 2017). Distinct mechanisms are linked to different sorption sites depending on the initial pH. The inclusion of amine (-NH2, > NH) and hydroxyl groups, each with its own acid–base characteristics and potential to bind metal ions via chelation and ion-exchange processes, provides a diverse set of sorbent interaction options with U(VI) (Galhoum et al. 2015a, b). However, according to the species distribution for U(VI) hydrolysis and polynuclear hydrolyzed uranyl species (Fig. 5b), uranyl ions in the suggested system will precipitate at higher pH values. As a result, for subsequent experiments, an ideal initial pH value of roughly pH: 4.0 was chosen.
Both (sorbent acid–base characteristics and the measured pHZPC) imply that the sorption process involves considerable pH changes (Fig. 5a: pHeq vs. pH0). The pHZPC section shows a positively charged surface in this range at pH0 8.48, as a result of reactive group deprotonation.
Log plot of the distribution ratio (D, in L g−1) versus pHeq is plotted in Fig. 5a (insert). It is a linear relationship. The stoichiometric ion-exchange with bound UO22+ species is related to the slope of this figure in ion-exchange operations. The slope of this plot is correlated in ion-exchange processes with the stoichiometric ion-exchange with bound UO22+ species. The ion-exchange mechanism is playing a substantial role in the management of the sorption process, as evidenced by the high level of fitness (R2: 0.963). In an acidic medium, the lone-pair electron of the > NH/–NH2 group is protonated, which can aid in ion exchange with UO22+ ions during the sorption process.
Equilibration time
Uptake kinetics were examined and compared at different temperatures (under a set of experimental conditions). As illustrated in Fig. 5c, all curves follow a similar tendency, regardless of the temperature and the pseudo-equilibrium may be attained after 40–50 min. The sorption capacities were 0.659, 0.603, and 0.553 mmolU g−1 at 298, 308, and 318 K, respectively, demonstrating that as the temperature was raised, the sorption decreased (i.e., exothermic nature). Thermodynamic parameters will be examined in more detail, later.
There are several steps in sorption process that may be identified: (a) in the rapid first stage (during the first 5 min): the sorbent removes roughly 51–59% of total sorption (qeq), (b) The second stage (which takes anywhere between 5 and 30 min) results in complementary sorption, which accounts for up to 94.1–98.7% of total sorption, and (c) the last slower step, after 40 min. the total percentage for 98.0–99.8%, and the sorbent removes about 0.1–2.0% of qeq after 40 to 50 min. The final stage is under the control of the intraparticle diffusion resistance through inner micropores. The sorbent has a large enough mesopores that allow uranium ions to pass through (Shannon radius for hydration [UO2(H2O)5]2+ is 1.08 Å (Persson 2010)) in accordance with textural analysis. To attain the actual equilibrium, it is required to wait 50 min. The fast kinetics is likely ascribed to the small size of sorbent particles (below 25 nm), high surface area (75.27 m2 g−1), and content of functional groups (amino and hydroxyl).
Figure AM3 displays the modeling of kinetic profiles using Weber and Morris simplified model of intraparticle diffusion resistance (Table AM1 for model equations) (Cheung et al. 2007; Kyzas et al. 2014). Three linear segments may be found, one for each of the three stages listed above. Essential characteristics are shown in Table AM5.
The intraparticle diffusion rate constants' values (Kid): both the first and second rate constants (Kid,1 and Kid,2) were higher and ordered as following: Kid,1 > Kid,2, implying the quick sorption on the most accessible active sites on the TA-MCS surface (hydroxyl and amine groups). Following that, the concentration gradient between the solution and the sorbent has diminished, slowing the mass transfer of uranyl ions to interior reactive groups (the structured material first interior layers and/or larger pores). Finally, the Kid,3-values were almost nil, as a result of the sorption–desorption equilibrium; assume that sorption occurs in micropores (the sorbent is nearing saturation) (Tien 1994; Cheung et al. 2007). This implies that the rate of sorption is influenced by both intraparticle diffusion and the boundary layer (Pan et al. 2020): this has been proven by textural features.
Two simplified kinetic models, the pseudo-first order rate equation (PFORE) and the pseudo-second order rate equation (PSORE), were used to describe the sorption/time data acquired (Table AM1) (Tien 1994). The fits of experimental profiles at three different temperatures are compared in Fig. 5c. The parameters of these models are summarized in Table AM5 (Figure AM3). Obviously, PSORE's experimental results (R2 > 0.999) was clearly superior to PFORE's (R2 > 0.994). Furthermore, the model's suitability (PFORE vs. PSORE) is determined by comparing the experimental (qeq,exp) and estimated values of the equilibrium sorption capacities (qeq,1 or qeq,2). The PSORE, unlike the PFORE, allows for a closer approximation to the experimental value: the Δqeq (underestimated by ranges 4.9–6.2%), whereas the PFORE (much overestimated by 7.7–17.0%).
The PSORE is often used to model chelation mechanisms, whereas the PFORE is utilized to model physical sorption mechanisms. Demonstrating that the PSORE was an excellent model for modeling UO22+ sorption with various materials and, in particular, chemisorption reactions involving ion-exchange and chelation mechanisms (Kyzas et al. 2014; Hubbe et al. 2019). Hubbe et al. (Hubbe et al. 2019) reviewed that the PSORE fit of kinetic profiles demonstrates that the process is really regulated by intraparticle diffusion, as previously described.
Based on the comparison of the PSORE constant (k2) apparent rate coefficients show sorption occurs faster at lower temperatures (Wang et al. 2015; Morshedy et al. 2021): k2-valuses (min−1 mg−1 min−1) were as follows: 68.112 × 10–2 > 55.487 × 10–2 > 40.491 × 10–4, for temperatures 298, 308, and 218 K, respectively. Moreover, from the k2 values: the half-sorption time (t0.5: the time it takes for ion-exchange reaction to achieve 50% of their maximal sorption capacity) was determined. The t0.5 might be employed as a metric of sorption rate, as described (t0.5 = 1/k2·q2) (Doğan et al. 2007; Pan et al. 2020). The t0.5 at 323 K (4.2 min) > 308 K (2.9 min) > 298 K (2.1 min) consistently with the observed k2-valuses and experimental results.
