
Abstract
Α low-cost and environmentally friendly clean-up technique is evaluated in this study, based on the use of a brewery by-product, malt spent rootlets, as potential biosorbent for U(VI) sequestration from aquatic systems. Uranium uptake was rapid (2.5 h at 25 °C), and MSR exhibited capability of removing U(VI) from effluents of high acidity (pH 1.5) and salinity (0.5 M NaCl and NaNO3). Maximum uptake was 157 mg U(VI) g−1 at 25 °C. The pseudo-second-order model gave the best fit for kinetic data, whereas film diffusion was the rate-controlling step. Langmuir adsorption isotherm was the best fitting model. Activation energy, thermodynamic data and the extent of sorption reversibility implied that sorption of U(VI) is predominantly chemical. FTIR studies showed that lignin moieties are mainly responsible for U(VI) uptake. Speciation modeling showed that only positively charged and uncharged uranium species can be retained on the biomass. Finally, desorption studies revealed that Na2CO3 was the most efficient eluent with 78 % of U(VI), previously bound on the biosorbent’s surface, recovered.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Excessive amounts of uranium have been released into the environment and more specifically in surface and groundwaters, as a consequence of a wide array of anthropogenic activities, such as nuclear industry applications, mining and phosphate fertilizer industry. Uranium is a severe, ecological threat due to its toxicity and some radioactivity (Bhatti and Hamid 2014). Common physicochemical techniques, such as electrochemical treatment, ion exchange, reverse osmosis, precipitation, have been employed for decades, for the treatment of heavy metal-bearing aquatic systems. Nevertheless, they show certain drawbacks such as high operational cost, generation of secondary wastes and low efficiency. Biotechnological approaches can cover those niches for removal and recovery of both toxic and precious metals (Esmaeili and Aghababai Beni 2015). Biosorption is the property of sorbents of biological origin to bind heavy metals and radionuclides from aquatic systems through a passive, metabolism-independent mechanism. The advantages of biosorption include, among others, high efficiency, especially in low pollutant concentrations, eco-friendliness, lower cost and regeneration of the biomass for consecutive cycles of use (Flouty and Estephane 2012). It is estimated that resin market sales in North America alone account for US$ 2 billion per year; however, only 15 % of the total sales are specially designed for heavy metal removal. The further development and commercialization of low-cost biosorbents could penetrate the existing market of resin R&D, and with a conservative estimate of 10 % penetration, this might account for US$ 30 million per year in North America only (Volesky 2001). The worldwide sales are estimated to a quadruple range of that of North America. Furthermore, the cost-effectiveness of biosorption as a clean-up technology is important in developing countries which face serious challenges due to increasing environmental pollution from industrial wastewaters and are interested in low-cost, easily applicables in large-scale remediation technologies (Gan and Li 2013).
Most uranium biosorption studies use biomaterials relatively abundant in nature, such as microbial species of algae (Cecal et al. 2012), fungi (Akhtar et al. 2009), yeasts (Anagnostopoulos et al. 2011), bacteria (Li et al. 2004). Inexpensive lignocellulosic wastes of the agricultural and food production have also been evaluated as biosorbents for the removal of toxic metals such as mercury (Boutsika et al. 2013) and Cr(III) (Ferraz et al. 2014), textile dyes such as Malachite Green (Chanzu et al. 2012) and metal dye (Ben Hamissa et al. 2008), and hydrophobic organic pollutants such as phenanthrene (Valili et al. 2013) from aqueous systems. Brewer’s spent grains (BSG) and malt spent rootlets (MSR) represent readily available, high-volume and low-cost by-products of the brewing industry. Their chemical composition varies according to barley variety and brewing practices and includes considerable amounts of fiber, protein, cellulose, lignin, phenolic compounds, etc.(Robertson et al. 2010; Waters et al. 2013). These wastes are mainly used without pre-treatment as low-cost (and low nutritional value) animal feed, compost/fertilizer and combustion fuel, while their disposal is a considerable source of environmental pollution. Alternative efforts to exploit them, thus creating added-value and environmental benefits, include the development of biosorbent materials suitable for aquatic systems and industrial effluent clean-up uses.
The present work explores the potential use of MSR for uranium sequestration from aqueous solutions. Basic parameters affecting uranium sorption, such as pH, contact time and temperature, were investigated. Furthermore, recovery experiments were carried out in an attempt to quantitatively reclaim uranium, given the wide array of its industrial applications. MSR have not been previously investigated for the removal of uranium or other radionuclides from aqueous systems. All experiments were carried out in the Radiochemistry Lab of the Department of Chemistry at University of Patras (Greece) during the period 2011–2012.
Materials and methods
MSR and surface analysis
MSR were supplied by the Athenian Brewery S.A. (Heineken Group; Achaia, Greece). They were washed with triply distilled water to remove impurities, oven-dried for 24 h at 70 °C and subsequently sieved. The fraction 0.18 < d<1.0 mm, where d is the biosorbent’s particle mean diameter (mm), was retained for all experiments carried out in the present work. Specific surface area and porosity of MSR were measured using N2 adsorption and applying the BET equation for five different samples (a Micrometrics Tristar 060 with Sample Degas System was used).
