
Abstract
Background and purpose
Hereditary spastic paraplegia (HSP) is a group of neurodegenerative diseases divided into pure and complex forms, with spasticity in lower limbs only, or associated with other neurologic and non-neurologic manifestations, respectively. Although widely reported in other populations, very little data exist in sub-Saharan Africa.
Methods
Patients with neurodegenerative features were evaluated over a 19-month period at the Department of Neurology, Teaching Hospital of Point “G”, Bamako, Mali. The diagnosis of HSP was considered based on family history and the absence of other known non-genetic causes. Genetic analysis including candidate gene and whole exome sequencing was performed and variant pathogenicity was tested using prediction tools and ACMG guidelines.
Results
Of the 170 families with hereditary neurological disorders enrolled, 16 had features consistent with HSP, a frequency of 9%. The average age of onset was 14.7 years with 46% starting before age 6. The male/female ratio was 2.6:1. Complex forms were seen in 75% of cases, and pure forms in 25%. Pyramidal findings were present in all patients. Associated features included mental retardation, peripheral neuropathy, epilepsy, oculomotor impairment and urinary urgency. Most patients were treated with a muscle relaxant and physical therapy, and restorative surgery was done in one. Genetic testing identified novel variants in three families (19%).
Conclusion
This study confirms the clinical variability of HSPs and adds African data to the current literature.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hereditary spastic paraplegia (HSP) is a neurodegenerative disorder with extensive clinical and genetic heterogeneity [1, 2]. It is caused by degeneration of the corticospinal tracts and sometimes the posterior columns of the spinal cord [3]. It manifests clinically by spasticity and progressive weakness of the lower limbs [1, 4]. The prevalence is variable and depends on the mode of inheritance and the geographic area. However, the pooled global average has been estimated to be 1.8 per 100,000 [5]. Population-based studies found a prevalence of 7.4 per 100,000 in southeast Norway [6] and 1.4 per 100,000 in Portugal [7] while the prevalence in the district of Sfax in southern Tunisia was found to be at least ~ 5.7 per 100,000 [8]. Clinically, HSPs present in two types: pure forms that are limited to spasticity in the lower limbs and complex forms when other neurological or extra-neurological manifestations are associated [3]. All modes of inheritance have been described [9, 10]; however, recessive forms are predominant in North Africa and the Mediterranean region whereas dominant HSPs are more common in North American and northern European populations [1, 10]. Cases have been most reported in North Africa (Morocco, Algeria, Tunisia, Egypt and Soudan) [11,12,13,14,15] but reports in the sub-Saharan African population are rare. In Mali, before this study, only one genetically confirmed family was reported [16]. We report here the clinical features of a cohort of 25 patients from 16 families with different types of HSP, for some of which the genetic diagnosis was available [17,18,19].
Methods
The study was approved by the Ethics Committees of Faculty of Medicine and Dentistry (Faculté de Médecine et d’Odonto-Stomatologie, Bamako, Mali). This was a longitudinal and descriptive study that took place over a period of 19 months (March 2014–September 2015) in the Department of Neurology of the Teaching Hospital of Point “G”, Bamako, Mali. Patients were recruited among our neurogenetics clinic cohort or were referred by neurologists working in other hospitals. All patients, available relatives, and guardians of minors and mentally disabled gave written consent before participating in this study. Patients with progressive spastic paraplegia and a family history and sporadic cases in which no other cause was found, were included in this study. All available unaffected family members were enrolled as controls for disease segregation. Patients were classified based on previously diagnostic criteria [4, 20, 21] as follow: (1) pure form spastic paraplegia, (2) spastic tetraparesis with earlier and greater severity in the lower limbs, or (3) spastic paraplegia as an early prominent sign (within the first 3 years of the disease) of a degenerative disease affecting several parts of the nervous system and (4) other causes of the presenting symptoms excluded. Pure HSP was determined with the presence in lower limbs of hyperreflexia, clonus, and plantar extensor response with or without weakness and urinary incontinence. Complex HSP was diagnosed when additional neurological (peripheral neuropathies, cerebellar ataxia features, cognitive decline, seizures, extra-pyramidal signs) or non-neurological (visual and hearing impairment) symptoms were involved. Socio-demographic data (gender, age at examination and at onset, ethnicity, region or country of origin, and profession), family history and disease features (walking problems, spasticity in lower limbs, progressive weakness, feet deformities, bladder urgency etc.) were collected from all participants. A pedigree was drawn for each family to explore the patterns of inheritance. Dominant (AD, X-linked) inheritance was determined when HSP is reported in more than one generation (the affected individual has one of the parents symptomatic). Families where the HSP trait skips a generation with several affected in one generation and where both parents were asymptomatic were determined as recessive (AR, X-linked). Single cases, with no family history with no other cause found were classified as sporadic HSP.