Activation energy for sorption
The Arrhenius equation (ln K2 = ln K0 − (Ea/RT)) was used to compute the UO22+ sorption activation energy, aimed toward determining the type of sorption (physical or chemical): where K2 is the PSORE overall rate constant (g mg−1 min−1), R is the gas constant (8.314 J mol−1 K−1), K0 is the Arrhenius constant (g mg−1 h−1), and Ea is the sorption activation energy (Ea, in kJ mol−1). The change of sorption capacity with time at different temperatures was examined to determine the activation energy (Fig. 5c) (Wang et al. 2015; Galhoum 2019). The average activation energy was determined to be 20.433 kJ mol−1, which might be attributed to intraparticle and surface diffusion processes. The apparent Ea is consistently less than 40 kJ mol−1, indicating that sorption occurs by physisorption (Bai et al. 2020; Chen et al. 2021). Physical sorption was generally a multilayered, fast, and reversible process governed by weak intra-particle bonds existing between the sorbate and the sorbent, e.g., van der Waals and π–π interaction; therefore, little energy was needed (Galhoum 2019). Contrary, the chemical sorption was monolayered, slow, and governed by strong interaction, due to the exchange or sharing of electron such as covalent and ionic bonds and this type of chemisorptions is deemed to be irreversible in the majority of cases, so higher activation energies were required (Casarin et al. 2018; Galhoum 2019). Furthermore, both processes may coexist (Galhoum 2019).
Conclusions coming from sorption kinetics, isotherms, and activation energy appear to contradict each other. Chemisorption is frequently associated with the PSORE and the Langmuir model (monolayered-chemisorption). However, in physisorption, the Arrhenius equation is used to compute the mean activation energy (Ea < 40 kJ mol−1) (Casarin et al. 2018; Galhoum 2019). This suggests a dual mechanism of physisorption (electrostatic forces) and chemisorption (ionic forces: coordination) owing to heterogeneous binding sites (Bai et al. 2020).
Sorption isotherms and thermodynamics
The sorption isotherms (qeq = f(Ceq)) show how the solute is distributed between the liquid (Ceq) and solid (qeq) phases (for various initial concentrations (C0), but at the same temperature and pH) (Morshedy et al. 2021). Figure 5d depicts data on UO22+ sorption isotherms at pH0: 4.0 and various temperatures (298–323 K). The isotherm is characterized by a gradual increase in sorption capacity, followed by sorbent saturation at maximum experimental sorption capacity up to 0.748, 0.698, 0.638, and 0.587 mmolU g−1 at 298, 308, 318, 323 K, respectively, consistently with the results of time effect. The plateau revealed that the sorbent's most active sites reacted with UO22+. Furthermore, the underlying curves at various temperatures show that temperature has a detrimental influence on sorption efficiency (i.e., exothermic feature).
Compared with other sorption materials, TA-MCS sorbent has excellent sorption performance, although a direct comparison is difficult due to the differences in experimental settings (Table 1). The TA-MCS has a good maximal sorption capacity, equilibrium time is 40 min, and a broad pH range (3.5–5.0) which indicates that sorbent is the appropriate candidate for uranium sorption.
Sorption isotherms can be modeled using Langmuir, Temkin and Freundlich equations (Table AM1) (Tien 1994; Araújo et al. 2018). Figure AM4 and Table AM6 show the computed parameters. The comparison of determination coefficients (R2) for the Langmuir generally gives slightly higher coefficients (R2 > 0.997) compared to Freundlich (R2 < 0.990). Furthermore, the sorption capabilities matched experimental data and both the Langmuir and Freundlich models (qm,L & KF) were persistently: shows that overestimates profiles (Δqeq,): for the Langmuir equation (~ 4.6–6.1%), while for the Freundlich (~ 1.1–5.2%). This difference may be interpreted as irregular disturbance of the experimental points. The Langmuir equation fits the isotherms because the sorption isotherms show an asymptotic tendency, whereas the Freundlich equation implies an exponential trend (El-Bohy et al. 2017). As a result, monolayer uniform sorption occurs, with a finite number of identical sites scattered over the sorbent surface (Araújo et al. 2018). Moreover, the Langmuir constant (bL) is proportional to the sorption energy; the higher the bL value, the higher the sorption energy, and the greater the sorbent-sorbate affinity (Rangabhashiyam et al. 2014). Table AM6 illustrates that both qmax and the bL values: slightly decrease with increasing temperature, the sorption is likely to be slightly exothermic. The bL values decreases (from 22.09 to 14.55) with increasing the temperature (from 298 to 328 K). This means that at room temperature, sorption is more favorable.
Another model is the Temkin isotherm supposes that sorption free energy is proportional to surface coverage (Tien 1994). The Temkin model constants are reported in Table AM6 and Figure AM4. The constant AT represents the sorbent initial sorption heat: the higher the AT, the greater the sorption heat, and the higher the sorbent's affinity for the sorbate (Tien 1994). The AT values reduced from 2384.16 to 1510.81 L/mmol and the energetic parameter (bT) decreases (from 0.0985 to 0.0796 J mol−1) with increasing the temperature from 298 to 328 K, as previously observed (such as comparison of bL and qm in the Langmuir model).
Based on the Langmuir isotherms, the thermodynamics might be examined. The corrected bL constant (as from L g−1 to L mol−1) and processing with no dimensions (adjusting for molality in the water) as determined by Lima et al. (2019). Thermodynamic parameters are calculated by the Van’t Hoff equation (ln bL = (−ΔH°/R)1/T + ΔS°/R) can be used to calculate the enthalpy (ΔH°) and entropy (ΔS°) changes (Lima et al. 2019; Morshedy et al. 2021), while the free energy (ΔG°) change may be determined from (ΔG° = ΔH° − TΔS°), where bL is the Langmuir constant. The ∆H°, and ∆S° values are obtained from the slope and intercept of the plot of ln bL versus 1/T (Fig. 5d).