FTIR and potentiometric titrations
The study of the MSR functional groups was performed by Fourier transform infrared analysis (FTIR) by the KBr disk method using a PerkinElmer (model 16PC FT-IR) spectrophotometer. The spectra were analyzed with the software IR Search Master 6.0. For potentiometric titrations, 0.5 g of MSR and 50 mL of NaNO3 0.1 M were placed in a double-wall water-jacketed reactor at 25 °C. Inert atmosphere was ensured by bubbling water vapor-saturated nitrogen through the solutions, which were homogenized by magnetic stirring. The adjustment of the suspension pH to 11 was achieved by the addition of 1 M NaOH solution into the beaker. Inert atmosphere is required in order to avoid the formation of carbonate in the solution, which occurs when atmospheric carbon dioxide is absorbed by an alkaline solution. Past the establishment of equilibrium in the suspension, identified by the stability of the solution pH, the titration was conducted by adding small aliquots of standard HNO3 solution with an electronic burette and the corresponding pH values were recorded. Past the end of the biomass titration, the supernatant was collected by centrifugation followed by membrane filtration (Millipore, GSTF 04700, pore diameter 0.22 μm,) under vacuum. An equal volume of the standard NaOH solution was added into the collected liquid phase, which was subsequently titrated in the same exact way as previously described. The purpose of the last step was to estimate the functional groups possibly leaching out from the MSR to the supernatant. The net surface H+ consumption was calculated at each pH value subtracting the H+ supernatant consumption (2nd titration) from the total H+ consumption (1st titration).
Sorption experiments
For sorption experiments, a stock solution was prepared from a standard 1000 mg U(VI) L−1 (Aldrich 207624, Atomic Absorption Standard Solution). Fresh working solutions were prepared by diluting appropriate portions of the stock solution with triply distilled water.
0.010 g of MSR were suspended in 10 mL of U(VI) solution, in polyethylene vials, with concentration 800 mg L−1 under constant ionic strength I = 0.1 M NaClO4 at different pH values (1.5–4.5) at 25 °C on a rotary shaker. The vials were equilibrated on a rotary shaker to keep the suspensions homogeneous throughout the sorption period. Past equilibration time of 24 h, the suspensions were centrifuged to separate the fluid phase from the solid substrate and a portion of the supernatant was analyzed for remaining uranium concentration by using Arsenazo III method. The effect of solid-to-solution ratio was studied for MSR concentration ranging from 1 to 12.5 mg mL−1, initial U(VI) concentration 800 mg L−1, I = 0.1 M NaClO4 at 25 °C for 24 h. In kinetic studies, the MSR were allowed to equilibrate with 10 mL of 800 mg U(VI) L−1 at 5, 15, 25 and 33 °C for different time periods. In equilibrium studies, the initial U(VI) concentration was in the range 25–800 mg L−1 at 5, 15, 25 and 33 °C. In all the above experiments, the ionic strength of the solution was kept constant at 0.1 M with NaClO4.
Effect of inorganic and organic ligands
In order to explore the effect of inorganic and organic ligands on uranium uptake, 0.01 g of MSR in 10 mL of 250 mg U(VI) L−1, pH 3 at 25 °C for 24 h under different concentrations of inorganic and organic ligands: 0.001–0.5 M NO3 − and Cl−, 0.001–0.02 M HCOO− and CH3COO−, 0.01–0.1 M C6H5O7 3− and 0.001–0.1 M C2O4 2−. Speciation diagrams were obtained by means of Hydra–Medusa Equilibrium Diagrams software.
Recovery and reversibility experiments
In order to study the potential recovery of uranium from the biomass, 0.010 g of MSR was suspended in 10 mL of 100 mg U(VI) L−1 solution at optimum pH for 24 h at 25 °C. Past 24 h of equilibration, the residual U(VI) concentration in the supernatant was determined and thus the adsorbed amount was accurately measured. Next, the samples were filtered and the biomass was washed three times with small portions of distilled water. For the investigation of uranium recovery, the biomass with the adsorbed U(VI) was transferred in new polyethylene vials, in which 10 mL of different solutions of 0.1 M was added: H2SO4, HNO3, HCl, HCOOH, CH3COOH, EDTA, NaOH, Na2CO3, NaCl and NaNO3. The vials were capped without pH adjustment, and the suspensions were equilibrated on a rotary shaker for 24 h at 25 °C. Finally, the desorbed U(VI) concentration in the aqueous phase was determined following the separation of the fluid from the solid substrate by filtration and quantitative analysis of the filtrate for U(VI).
A series of additional experiments were done in order to examine the reversibility of adsorption of U(VI) by suspending the adsorbent loaded with the adsorbate in 10 mL 0.1 M NaClO4 for a period of 7 days at 25 °C.