An extensive neurological examination was done to confirm the spastic paraplegia phenotype, and based on the clinical diagnosis, laboratory testing was done. Human T-cell leukemia virus type 1 (HTLV-1) and HIV serologies, vitamin B-12 levels, brain and spinal cord imaging including magnetic resonance imaging (MRI), CT-scan and X-rays were done where possible to consolidate the diagnosis or to exclude non-genetic causes. The HTLV-1 testing was done in a private laboratory in France for serology and at the National Institutes of Health for the PCR. In some cases, specific neurological evaluations such as electroencephalography (EEG) and nerve conduction studies (NCS) were done to confirm epilepsy when patients presented an active seizures and peripheral nerve involvement when abnormal distal motor and sensory examination was found, respectively. For further phenotypic characterization, other organ systems were assessed by specialists including ophthalmologists for patients with uncoordinated eyes movement or visual impairment, otorhinolaryngologists when hearing impairment or problems with swallowing were seen, and cardiologists for suspected heart abnormalities. Patients with a family history of another neurological or non-neurological disease, brain and spinal cord tumors and injuries, infectious and inflammatory damage in central nervous system and lower limb dysfunction due to other diseases were excluded. The selection process is summarized in Fig. 1.

Summary of HSPs families selection process filtering. Diagram of the selection process of the 16 HSP families (Green shade) (Color figure online)
DNA was extracted from peripheral blood in all available family members for genetic analysis. Candidate genes including ALS2, ERLIN2 were sequenced in our laboratories and spastic paraplegia Next-Generation DNA sequencing gene panel (58 genes, SPG4 deletion, and mtDNA) was ordered from a CLIA-certified laboratory (Medical Neurogenetics, Atlanta, GA) in 10 index patients. The Illumina platform Hiseq 1500 was used with Agilent SureSelect capture reagent with an estimated 99.9% coverage of the regions of interest. Variants identified were classified according to American College of Medical Genetics and Genomics (ACMG) recommendations [22]. DNA from available family members was sequenced for segregation analysis. Whole exome sequencing (WES) was done in families negative for targeted genetic testing and homozygous mapping was completed in selected families with suspected autosomal recessive inheritance patterns. In a suspected familial ALS, C9ORF72 repeat expansion testing was performed through GeneDx (Gaithersburg, MD).
Symptomatic treatment was offered to all patients. This included centrally acting muscle relaxants, physical therapy, and restorative surgery as needed. A 6-month follow-up was done to assess the disease progression using some items from the previously described Spastic Paraplegia Rating Scale (SPRS), including spasticity, weakness, contractures, bladder urgency and other symptoms [22].
Results
Socio-demographic profile of patients
Over the period of study, 170 families with suspected hereditary conditions were enrolled in our research protocol. A total of 21 families were seen in clinic for progressive spastic paraplegia features. Sixteen families met the enrollment criteria for HSP, making HSPs the third most common hereditary neurological disorder in our cohort with a frequency of 9%. All participants were from Mali, except one Fulani family originating from Guinea, a neighboring country with similar ethnic distribution. Male patients were predominant with a sex ratio of 2.6:1. The mean age at first visit was 25.8 years, ranging from 1 to 68 years, and two equal peaks were seen in the age ranges (1–10 and 11–20 years) with 6 cases each. Over eight Malian ethnic groups were represented in this study. More than a third of the families were from the Bambara ethnic group (38%). The other ethnic groups with more than one family were Fulani, Soninkés and Sénoufo, which were represented by two families each. Table 1 summarizes the patients’ epidemiological data.
Clinical findings
Of the 21 families seen for suspected HSP, five families were excluded based on laboratory testing detailed below. The remaining 16 families totaling 25 patients met the enrollment criteria for HSP. The mean age of onset was 14.7 years ranging from 6 months to 52 years, and the disease started before 6 years old in 10 patients (42%). Figure 2 represents the age of onset.

Patients age at onset of the disease (year). Distribution of patients according to the age at symptom onset are indicated by symbols for each group
The clinical presentations varied widely within some families, with index patients having severe motor impairment while some older affected siblings reported no symptoms before examination. All index patients had walking difficulty, ranging from stiffness in gait to inability to walk. One participant had no medical complaints was found to be affected during neurological examination when he was being enrolled as a control for his son’s disease investigation. Similarly, affected relatives were reported in seven families. A recessive pattern of inheritance was suggested in eight families in which consanguinity was reported (50%) and dominant inheritance in three families (19%). Figure 3A, B shows pedigrees for two families in this study: Family #1 shows consanguinity between parents with probable autosomal recessive inheritance pattern and Family #2 shows dominant inheritance.

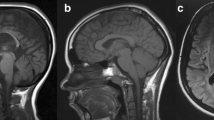
Phenotypic features of some families/patients. Pedigree of a family showing A consanguinity between unaffected parents with autosomal recessive (Family#1) and B dominant or maternal inheritance pattern (Family#2). The arrows identify the probands. Ages at examination are shown at the top of each symbol, and asterisks (*) identify those seen in clinic. Photographs of skeletal deformities from two patients. Patient#1 (7-year-old girl, V.4 from family #1) C equinus foot deformities and D X-ray imaging showing scoliosis of the spine (green bracket). Patient#2 (9-year-old boy) E foot deformities (“club foot”) before Achille’s tendon lengthening surgery, and Patient#3 (25-year-old female) F brain MRI showing thin corpus callosum (white arrow) and cerebellar atrophy (orange arrow) (Color figure online)
In 31% of cases, the disease pattern was considered sporadic because there was no known consanguinity and no other similar cases in the family.