Table AM7 summarizes the thermodynamic parameters. The exothermic nature is confirmed by ∆H°'s negative sign and the reaction is more favorable at lower temperatures. Since, the ∆H° values are less than 40 kJ mol−1, suggesting that there are physical forces existing in UO22+ sorption process. The positive ΔS° value showed that the interaction between UO22+ and TA-MCS would enhance entropy, resulting in more degrees of freedom for solute molecules (Bai et al. 2020). The negative ΔG° values (in the same range from − 24.80 to − 25.81 kJ mol−1) imply that the sorption process is spontaneous. The absolute ΔG° values is proportional with the temperature, indicating the more spontaneous sorption process (Huang et al. 2019). Moreover, Table AM7 indicates that the reaction is controlled by entropic changes than by enthalpy changes (│ΔH°│ < │TΔS°│) (with values very close).
Effect of sorbent dose
The impact of sorbent dose (SD: 10–150 mg/100 mL) on uranyl ion sorption and removal efficiencies is shown in Figure AM5. The removal efficiency improved from (10 to 150 mg/100 mL, or 0.10 to 1.5 g/L) with increasing sorbent dosage, reaching a maximum (99.8%) at 1.5 g/L; however, the sorption capacity decreased dramatically. This likely due to the growing number of accessible binding sites (Al-Ghamdi et al. 2022). As the sorbent dose was increased, sorbent molecules partly aggregated and formed bridging bonds; reducing the effective surface area and active sites for metal ions binding (Guo and Yu 2014). Depending on the process's goal, decontamination of the effluent (lowest residual concentrations needed) necessitates a high sorbent dosage, whereas metal valorization necessitates metal concentration in the sorbent (i.e., lesser sorbent dosages).
Metal desorption and sorbent recycling
Uranium concentration for final recovery should be enhanced, and the sorbent should be regenerated and recyclable, to reduce the cost-effectiveness. In the case of magnetic chitosan particles loaded with uranyl ions, NaHCO3 and acidic urea solutions were tested for metal elution (Galhoum et al. 2017; Imam et al. 2018). Sorption/desorption was performed repeatedly using acidified urea (0.25 M at pH 2.5) by H2SO4 as an eluent and the proposed desorption mechanism was depicted in Scheme 2 (Al-Ghamdi et al. 2022). Table 2 reports eight successive sorption/desorption cycles: both the sorption and desorption efficiencies decrease somewhat at each sorption stage. At the end of 8th cycle, a limited loss in efficiency does not exceed 14.7%, and 8.6% for sorption and desorption process, respectively. This indicated that the sorbent is durable, reusable, and stable.
Study of chemical stability
The TA-MCS nanocomposite's chemical stability was evaluated: the TA-MCS was treated and leached using conventional chemicals like HCl, H2SO4, HNO3, and NaOH solutions (under the experimental conditions: 0.01 g/10 mL, temperature 26 °C, and time 24 h) (Kawady et al. 2022; Bai et al. 2020). After centrifuging, the TA-MCS samples (both raw and treated samples after the leaching tests) were kept at 60 °C for 4 h. The weight losses were calculated using the formula (wt% = 100(m0 − mf)/m0), where m0 and mf represents the dry mass before and after treated (Kawady et al. 2022):were shown to be ~ 3–7% in comparison to raw TA-MCS under the stated conditions. Using HNO3 solutions revealed the largest weight loss, likely due to dissolving of the Fe3O4 at the surface.
Moreover, utilizing FTIR spectroscopy, the material's chemical and functional stability was further evaluated after eight sorption/desorption cycles (see “FTIR characterization” section), and the slight decrease in sorption/desorption performances confirms and supports the material's stability. This indicated the TA-MCS maintains notably stable and durable.
Selectivity tests
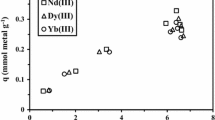
The selective sorption of UO22+ from a simulated nuclear industrial effluent (in multi-component equimolar solutions) was examined. In terms of sorption capacities (qeq, mmol/g), distribution ratio (D, L/g) and selectivity coefficient (SCmetal/U = Dmetal/DU) data were analyzed (Al-Ghamdi et al. 2022) (Fig. 6). The sorption capacity is 0.603 mmolU g−1, which is significantly lower than that of synthetic solutions (0.752 mmolU g−1). This suggests that excessive concentrations of co-ions lower maximum sorption by 19.81%; considering the significant abundance of coexisting ions, and the sorbent has a strong preference for UO22+. Despite the solution complexity, potential changes in metal speciation (because of nitrate anion), and the possible competitive impact of other metal ions. Other co-metal ions attach to the particular reactive sites through various stoichiometric ratios and/or distinct functional groups, resulting in a total sorption capacity of 1.154 mmol metal g−1 greater than that of UO22+ in synthetic solutions. The distribution ratio and the selectivity coefficients were calculated: highlight more efficiently the selectivity for the target metal. Hence, U(VI) has higher D values (1.58 L/g) and SCmetal/U values than other co-ions; the sorbent has preferential affinity and competitiveness for highly selective for uranium sorption.