Results and discussion
Uranium concentration was determined spectrophotometrically (JENWAY 63000 UV/vis) using the Arsenazo III method. The metal uptake, q, was calculated from the difference between the initial and the final metal concentration using Eq. (1):
The percentage of metal removal, R (%), was calculated from Eq. (2):
In each experiment, the mean values of q or R (%) were estimated from triplicate experiments.
Statistical analyses were performed with statistical software SPSS 17.0. Statistically equivalent values (p > 0.05, Student’s t test) are denoted by same superscripts in the tables that follow.
Specific surface area, porosity and potentiometric titrations
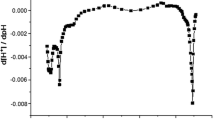
The specific surface area of MSR was found to be 0.1 m2 g−1 and porosity was 0.6 mm3 g−1. Low values of the specific surface areas have been reported elsewhere for lignocellulosic materials of biological origin, such as 1.05 m2 g−1 for wood sawdust (Sciban et al. 2007), 0.376 m2 g−1 for grape stalks (Martínez et al. 2006), and 1.5 and 1.6 for delignified rice husk and mango tree sawdust, respectively (Kumar et al. 2014). The previously described titration procedure with acid led to a potentiometric curve in which the concentration of H+ (denoted as [H+]) “consumed” by the biomass may be seen. In differential potentiometric curve charts, the reversed peaks correspond to the −pK values of the functional groups present on MSR surface, which may be ionized providing acid–base sites, and the area under the peaks of the differential curve is proportional to the concentration of the respective functional groups (Bourikas et al. 2006). In the case of MSR, four distinct areas were observed (Fig. 1a): The first one lies between pH values 2.8–5, the second between pH values 6–7.5, the third at pH 8–9.5, and the last one between pH values 9.5–11. Therefore, groups of both acidic and basic nature are present on MSR surface, which can be attributed to carboxyl and hydroxyl groups of the lignin aromatic structure. More specifically, the area under the 2.8–5 peak corresponds to H+ consumption of 7.4 × 10−2 mol L−1, whereas the peaks of 8–9.5 and 9.5–11 show consumption of 1.3 × 10−3 and 1.7 × 10−3 mol L−1 H+. Therefore, it can be concluded that carboxyl groups that exhibit acidic nature are present to a greater extent on the surface of MSR, as opposed to hydroxyl groups, whose consumption of H+ is 25 times less.

Differential potentiometric curve for suspensions of MSR, T = 25 °C, ionic strength I = 0.1 M NaNO3 (a). FTIR spectra of MSR before sorption (black line) and after uranium uptake (gray line) (U(VI) concentration 100 mg L−1, pH 4.5, 25 °C, 24 h) (b)
Effect of pH
The adsorption results showed that uranium uptake progressively increased from pH 1.5 to 4.5 (Online Source 1). The effect of pH at values higher than 4.5 was not studied, in order to avoid uranium precipitation as predicted by the speciation diagram (Fig. 2). The percentage of uranium uptake for the pH values studied, varied between 5 and 20 %. The error bars indicate the standard deviation estimated from triplicate experiments. MSR were able to remove a significant amount of uranium ions under strong acidic conditions, as low as pH 1.5, which could be important for recovery of uranium and similar cations from acidic aquatic systems like mining effluents (Laus et al. 2007). At low pH values, most functional groups present on MSR are protonated and undissociated, having positive (–NH3 +) or neutral charge (–COOH), respectively, and hence, the approach of positively charged species is not favored by electrostatic attractions. Increasing the solution pH, the negative charge density increases due to the increasing deprotonation of the functional groups (–COO−) and the approach of positively charged species is favored, resulting in higher metal loading. Potentiometric titrations revealed that despite the presence of groups with basic nature, such as hydroxyl groups, a major portion of MSR surface functional groups are of acidic nature.

Uranium speciation as a function of pH; C i = 800 U(VI) mg L−1, 25 °C, I = 0.1 M NaClO4, according to Hydra–Medusa, Chemical Equilibrium Diagrams
FTIR studies
FTIR spectra of the adsorbent were obtained before (raw material) and after U(VI) uptake, in an attempt to obtain a better idea of the results of the interaction between adsorbate and adsorbent. In the FTIR spectra shown in Fig. 1b, the band at 3300 cm−1 corresponds to the O–H stretching of bound hydroxyl groups, while the band at 2920 cm−1 corresponds to C–H stretching of aliphatic groups (Pedro Silva et al. 2004). The band at 1380 cm−1 is attributed to C–H bending, the one at 1058 cm−1 is assigned to C–O bond of the lignin structure, and the one at 1246 cm−1 is assigned to guaiacyl lignin (Martínez et al. 2006; Xu et al. 2013). Following U(VI) sorption, some of the aforementioned bands shifted and/or were suppressed, an indication that the aromatic structures of lignin are involved in U(VI) binding.