Sorption capacities, distribution coefficients, and selectivity coefficient (SCmetal/U) in selectivity tests (SD: 0.2 g/L, C0: 0.5 mmol/L, pH0: 4.0, T: 298 ± 1 K, time: 60 min)
Figure AM6 depicts the relative percentages of various metal ions in the initial tested pregnant liquor and the two compartments (pH 4.0: treated liquor and sorbent): there is a significant difference in the solution before and after sorption. This is the first proof that the selectivity is quite high for uranium. In comparison to the liquor original composition, the percentages in the sorbent reflect a wide range of trends: UO22+ exhibits a significant enrichment (from 10.0% to 52.0%), as well as, and to a lesser extent for ƩREEs 0.149 mmol RE g−1 represented ~ 12.9% of the total capacity. Alternatively, the sorption capacity of other metal ions represents ~ 33.8% ~ 13.5% of the total sorption capacity ≈ 0.401 mmol g−1, including 0.109 mmol g−1 of Co(II) which represents about 9.4% of the total sorption capacity. As a result, the TA-MCS-overall sorbent's selectivity for UO22+ decreases. The selectivity coefficient was scaled using UO22+ as a reference since it has the greatest affinity worldwide. Figure 6 depicts the plot of selectivity coefficients (over UO22+); metal ions can be ordered according to their selectivity coefficients.: U(VI) (1.0) > Co(II) (0.14) > Cd(II) (0.12) > Sr(II) (0.10) > Ba(II) (0.09) > Cs(I) (0.09) > Yb(III) (0.08) > Eu(III) (0.06) > Ce(III) (0.04) > La(III) (0.03). The relationships between the atomic number, the enrichment factor (EF = molar percentage ratio on the sorbent relative to that in the liquor and the distribution coefficient (D) are shown in Figure AM7a, reveals obvious trends: the enrichment factor for U(VI) is 5.22, and raises as atomic number increases. Other co-metal ions display a reciprocal trend (REs) with increasing atomic number (0.94 > EF > 0.5), whereas rare earth ions show a reciprocal trend (REs) with increasing atomic number (0.5 > EF > 0.20) (Figure AM7a). Obviously, the linear curves clearly exclude uranium: U is commonly connected with REEs, due to its atomic number (92). The distribution coefficients, which are substantially greater for various co-metal ions than for REs(III) present in the solution, show the same results (Figure AM7b). This indicates that for the Res, both EF and D increase with increasing atomic number, contrary to the other co-metal ions follow a reciprocal trend (Figure AM7a&b).
In fact, comparing the chemical characteristics of several metal ions in Table AM8 shows that the ionic radii and electronegativity (Pauling units) of the uranium element are close to those of REs elements, but the atomic mass is substantially different. U point has shifted to its "equivalent atomic number" deviates significantly from the REs' linear trends (Figure AM7c). The association between the enrichment factor and the REs and other co-metal ions electronegativity or hydrated ionic radius is seen in Figure AM7c&d. In general, the hydrated ionic radius and the enrichment factor are inversely related (after exclusion of U). The electronegativity of REs(III) contributes to the enrichment factor in an irregular manner, while for the ER of other metal ions improves with electronegativity and falls as the hydration ionic radius increases.
Graphical simulation and nonlinear regression modeling
Recent developments in multimedia have increased the quantity of three-dimensional (3D) data that are available. Therefore, creating useful methods for their representation and processing is necessary for effective data management. Recently, nonlinear models have been the more popular option, not only for accuracy needs but also for improving the model's utility (Boyadjiev 2010; Fouda et al. 2022). The system criteria are successfully realized as a result of the emphasis on accuracy during model simulation. The latter is accomplished using MATLAB software together with a nonlinear regression approach and graphical simulation.
The description considers the general distribution of relation between individual points (qeq vs. initial pH and initial concentration), to evaluate how effective the suggested strategy is. Since Langmuir model best fits the data, the generic Langmuir isotherm model's nonlinear equation (q = qm KCn/(1 + KC)m) was used to depict several 3D (Tien 1994; Boyadjiev 2010). Equation 6 shows the relationship between the U(VI) sorption and the initial pH and U(VI) concentration (Boyadjiev 2010). By generalizing Langmuir to include initial pH and initial concentrations (Eq. 6):
Gibbs free energy change (ΔG) was utilized to fitting the recommended mathematical solution (in Eq. 6 via using k1 and k2 parameters), as follows (Eq. 7):
where f(pH,C) = qeq (the resultant and required sorption capacity at certain initial pH and concentration. qm (0.747 mmolU g−1), pH and C are the maximum sorption capacity, initial pH and initial concentration, respectively, under investigation (Boyadjiev 2010). Exponentials (a, k1, m1, n1, f1, e1, g1, m2, n2, k2, f2, e2, g2) are the constant powers of the parameters, k (the Langmuir constant corrected according to Lima et al. (2019)) and k1 and k2 (both parameters obtained from Eq. 6).
Based on the sorption results at varied initial pHs and concentrations, the experimental data were graphed in 3D space using MATLAB software (Fig. 7), in accordance with the methodical technique outlined in Eq. 6. This equation can be applied over the initial pH range (pH0: 2.5–5.7) and initial concentration range (C0: 0.093–1.253 mmolU L−1): all of these ranges provided good fits for the equation, except the pH0: 1.3. So, this point (pH0: 1.3) was eliminated and excluded, as it was unsuitable and unfitted for this equation. In addition, Fig. 7 depicts a vacant space area on the figure's surface denotes that the outcomes in this case cannot be applied here.

Generalized Langmuir model including: U(VI) sorption versus initial pH, and initial U(VI) concentration
Practically, this mathematical equation generally correlates better with experimental profiles: this fitting is supported by SSE, R2 and Adj R2 terms (Boyadjiev 2010). Moreover, the computed free energy (ΔG: − 242.8 kJ mol−1 at room temperature ~ 298 K) is significantly consistently with experimental observations thermodynamically both in sign and value (see Table AM7).
According to Eq. 6, it was indicated that there were two parallel responses, typically referred to the PSORE. Ion-exchange and chelation mechanism were the two parallel reactions. The first ion-exchange mechanism is the initial reaction started with qm (likely mild sorption reaction), and followed by the main sorption chelation mechanism (started with qm-a). This is consistent with the outcomes of the previous experiments. Moreover, all parameters were presented in Fig. 7; evidently that: (m2: 13.57 > m1: 6.288) ≫ (f2: 11.29 > f1: 6.395) ≫ (n2: 3.33 > n1: 1.898) > (e2: 2.439 > e1: 2.164) ≫ (k2: 84.57 × 10–5 > k1: 25.43 × 10–5), whereas, for (g1: 2.303 > g2: 1.279). This might support the suggestion that the chelation was more dominant than the ion-exchange mechanism.
Figure 7 illustrates how the function-power is: the higher order of exponentials magnitude, the more reaction dependence on (Fouda et al. 2022): the initial pH, and initial concentration. Based on m and f values, the most crucial aspect is the initial pH. The second main practical component is the C0, from the n and e values. This is consistent with the experimental results.
The validation of the mathematical method is evaluated by the degree to which the mathematical results agree with the experimental results via the error percentage (Error, % = (qeq,exp. − qeq,Math) × 100)/qeq,exp.). Table 3 displays the outcomes of the validation and comparison of the mathematical equation for the initial pH range (2.0 to 5.7) and initial concentrations (C0: 0.2–1.3 mmol/L). The accuracy (Δ) of the mathematical method is determined, and ∆was found to be overestimated by (∆ < 2.24%) and underestimated by (∆ < 1.88%).
Conclusion
Diethylene-triamine effectively functionalized a hybrid mesoporus magnetic-chitosan nanocomposite to develop a TA-MCS sorbent for highly selective and efficient uranyl ions sorption from aqueous solution and radioactive effluents. The physiochemical parameters of nanocomposites were determined using a variety of analytical techniques, including CHN, XRD, BET, pHzpc, TEM, XPS, FTIR, and VSM. The mesoporus TA-MCS sorbent has a unipore structure (27.53 nm) and a significant surface-to-volume ratio (75.28 m2 g−1 and 0.57 cm3 g−1, respectively). Batch experiments revealed that sorption equilibrated in 40 min at 298 K, with a sorption capacity of 0.76 mmolU g−1 throughout a pH0 range of 3.5–5.0. The sorption followed pseudo-second-order and Langmuir models, indicating monolayer chemical sorption. The sorption activation energy (Ea) was found to be 20.43 kJ mol−1, which might indicate two different sorption processes (intraparticle and surface diffusion). According to a comprehensive investigation (FTIR and XPS analysis) of reaction mechanism (Surface interaction speculation): the principal reactive groups that absorbed UO22+ on sorbent surfaces were identified as amino and hydroxyl functional groups via ion-exchange, electrostatic attraction, chelation, and reduction mechanisms. The temperature dependence revealed that the reaction was spontaneously exothermic. Using an acidic urea solution (0.25 M), uranyl ions may be effectively eluted and recycled from a loaded TA-MCS nanocomposite, demonstrating that the sorbent exhibits excellent sorption, durability, and reusability. Competitive sorption tests revealed that extracting uranium from a simulated nuclear waste solution was more selective and effective than removing rare earths (La, Ce, Eu, and Yb) and other base co-metal ions (Cd, Co, Cs, Ba, and Sr). The nanocomposites' superparamagnetic characteristics (Ms: 30.73 emu g−1) suggested that the sorbent may be easily recovered from aqueous solution. Finally, an efficient mathematical expression has been developed and applied for assessing the sorption capacity at a certain initial pH and concentrations.
References
Al-Ghamdi AA, Galhoum AA, Alshahrie A, Al-Turki YA, Al-Amri AM, Wageh S (2022) Mesoporous magnetic cysteine functionalized chitosan nanocomposite for selective uranyl ions sorption: experimental, structural characterization, and mechanistic studies. Polymers 14(13):2568. https://doi.org/10.3390/polym14132568
Araújo CST, Almeida ILS, Rezende HC, Marcionilio SMLO, Léon JJL, de Matos TN (2018) Elucidation of mechanism involved in adsorption of Pb(ii) onto lobeira fruit (Solanum lycocarpum) using Langmuir, Freundlich and Temkin isotherms. Microchem J 137:348–354. https://doi.org/10.1016/j.microc.2017.11.009
Bai J, Ma X, Yan H, Zhu J, Wang K, Wang J (2020) A novel functional porous organic polymer for the removal of uranium from wastewater. Microporous Mesoporous Mater 306:110441. https://doi.org/10.1016/j.micromeso.2020.110441
Benettayeb A, Morsli A, Elwakeel KZ, Hamza MF, Guibal E (2021) Recovery of heavy metal ions using magnetic glycine-modified chitosan—application to aqueous solutions and tailing leachate. Appl Sci 11(18):8377. https://doi.org/10.3390/app11188377
Boyadjiev C (2010) Theoretical chemical engineering: modeling and simulation. Springer, Berlin
Casarin J, Gonçalves Junior AC, Segatelli MG, Tarley CRT (2018) Insight into the performance of molecularly imprinted poly(methacrylic acid) and polyvinylimidazole for extraction of imazethapyr in aqueous medium. Chem Eng J 343:583–596. https://doi.org/10.1016/j.cej.2018.03.030
Chen Y, Tang J, Wang S, Zhang L (2021) High selectivity and reusability of coordination polymer adsorbents: synthesis, adsorption properties and activation energy. Microporous Mesoporous Mater 324:111309. https://doi.org/10.1016/j.micromeso.2021.111309
Cheung WH, Szeto YS, McKay G (2007) Intraparticle diffusion processes during acid dye adsorption onto chitosan. Biores Technol 98(15):2897–2904. https://doi.org/10.1016/j.biortech.2006.09.045