Effect of contact time
Several mathematical models have been used to describe sorption data, which, in bulk, fall under two categories: sorption reaction models and sorption diffusion models. All these models describe the kinetic process of sorption; however, they are quite different in nature. Reaction models describe the whole process of sorption based on chemical reaction kinetics, whereas the diffusion models take into consideration diffusion phenomena that take place in the liquid film surrounding the sorbent particles, as well as within the pores of the material.
Reaction-based kinetics
Kinetic experiments were carried out for a ratio of 10 mg of MSR:10 mL U(VI) solution, with 800 mg U(VI)L−1, pH 4.5 at 5, 15, 25 and 33 °C. Kinetic data are compiled in Fig. 3.

Kinetic data for the sorption of U(VI) on MSR at 5, 15, 25 and 33 °C (C i = 800 mg U(VI) L−1, pH 4.5, 25 °C)
The reaction-based kinetic models used in this study were the pseudo-first-order, the pseudo-second-order and Elovich models. Pseudo-first-order model is a first-order rate equation that describes the kinetics of liquid–solid phase adsorption, whereas the pseudo-second-order model describes the existence of two chemical equations, with the slowest being the rate-limiting step, and Elovich model is perceived to describe chemisorption phenomena (Rudzinski and Panczyk 2000). Pseudo-first-order model can be described in its linear form by Eq. (3):
Assuming that the rate of uranium sorption is a second-order process, the pseudo-second-order kinetics may be expressed as:
The Elovich equation is described by Eq. 5 (Ho et al. 2000):
For the comparison of the quality of the fit of the kinetic data to the proposed models, the normalized standard deviation, Δq (%), was used:
The results are summarized in Table 1. According to the estimated Δq (%) and R 2 values, the pseudo-second-order model seems to be the best fitting model of all three. The Elovich model provides a good fit as well. On the other hand, pseudo-first-order model fails to describe the experimental data.
Diffusion-based models
The prediction of the rate-limiting step for a sorption process is an important factor. The process is usually assumed to consist of four consecutive steps: transport of the solute from the bulk solution through a liquid film to the exterior surface of the adsorbent (film diffusion), binding of adsorbate molecules on the active sites on the surface of the sorbent, transport of the solute within the pores of the sorbent (intraparticle diffusion) and binding of the solute molecules on the active sites distributed within the sorbent particles (Lazaridis and Asouhidou 2003). Generally, the steps involving binding are rapid and can be neglected when evaluating the rate-determining step of the sorption process. The Morris–Weber model (Weber and Morris 1963) is the most commonly used criterion to identify the mechanism involved and to distinguish between film or intraparticle diffusion. It is described by Eq. (7):
The intercept reflects the boundary layer effect. Linear correlation of q versus t 1/2 designates the presence of intraparticle diffusion, and if C = 0, intraparticle diffusion controls the process’ rate, whereas if C ≠ 0, intraparticle diffusion is not the rate-controlling step, but film diffusion is involved in the metal sorption process (Al-Degs et al. 2006; Lang et al. 2013). Clearly, there is a linear dependence of q on t 1/2 at all temperatures studied, suggesting that intraparticle diffusion might be involved in the sorption of U(VI) by MSR (Fig. 4). Multilinearity is also observed, which is the case when multiple mechanisms with multiple rate constants are involved. The first linear part can be attributed to the intraparticle diffusion, and the second linear part, almost parallel to x axis, represents an apparent saturation where diffusion plays no longer any role. For all temperatures studied, none of the linear parts crossed the origin, denoting that intraparticle diffusion is not the rate-controlling step.

Morris–Webber plots for uranium sorption on MSR at different temperatures; C i = 800 U(VI) mg L−1, pH 4.5, I = 0.1 M NaClO4
In order to evaluate whether external mass transfer or intraparticle diffusion exerts greater influence on the rate of metal sorption, the Boyd film diffusion model was employed, which in its simplified form can be expressed as (Eq. 8):
If the plot of Bt versus t is linear and passes through the origin, then intraparticle diffusion controls the rate of mass transfer. If the plot is nonlinear or linear, but does not pass through the origin, then film diffusion controls the adsorption rate (Shinde et al. 2012). Linear plots do not pass through the origin for all temperatures (data not shown); more specifically, intercept was found to be 0.6 ± 0.1, 0.7 ± 0.1, 0.8 ± 0.1 and 1 ± 0.1 at 5, 15, 25 and 33 °C, respectively. Hence, it seems that film diffusion is likely the rate-controlling step which is rather expected, as intraparticle diffusion depends mostly on porosity and MSR do not exhibit remarkable porosity.