Coates J (2000) Interpretation of infrared spectra: a practical approach. Encyclopedia of analytical chemistry. Wiley, Chochester
Doğan M, Özdemir Y, Alkan M (2007) Adsorption kinetics and mechanism of cationic methyl violet and methylene blue dyes onto sepiolite. Dyes Pigm 75(3):701–713. https://doi.org/10.1016/j.dyepig.2006.07.023
El-Bohy MN, Abdel-Monem YK, Rabie KA, Farag NM, Mahfouz MG, Galhoum AA, Guibal E (2017) Grafting of arginine and glutamic acid onto cellulose for enhanced uranyl sorption. Cellulose 24(3):1427–1443. https://doi.org/10.1007/s10570-017-1193-1
Elwakeel KZ, Atia AA (2014) Uptake of U(VI) from aqueous media by magnetic Schiff’s base chitosan composite. J Clean Prod 70:292–302. https://doi.org/10.1016/j.jclepro.2014.02.017
Esfandiari N, Kashefi M, Mirjalili M, Afsharnezhad S (2020) Role of silica mid-layer in thermal and chemical stability of hierarchical Fe3O4-SiO2-TiO2 nanoparticles for improvement of lead adsorption: kinetics, thermodynamic and deep XPS investigation. Mater Sci Eng B 262:114690. https://doi.org/10.1016/j.mseb.2020.114690
Fouda SR, El-Sayed IE, Attia NF, Abdeen MM, Abdel Aleem AAH, Nassar IF, Mira HI, Gawad EA, Kalam A, Al-Ghamdi AA, Galhoum AA (2022) Mechanistic study of Hg(II) interaction with three different α-aminophosphonate adsorbents: insights from batch experiments and theoretical calculations. Chemosphere 304:135253. https://doi.org/10.1016/j.chemosphere.2022.135253
Fraga TJM, de Lima LEM, de Souza ZSB, Carvalho MN, Freire EMPL, Ghislandi MG, da Motta MA (2019) Amino-Fe3O4-functionalized graphene oxide as a novel adsorbent of methylene blue: Kinetics, equilibrium, and recyclability aspects. Environ Sci Pollut Res 26(28):28593–28602. https://doi.org/10.1007/s11356-018-3139-z
Galhoum AA (2019) Facile synthesis of functionalized polyglycidyl methacrylate-magnetic nanocomposites for enhanced uranium sorption. RSC Adv 9(66):38783–38796. https://doi.org/10.1039/C9RA06874K
Galhoum AA, Eisa WH, El-Tantawy El-Sayed I, Tolba AA, Shalaby ZM, Mohamady SI, Muhammad SS, Hussien SS, Akashi T, Guibal E (2020) A new route for manufacturing poly(aminophosphonic)-functionalized poly(glycidyl methacrylate)-magnetic nanocomposite-application to uranium sorption from ore leachate. Environ Pollut 264:114797. https://doi.org/10.1016/j.envpol.2020.114797
Galhoum AA, Hassan KM, Desouky OA, Masoud AM, Akashi T, Sakai Y, Guibal E (2017) Aspartic acid grafting on cellulose and chitosan for enhanced Nd(III) sorption. React Funct Polym 113:13–22. https://doi.org/10.1016/j.reactfunctpolym.2017.02.001
Galhoum AA, Mafhouz MG, Abdel-Rehem ST, Gomaa NA, Atia AA, Vincent T, Guibal E (2015a) Cysteine-functionalized chitosan magnetic nano-based particles for the recovery of light and heavy rare earth metals: uptake kinetics and sorption isotherms. Nanomaterials (basel) 5(1):154–179. https://doi.org/10.3390/nano5010154
Galhoum AA, Mahfouz MG, Gomaa NA, Abdel-Rehem SS, Atia AA, Vincent T, Guibal E (2015b) Cysteine-functionalized chitosan magnetic nano-based particles for the recovery of Uranium(VI): uptake kinetics and sorption isotherms. Sep Sci Technol 50(18):2776–2789. https://doi.org/10.1080/01496395.2015.1085405
Gouder T, Eloirdi R, Caciuffo R (2018) Direct observation of pure pentavalent uranium in u2o5 thin films by high resolution photoemission spectroscopy. Sci Rep 8(1):8306. https://doi.org/10.1038/s41598-018-26594-z
Grosvenor AP, Kobe BA, Biesinger MC, McIntyre NS (2004) Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds. Surf Interface Anal 36(12):1564–1574. https://doi.org/10.1002/sia.1984
Guibal E, Roulph C, Le Cloirec P (1992) Uranium biosorption by a filamentous fungus Mucor miehei pH effect on mechanisms and performances of uptake. Water Res 26(8):1139–1145. https://doi.org/10.1016/0043-1354(92)90151-S
Guo J, Yu J (2014) Sorption characteristics and mechanisms of Pb(II) from aqueous solution by using bioflocculant mbfr10543. Appl Microbiol Biotechnol 98(14):6431–6441. https://doi.org/10.1007/s00253-014-5681-z
Huang S, Jiang S, Pang H, Wen T, Asiri AM, Alamry KA, Alsaedi A, Wang X, Wang S (2019) Dual functional nanocomposites of magnetic mnfe2o4 and fluorescent carbon dots for efficient U(VI) removal. Chem Eng J 368:941–950. https://doi.org/10.1016/j.cej.2019.03.015
Huang Z, Li Z, Zheng L, Zhou L, Chai Z, Wang X, Shi W (2017) Interaction mechanism of uranium(VI) with three-dimensional graphene oxide-chitosan composite: Insights from batch experiments, IR, XPS, and EXAFS spectroscopy. Chem Eng J 328:1066–1074. https://doi.org/10.1016/j.cej.2017.07.067
Hubbe MA, Azizian S, Douven SJB (2019) Implications of apparent pseudo-second-order adsorption kinetics onto cellulosic materials: a review. BioResources 14:7582–7626. https://doi.org/10.15376/BIORES.14.3.7582-7626
Imam EA, El-Tantawy El-Sayed I, Mahfouz MG, Tolba AA, Akashi T, Galhoum AA, Guibal E (2018) Synthesis of α-aminophosphonate functionalized chitosan sorbents: effect of methyl vs phenyl group on uranium sorption. Chem Eng J 352:1022–1034. https://doi.org/10.1016/j.cej.2018.06.003