Activation energy, E a
For the determination of the phenomenal activation energy, E a, Arrhenius equation was used (Eq. 9):
Activation energy provides an estimate about the mechanism of sorption: physisorption and chemisorption. In physisorption, the phenomenon is reversible to a considerable degree since the bonds formed are weak and the energy requirements are small. On the other hand, in chemisorption, U(VI) is retained by chemical bonds and the phenomenon is little or no reversible. Activation energy in physical sorption phenomena is usually 4 kJ mol−1 or smaller, whereas in chemisorption, values span between 10 and 80 kJ mol−1 (Ucun et al. 2008). E a was calculated to be 38 ± 3 kJ mol−1 (R 2 = 0.99), indicating that sorption of U(VI) by MSR is predominantly of chemical nature. Physical sorption involves retention of the metal by an active group through weak bonds (e.g., electrostatic interaction, van der Waals) and is largely reversible, whereas in chemisorption, the binding is strong and the reversibility is relatively low.
Reversibility
The objective of these experiments was to assess the degree of reversibility of the U(VI) uptake by MSR. The percentage of uranium released in the aqueous phase past equilibration for 7 days was estimated to be 9 ± 2 %. This finding suggests that U(VI) is strongly retained by MSR and the phenomenon is reversible to a small degree. This comes into agreement with the assumption made based on the activation energy that the formation of chemical bonds plays a dominant role in U(VI) sorption by MSR.
Effect of initial concentration: isotherm modelling
The effect of the initial U(VI) concentration was studied for concentrations in the range 25–800 mg L−1, pH 4.5, for equilibrium time of 6 h at 5, 15, 25 and 33 °C (Fig. 5).

Isotherm data for U(VI) sorption on MSR at different temperatures; C i range was 25–800 mg L−1, pH 4.5, I = 0.1 M NaClO4, equilibration time 6 h
Isotherms were analyzed in terms of Langmuir, Freundlich and Dubinin–Radushkevich models. These models are among the most widely used in order to describe isotherm data, despite the fact that they differ in nature. Langmuir model assumes that sorption takes place in a homogeneous way onto limited sites and sorption takes place in a monolayer (Anagnostopoulos et al. 2012). Freundlich is a semi-empirical, simple model, and Dubinin–Radushkevich is a more general model which does not assume homogeneous surface or constant sorption potential (Foo and Hameed 2010). Langmuir, Freundlich and Dubinin–Radushkevich models are described by Eqs. 10, 11 and 12, respectively.
where \(\upvarepsilon = \, R \cdot T \cdot \ln \left( {1 \, + \, 1/C_{\text{fin}} } \right)\). The Freundlich constant, K F , is indicative of sorption capacity, and n is a constant, indicative of sorption intensity. Generally, higher n values represent stronger sorption intensity (Anayurt et al. 2009). K′ is an adsorption energy-related constant that can lead to the calculation of mean adsorption energy E (kJ mol−1), using Eq. 13:
For E < 8 kJ mol−1, the process is most likely to be of physical nature, while if E is between 8 and 16 kJ mol−1, ion exchange or chemisorption is strongly involved in the process (Kumar et al. 2011). The parameters of each model applied were summarized in Table 2.
Freundlich model presented the lowest applicability among the models. On the other hand, the Langmuir model fits very well with the experimental data and the predicted maximum uptake q max decreases with the temperature implying that the nature of the phenomenon is exothermic. The mean adsorption energy values calculated by Dubinin–Radushkevich model imply that the process is predominantly of chemical nature. On the other hand, the maximum uptake values predicted by Dubinin–Radushkevich model deviate to a significant degree from the experimental values, denoting that neither model is appropriate to fully interpret experimental data.
Thermodynamic calculations
Thermodynamic calculations were performed applying the distribution coefficient of the Langmuir model, K L (L mol−1), in van’t Hoff equation (Eq. 14):
The values of ΔΗ o and ΔS o were found to be −20 ± 2 kJ mol−1 and 13 ± 1 J K−1 mol−1, respectively. The negative value of ΔΗ o verifies the exothermic nature of the process, whereas the positive value of ΔS o denotes the increase in disorder at the solid–solution interface during sorption. The estimated ΔΗ o lies on the borderline between physical and chemical sorption (Liu and Xu 2007). This result confirms the complex nature of biosorption, since it implies that despite most evidence pointing toward chemisorption, other mechanisms might be involved as well, e.g., physisorption.
Effect of inorganic and organic ligands
In the range of concentration of inorganic and organic ligands studied, speciation of uranium changes drastically with the formation of various uranium–ligand complexes. The ability of MSR to sequester U(VI) from aqueous solutions remains unaffected throughout a wide range of concentration of nitrates, chlorides, formates and acetates (Fig. 6a, b). MSR seem to be a promising biosorbent for U(VI) removal even from complicated aquatic systems that exhibit high salinity. The results imply that there is no selectivity and all positively charged uranium species, regardless of size and valence, are equally retained by the active sites of the sorbent. U(VI) removal values were found to be ~30 % for the range of nitrate and chloride studied, ~70 % for the range of formate studied and ~60 % for the range of acetate (values for each ligand category were statistically equivalent, p > 0.05, Student’s t test). Interestingly, when the concentration of citrates ranged from 0.001 to 0.03 M, U(VI) removal was found to be zero. Speciation remained stable throughout the above-mentioned range, UO2(cit)− 20 % and UO2(cit) 2−2 (80 %), implying that uranium is stabilized in the aqueous phase through complexation with citrate ligands. Oxalates concentrations ranged from 0.001 to 0.1 M, and the results are shown in Table 3. There seemed to be selectivity of MSR binding sites toward uncharged UO2(ox) against the negatively charged UO2(ox) 2−2 . Moreover, when U(VI) is present only under negative charge (Νa2C2O4 0.1 M), there is practically no sorption. All values are statistically different (p < 0.05, Student’s t test). Therefore, citrate and oxalate studies revealed that the negatively charged are most likely stabilized in the aqueous phase through complexation and subsequently are not retained by the biomass.