Jun B-M, Lee H-K, Park S, Kim T-J (2021) Purification of uranium-contaminated radioactive water by adsorption: a review on adsorbent materials. Sep Purif Technol 278:119675. https://doi.org/10.1016/j.seppur.2021.119675
Kawady NA, Elkattan M, Salah M, Galhoum AA (2022) Fabrication, characterization, and gamma ray shielding properties of PVA-based polymer nanocomposite. J Mater Sci 57(24):11046–11061. https://doi.org/10.1007/s10853-022-07213-9
Kharissova OV, Dias HVR, Kharisov BI (2015) Magnetic adsorbents based on micro- and nano-structured materials. RSC Adv 5(9):6695–6719. https://doi.org/10.1039/C4RA11423J
Kong H, Song J, Jang J (2010) One-step fabrication of magnetic γ-Fe2O3/polyrhodanine nanoparticles using in situ chemical oxidation polymerization and their antibacterial properties. Chem Commun 46(36):6735–6737. https://doi.org/10.1039/C0CC00736F
Kuroda Y, Kubo M (1959) Ch2 rocking vibrations and the structure of polyethylene glycols. J Polym Sci 36(130):453–459. https://doi.org/10.1002/pol.1959.1203613039
Kyzas GZ, Travlou NA, Deliyanni EA (2014) The role of chitosan as nanofiller of graphite oxide for the removal of toxic mercury ions. Colloids Surf B 113:467–476. https://doi.org/10.1016/j.colsurfb.2013.07.055
Lima EC, Hosseini-Bandegharaei A, Moreno-Piraján JC, Anastopoulos I (2019) A critical review of the estimation of the thermodynamic parameters on adsorption equilibria. Wrong use of equilibrium constant in the van’t Hoof equation for calculation of thermodynamic parameters of adsorption. J Mol Liq 273:425–434. https://doi.org/10.1016/j.molliq.2018.10.048
Liu Y, Fang C, Zhang S, Zhong W, Wei Q, Wang Y, Dai Y, Wang Y, Zhang Z, Liu Y (2020) Effective adsorption of uranyl ions with different mos2-exposed surfaces in aqueous solution. Surf Interfaces 18:100409. https://doi.org/10.1016/j.surfin.2019.100409
Marczenko Z, Ramsay CG, Jurecki M (1976) Spectrophotometric determination of elements. E. Horwood, New York
Morshedy AS, Galhoum AA, Aleem A, Abdel Aleem H, Shehab El-din MT, Okaba DM, Mostafa MS, Mira HI, Yang Z, El-Sayed IET (2021) Functionalized aminophosphonate chitosan-magnetic nanocomposites for Cd(Ii) removal from aqueous solutions: performance and mechanisms of sorption. Appl Surf Sci 561:150069. https://doi.org/10.1016/j.apsusc.2021.150069
Muthuselvi C, Pandiarajan SS, Ravikumar B, Athimoolam S, Srinivasan N, Krishnakumar RV (2018) Ft-ir and ft-raman spectroscopic analyzes of indeno quinoxaline derivative crystal. Asian J Appl Sci 11:83–91
Oh SY, Yoo DI, Shin Y, Kim HC, Kim HY, Chung YS, Park WH, Youk JH (2005) Crystalline structure analysis of cellulose treated with sodium hydroxide and carbon dioxide by means of X-ray diffraction and FTIR spectroscopy. Carbohydr Res 340(15):2376–2391. https://doi.org/10.1016/j.carres.2005.08.007
Oshita K, Sabarudin A, Takayanagi T, Oshima M, Motomizu S (2009) Adsorption behavior of uranium(VI) and other ionic species on cross-linked chitosan resins modified with chelating moieties. Talanta 79(4):1031–1035. https://doi.org/10.1016/j.talanta.2009.03.035
Oshita K, Takayanagi T, Oshima M, Motomizu S (2007) Adsorption behavior of cationic and anionic species on chitosan resins possessing amino acid moieties. Anal Sci 23(12):1431–1434. https://doi.org/10.2116/analsci.23.1431
Pan S-Y, Syu W-J, Chang T-K, Lee C-H (2020) A multiple model approach for evaluating the performance of time-lapse capsules in trapping heavy metals from water bodies. RSC Adv 10(28):16490–16501. https://doi.org/10.1039/D0RA03017A
Pearson RG (1966) Acids and bases: hard acids prefer to associate with hard bases, and soft acids prefer to associate with soft bases. Science 151(3707):172–177. https://doi.org/10.1126/science.151.3707.172
Persson I (2010) Hydrated metal ions in aqueous solution: How regular are their structures? Pure Appl Chem 82(10):1901–1917. https://doi.org/10.1351/PAC-CON-09-10-22
Pourmortazavi SM, Sahebi H, Zandavar H, Mirsadeghi S (2019) Fabrication of fe3o4 nanoparticles coated by extracted shrimp peels chitosan as sustainable adsorbents for removal of chromium contaminates from wastewater: the design of experiment. Compos B Eng 175:107130. https://doi.org/10.1016/j.compositesb.2019.107130
Qiang S, Wang J, Wang Y, Yuan L, Shi L, Ding Z, Wang W, Liang J, Li P, Fan Q (2022) Analysis of the uranium chemical state by XPS: Is what you see real? Appl Surf Sci 576:151886. https://doi.org/10.1016/j.apsusc.2021.151886
Rangabhashiyam S, Anu N, Giri Nandagopal MS, Selvaraju N (2014) Relevance of isotherm models in biosorption of pollutants by agricultural byproducts. J Environ Chem Eng 2(1):398–414. https://doi.org/10.1016/j.jece.2014.01.014
Rashad MM, El-Sayed IE, Galhoum AA, Abdeen MM, Mira HI, Elshehy EA, Zhang S, Lu X, Xin J, Guibal E (2021) Synthesis of α-aminophosphonate based sorbents—influence of inserted groups (carboxylic vs. amine) on uranyl sorption. Chem Eng J 421:127830. https://doi.org/10.1016/j.cej.2020.127830
Ritcey GM, Ashbrook AW (1984) Solvent extraction: principles and applications to process metallurgy. Elsevier, Amsterdam
Sen T, Mishra S, Shimpi NG (2016) Synthesis and sensing applications of polyaniline nanocomposites: a review. RSC Adv 6(48):42196–42222. https://doi.org/10.1039/C6RA03049A
Shahrashoub M, Bakhtiari S, Afroosheh F, Googheri MS (2021) Recovery of iron from direct reduction iron sludge and biosynthesis of magnetite nanoparticles using green tea extract. Colloids Surf A 622:126675. https://doi.org/10.1016/j.colsurfa.2021.126675
Sharma JK, Srivastava P, Singh G, Virk HS (2016) Nanoferrites of transition metals and their catalytic activity. Solid State Phenom 241:126–138. https://doi.org/10.4028/www.scientific.net/SSP.241.126
Sing KSW (1985) Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (recommendations 1984). Pure Appl Chem 57(4):603–619. https://doi.org/10.1351/pac198557040603
Song Q, Ma L, Liu J, Bai C, Geng J, Wang H, Li B, Wang L, Li S (2012) Preparation and adsorption performance of 5-azacytosine-functionalized hydrothermal carbon for selective solid-phase extraction of uranium. J Colloid Interface Sci 386(1):291–299. https://doi.org/10.1016/j.jcis.2012.07.070
Stoia M, Istratie R, Păcurariu C (2016) Investigation of magnetite nanoparticles stability in air by thermal analysis and ftir spectroscopy. J Therm Anal Calorim 125(3):1185–1198. https://doi.org/10.1007/s10973-016-5393-y
Tien C (1994) Adsorption calculations and modeling. Butterworth-Heinemann, Boston
Wagner CE, Marshall PA (2010) Synthesis of 2,5-dichloro-2,5-dimethylhexane by an SN1 reaction. J Chem Educ 87(1):81–83. https://doi.org/10.1021/ed8000057
Wang J-S, Peng R-T, Yang J-H, Liu Y-C, Hu X-J (2011) Preparation of ethylenediamine-modified magnetic chitosan complex for adsorption of uranyl ions. Carbohyd Polym 84(3):1169–1175. https://doi.org/10.1016/j.carbpol.2011.01.007
Wang J, Liu G, Li T, Zhou C (2015) Physicochemical studies toward the removal of Zn(II) and Pb(II) ions through adsorption on montmorillonite-supported zero-valent iron nanoparticles. RSC Adv 5(38):29859–29871. https://doi.org/10.1039/C5RA02108A
Wang J, Ma X, Qu F, Asiri AM, Sun X (2017) Fe-doped ni2p nanosheet array for high-efficiency electrochemical water oxidation. Inorg Chem 56(3):1041–1044. https://doi.org/10.1021/acs.inorgchem.6b02808
Wang S, Wang L, Li Z, Zhang P, Du K, Yuan L, Ning S, Wei Y, Shi W (2021) Highly efficient adsorption and immobilization of U(VI) from aqueous solution by alkalized MXene-supported nanoscale zero-valent iron. J Hazard Mater 408:124949. https://doi.org/10.1016/j.jhazmat.2020.124949
Wang Y, Gu Z, Yang J, Liao J, Yang Y, Liu N, Tang J (2014) Amidoxime-grafted multiwalled carbon nanotubes by plasma techniques for efficient removal of uranium(VI). Appl Surf Sci 320:10–20. https://doi.org/10.1016/j.apsusc.2014.08.182
Willianms R (2017) Pka data compiled by R. Willianms, pp 1–33
Winsett J, Moilanen A, Paudel K, Kamali S, Ding K, Cribb W, Seifu D, Neupane S (2019) Quantitative determination of magnetite and maghemite in iron oxide nanoparticles using mössbauer spectroscopy. SN Appl Sci 1(12):1636. https://doi.org/10.1007/s42452-019-1699-2
Xie Y, Chen C, Ren X, Wang X, Wang H, Wang X (2019) Emerging natural and tailored materials for uranium-contaminated water treatment and environmental remediation. Prog Mater Sci 103:180–234. https://doi.org/10.1016/j.pmatsci.2019.01.005
Xu J, Chen M, Zhang C, Yi Z (2013) Adsorption of uranium(VI) from aqueous solution by diethylenetriamine-functionalized magnetic chitosan. J Radioanal Nucl Chem 298(2):1375–1383. https://doi.org/10.1007/s10967-013-2571-2
Yamaura M, Camilo RL, Sampaio LC, Macêdo MA, Nakamura M, Toma HE (2004) Preparation and characterization of (3-aminopropyl)triethoxysilane-coated magnetite nanoparticles. J Magn Magn Mater 279(2):210–217. https://doi.org/10.1016/j.jmmm.2004.01.094
Yang A, Yang P, Huang CP (2017) Preparation of graphene oxide–chitosan composite and adsorption performance for uranium. J Radioanal Nucl Chem 313(2):371–378. https://doi.org/10.1007/s10967-017-5329-4
Zhang X, Jiao C, Wang J, Liu Q, Li R, Yang P, Zhang M (2012) Removal of uranium(VI) from aqueous solutions by magnetic Schiff base: kinetic and thermodynamic investigation. Chem Eng J 198–199:412–419. https://doi.org/10.1016/j.cej.2012.05.090
Zhang Z, Dong Z, Wang X, Ying D, Niu F, Cao X, Wang Y, Hua R, Liu Y, Wang X (2018) Ordered mesoporous polymer–carbon composites containing amidoxime groups for uranium removal from aqueous solutions. Chem Eng J 341:208–217. https://doi.org/10.1016/j.cej.2018.02.044
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Editorial responsibility: Parveen Fatemeh Rupani.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Alghamdi, N.A. Mesoporous magnetic-polyaminated-chitosan nanocomposite for selective uranium removal: performance and mechanistic studies. Int. J. Environ. Sci. Technol. 20, 8319–8340 (2023). https://doi.org/10.1007/s13762-022-04565-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13762-022-04565-2