Dependence of U(VI) removal on nitrate and chloride concentrations (a) and on formate and acetate concentrations (b) by MSR, 250 mg U(VI) L−1, pH 3 at 25 °C for 24 h. Error bars represent relative standard deviation calculated from triplicate samples
Recovery experiments
In order to determine the most suitable agent for the quantitative recovery of U(VI), different alkalis, acids and salts were used at 0.1 M concentration. The contact time between metal-loaded biomass and the desorption solution was 24 h at 25 °C. The results are summarized in Table 4.
Assuming that the higher the affinity of a ligand to form complexes with uranium, the easier the removal of the sorbent-bound metal, then the highest recovery can be expected by carbonates, followed by sulfates and to a lesser degree nitrates and chlorides (Bogolepov et al. 2009). The experimental findings obtained were consistent, since Na2CO3 was found to be the most efficient desorbing agent (78 % uranium recovery), followed by H2SO4 (47 % uranium recovery) and HNO3 and HCl, which showed statistically similar values (35 ± 3 and 39 ± 3 %, respectively, p > 0.05 Student’s t test). A large recovery difference was found between HCl–HNO3 and the equivalent salts NaCl–NaNO3 of the same concentration, namely 35 and 39 % for the pair HCl–HNO3 and 12 and 4 % for NaCl–NaNO3. This was attributed to the large difference in H+ concentration of the desorption agents, because when acids were used, apart from the ability of the ligand to “extract” U(VI) by forming complexes, the binding sites were protonated, and as a result, significantly higher desorption is anticipated (Naja et al. 2009). Organic acids, EDTA and NaOH, showed a rather poor recovery (Table 4). EDTA and NaOH showed the same statistically desorbing efficiency, 26 ± 4 and 24 ± 2, respectively. Although EDTA forms very stable complexes with uranium and therefore a higher recovery was expected, the low recovery may be due to the difficulty of the EDTA molecule approaching the surface, because of its relatively large size.
The strategy of choosing a desorbing agent should take into account various factors such as recovery efficiency, possible alteration of the biomass and cost. In the case of U(VI) uptake by MSR, sodium carbonate seems to be the most suitable desorbing agent, due to the higher affinity of carbonates in the aqueous phase to form complex with uranium on the surface of the material, whereas sulfates, nitrates and chloride exhibit much smaller complex formation constant with uranium. Furthermore, sodium carbonate has not been reported in the literature to incur any morphological alterations in the biomass, as opposed to inorganic acids, which would render the reusability of the sorbent limited due to the decline of metal binding efficiency. Nevertheless, further experiments of continuous sorption–desorption cycles might be needed in order to derive certain conclusions about the amount of times the biomass can be reused.
Conclusion
MSR are a promising biosorbent for the decontamination of U(VI)-bearing aquatic systems due to high sorption capacity and fast kinetics. It is a low-cost, easily accessible material that exhibits the ability to sequester significant uranium amounts from highly acidic environments and effluents rich in salts and organic ligands. Kinetic, thermodynamic and reversibility experiments verified the complex nature of the phenomenon, suggesting that despite that chemisorption seems to play a dominant role in the sorption process, other mechanisms might also be involved. Significant recovery of uranium from MSR can be achieved with mild desorption agents such as Na2CO3 0.1 M. Finally, species calculations indicated that MSR were able to retain positively charged and neutral uranium species, whereas negatively charged uranium complexes were mobile and retention by the material was minimal.
References
Akhtar K, Khalid AM, Akhtar MW, Ghauri MA (2009) Removal and recovery of uranium from aqueous solutions by Ca-alginate immobilized Trichoderma harzianum. Bioresour Technol 100:4551–4558
Al-Degs YS, El-Barghouthi MI, Issa AA, Khraisheh MA, Walker GM (2006) Sorption of Zn(II), Pb(II), and Co(II) using natural sorbents: equilibrium and kinetic studies. Water Res 40:2645–2658
Anagnostopoulos V, Bekatorou A, Symeopoulos B (2011) Contribution to interpretation of metal uptake dependence upon the growth phase of microorganisms. The case of uranium (VI) uptake by common yeasts, cultivated at different temperatures, with or without aeration. J Radioanal Nucl Chem 287:665–671
Anagnostopoulos V, Manariotis ID, Karapanagioti HK, Chrysikopoulos CV (2012) Removal of mercury from aqueous solutions by malt spent rootlets. Chem Eng J 213:135–141
Anayurt RA, Sari A, Tuzen M (2009) Equilibrium, thermodynamic and kinetic studies on biosorption of Pb(II) and Cd(II) from aquous solutions from macrofungus (Lactarius scrobiculatus) biomass. Chem Eng J 151:255–261
Ben Hamissa AM, Ncibi MC, Mahjoub B, Seffen M (2008) Biosorption of metal dye from aqueous solution onto Agave americana (L.) fibres. Int J Environ Sci Technol 5:501–508
Bhatti HN, Hamid S (2014) Removal of uranium(VI) from aqueous solutions using Eucalyptus citriodora distillation sludge. Int J Environ Sci Technol 11:813–822
Bogolepov AA, Kobets SA, Pshinko GN (2009) Desorption of U(VI) from montmorillonite with aluminum and iron hydroxides deposited on its surface. Radiochemistry 51:301–307
Bourikas K, Kordulis C, Lycourghiotis A (2006) How metal (hydr)oxides are protonated in aqueous media: the (n + 1) rule and the role of the interfacial potential. J Colloid Interface Sci 296:389–395
Boutsika L, Karapanagioti H, Manariotis I (2013) Aqueous mercury sorption by biochar from malt spent rootlets. Water Air Soil Pollut 225:1–10
Cecal A, Humelnicu D, Rudic V, Cepoi L, Ganju D, Cojocari A (2012) Uptake of uranyl ions from uranium ores and sludges by means of Spirulina platensis, Porphyridium cruentum and Nostok linckia alga. Bioresour Technol 118:19–23
Chanzu H, Onyari J, Shiundu P (2012) Biosorption of malachite green from aqueous solutions onto polylactide/spent brewery grains films: kinetic and equilibrium studies. J Polym Environ 20:665–672
Esmaeili A, Aghababai Beni A (2015) Biosorption of nickel and cobalt from plant effluent by Sargassum glaucescens nanoparticles at new membrane reactor. Int J Environ Sci Technol 12:2055–2064
Ferraz AI, Amorim C, Tavares T, Teixeira JA (2014) Chromium(III) biosorption onto spent grains residual from brewing industry: equilibrium, kinetics and column studies. Int J Environ Sci Technol 12:1591–1602
Flouty R, Estephane G (2012) Bioaccumulation and biosorption of copper and lead by a unicellular algae Chlamydomonas reinhardtii in single and binary metal systems: a comparative study. J Environ Manag 111:106–114
Foo KY, Hameed BH (2010) Insights into the modelling of adsorption isotherm systems. Chem Eng J 156:2–10
Gan PP, Li SFY (2013) Biosorption of elements. In: Hunt A (ed) Element recovery and sustainability. The Royal Society of Chemistry, Cambridge
Ho YS, Ng JCY, McKay G (2000) Kinetics of pollutant sorption by biosorbents: review. Sep Purif Rev 29:189–232
Kumar PS, Ramalingam S, Kirupha SD, Murugesan A, Vidhyadevi T, Sivanesan S (2011) Adsorption behavior of nickel(II) onto cashew nut shell: equilibrium, thermodynamics, kinetics, mechanism and process design. Chem Eng J 167:122–131
Kumar MN, Gialleli A-I, Masson JB, Kandylis P, Bekatorou A, Koutinas AA, Kanellaki M (2014) Lactic acid fermentation by cells immobilised on various porous cellulosic materials and their alginate/poly-lactic acid composites. Bioresour Technol 165:332–335
Lang W, Buranaboripan W, Wongchawalit J, Parakulsuksatid P, Vanichsriratana W, Sakairi N, Pathom-aree W, Sirisansaneeyakul S (2013) Biosorption of lead from acid solution using chitosan as a supporting material for spore forming-fungal biomass encapsulation. Int J Environ Sci Technol 10:579–590
Laus R, Geremias R, Vasconcelos HL, Laranjeira MC, Favere VT (2007) Reduction of acidity and removal of metal ions from coal mining effluents using chitosan microspheres. J Hazard Mater 149:471–474
Lazaridis NK, Asouhidou DD (2003) Kinetics of sorptive removal of chromium(VI) from aqueous solutions by calcined Mg–Al–CO3 hydrotalcite. Water Res 37:2875–2882
Li P-F, Mao Z-Y, Rao X-J, Wang X-M, Min M-Z, Qiu L-W, Liu Z-L (2004) Biosorption of uranium by lake-harvested biomass from a cyanobacterium bloom. Bioresour Technol 94:193–195
Liu Y, Xu H (2007) Equilibrium, thermodynamics and mechanisms of Ni2+ biosorption by aerobic granules. Biochem Eng J 35:174–182
Martínez M, Miralles N, Hidalgo S, Fiol N, Villaescusa I, Poch J (2006) Removal of lead(II) and cadmium(II) from aqueous solutions using grape stalk waste. J Hazard Mater 133:203–211
Naja G, Murphy V, Volesky B (2009) Biosorption, metals. encyclopedia of industrial biotechnology bioprocess, bioseparation and cell technology. Wiley, New York
Pedro Silva J, Sousa S, Rodrigues J, Antunes H, Porter JJ, Gonçalves I, Ferreira-Dias S (2004) Adsorption of acid orange 7 dye in aqueous solutions by spent brewery grains. Separ Purif Technol 40:309–315
Robertson JA, I’Anson KJA, Treimo J, Faulds CB, Brocklehurst TF, Eijsink VGH, Waldron KW (2010) Profiling brewers’ spent grain for composition and microbial ecology at the site of production. LWT Food Sci Technol 43:890–896
Rudzinski W, Panczyk T (2000) Kinetics of isothermal adsorption on energetically heterogeneous solid surfaces: a new theoretical description based on the statistical rate theory of interfacial transport. J Phys Chem 104:9149–9162
Sciban M, Radetic B, Kevresan Z, Klasnja M (2007) Adsorption of heavy metals from electroplating wastewater by wood sawdust. Bioresour Technol 98:402–409
Shinde NR, Bankar AV, Kumar AR, Zinjarde SS (2012) Removal of Ni (II) ions from aqueous solutions by biosorption onto two strains of Yarrowia lipolytica. J Environ Manag 102:115–124
Ucun H, Bayhan YK, Kaya Y (2008) Kinetic and thermodynamic studies of the biosorption of Cr(VI) by Pinus sylvestris Linn. J Hazard Mater 153:52–59
Valili S, Siavalas G, Karapanagioti HK, Manariotis ID, Christanis K (2013) Phenanthrene removal from aqueous solutions using well-characterized, raw, chemically treated, and charred malt spent rootlets, a food industry by-product. J Environ Manag 128:252–258
Volesky B (2001) Detoxification of metal-bearing effluents: biosorption for the next century. Hydrometallurgy 59:203–216
Waters DM, Kingston W, Jacob F, Titze J, Arendt EK, Zannini E (2013) Wheat bread biofortification with rootlets, a malting by-product. J Sci Food Agric 93:2372–2383
Weber WJ, Morris JC (1963) Kinetics of adsorption on carbon from solution. J Sanit Eng Div 89:31–60
Xu F, Yu J, Tesso T, Dowell F, Wang D (2013) Qualitative and quantitative analysis of lignocellulosic biomass using infrared techniques: a mini-review. Appl Energy 104:801–809
Acknowledgments
This research has been cofinanced by the European Union (European Regional Development Fund—ERDF) and Greek national funds through the Operational Program “Regional Operational Programme” of the National Strategic Reference Framework (NSRF)—Research Funding Program: Support for research, technology and innovation actions in Region of Western Greece (D.237.007).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Nomenclature
- Bt
-
Dimensionless constant of Boyd model
- C
-
Vertical axis intercept of Morris–Weber model (mg g−1)
- C i
-
U(VI) initial concentration (mg L−1 or mol L−1)
- C fin
-
U(VI) final concentration (mg L−1 or mol L−1)
- E a
-
Activation energy (kJ mol−1)
- k 1
-
Rate constant of pseudo-first-order reaction kinetic model (min−1)
- k 2
-
Rate constant of pseudo-second-order reaction kinetic model (g mg−1 min−1)
- k d
-
Intraparticle diffusion rate constant (mg g−1 min−1/2)
- K L
-
Langmuir constant (L mol−1)
- K F
-
Freundlich constant
- K′
-
Dubinin–Radushkevich mean adsorption energy constant (mol2 kJ−2)
- m
-
Dry weight of biomass (g)
- q
-
U(VI) uptake per biosorbent mass unit (mg g−1 or mol g−1)
- q e
-
U(VI) bound per biosorbent mass unit at equilibrium (mg g−1 or mol g−1)
- q t
-
U(VI) bound per biosorbent mass unit at time t (mg g−1 or mol g−1)
- q max
-
Maximum U(VI) uptake as predicted by isotherm models (mol g−1)
- R
-
Gas constant (J K−1 mol−1)
- T
-
Temperature (K)
- R(%)
-
Percentage removal of U(VI) from the aqueous solution
- V
-
Solute volume (L)
- α
-
Initial sorption rate for Elovich model (mg g−1 min−1)
- β
-
Constant related to the extent of surface coverage and activation energy for chemisorptions for Elovich model (g mg−1)
- ΔΗ o
-
Apparent enthalpy change (kJ mol−1)
- ΔS o
-
Apparent entropy change (J mol−1 K−1)
Rights and permissions
About this article

Cite this article
Anagnostopoulos, V., Symeopoulos, B., Bourikas, K. et al. Biosorption of U(VI) from aqueous systems by malt spent rootlets. Kinetic, equilibrium and speciation studies. Int. J. Environ. Sci. Technol. 13, 285–296 (2016). https://doi.org/10.1007/s13762-015-0872-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13762-015-0872-4