Neurological examination showed pure spastic paraplegia in four patients from four families who presented with a pyramidal syndrome consisting of spasticity, brisk reflexes, and plantar extension response, and 21 patients from 12 families had additional neurological and non-neurological manifestations consistent with complex spastic paraplegia. A mild to moderate motor deficit was found in 13 of our patients (52%). Four patients from two families reported bladder urgency. Skeletal deformities including pes cavus, scoliosis, and wrist, elbow and knee contractures were seen in 13 patients (52%) from seven families. Ten of these patients with skeletal deformities had early age onset of disease (< 6 years). Images of foot deformities and curvature of the spine (scoliosis) from two patients are shown in Fig. 3C, D from patient#1 and patient#2 in Fig. 3E.
Cognitive impairment was first common neurological phenotype present in eight patients (32%) from four families. Five of these patients lost their speech progressively, and three presented later with psychiatric symptoms including aggressiveness, delirium, and confabulation. Brain MRI showed thinning of the corpus callosum and cerebellar atrophy in three of the six patients who had this test. Figure 3F shows an MRI scan from one these patients. The same pattern was previously reported in two of our patients [13, 14]. Peripheral neuropathy was the second commonly associated finding, seen in six patients (24%) from five families. Nerve conduction studies confirmed peripheral nerve involvement in four patients from three families consistent with demyelinating and axonal sensory motor polyneuropathy. Bulbar symptoms including problems with choking on food or liquid, swallowing, and phonation were seen in four patients (16%) from two families. These features were often associated with peripheral neuropathy as was bilateral sensorineural hearing impairment found in four of them. The fourth commonly associated finding was cerebellar ataxia, which was seen in three patients (12%) from three families. These patients presented with tremor, ataxic gait, and loss of coordination. Generalized tonic–clonic epilepsy was seen in three patients (12%) from two families. EEG confirmed active seizures in two and showed slow waves in the third. One patient had extra-pyramidal features (rigidity in the arms). Ophthalmological examination was otherwise normal in the proband from a family with oculomotor impairment (strabismus). The clinical findings are summarized in Table 2.
Laboratory findings
Vitamin B12 levels were normal in all 16 probands from each family. HTLV-1 testing was performed in all patients and was positive in three adult patients. These results were confirmed by PCR that was highly suggestive of HTLV-1 infection. Only one mother was available for testing and was also positive with lower titer, despite being unaffected. HIV testing was negative where done. Spinal CT-scan showed a tumor in one patient and evidence of traumatic injury in another with a history of failing from a tree on his neck. Although these five patients presented with progressive spastic paraparesis, they were excluded from our study.
Genetic findings
HSP gene panel testing was done in ten index patients and showed pathogenic variants in three families. Two of these variants were novel and were located in KIF5A causing SPG10 (a heterozygous variant c.1086G > C, p.Lys362Asn), and the spatacsin gene causing SPG11 (a homozygous variant c.4636-297_4743 + 304del), and one was previously reported FA2H causing SPG35 (a homozygous variant c.786 + 1G > A) [17,18,19]. All the three variants segregated with the disease within the families. In addition, C9ORF72 repeat expansion testing was negative in one patient with suspected familial ALS. Further genetic testing including whole exome sequencing (WES) is ongoing for all the remaining families.
In annex socio-demographic, clinical and genetic data details of the families and patients enrolled in this study in Table 3.
Therapeutic approaches and evolution
Ten ambulant adults and children age of 12 years or older patients were offered a muscle relaxant (baclofen) to reduce muscle spasms, with a low starting dose of 5 mg (mg) three times a day to minimize side effects. Then, the daily dosage was progressively increased until the desired response was reached, however, a maximum daily dosage of 80 mg (20 mg, four times a day) was not exceeded for adults and 40–60 mg for children older than 12 years old. Physical therapy (PT) was done three times a week in nine of 25 patients to improve their motility. Ambulatory patients reported an improvement in stiffness and walking, and those who were wheelchair-bound had a reduction in joint contractures and improved mobility. However, no major improvement was objectively observed during the follow-up check-ups. One patient had Achille’s tendon elongation, which has resulted in nearly normal walking. The disease progression was slow in all patients. However, spasticity and involuntary spasms persisted in all the patients despite the treatment, and a few months later the patient who underwent surgery had decreased ambulation with or without ankle–foot orthoses. Eleven ambulatory patients presented slight weakness and muscle atrophies in limbs after a year of follow-up. Overall, patients’ symptoms were gradually getting worse overtime and bedsores were seen in eight out of 13 patients who never walked or lost mobility. The four patients who had urinary incontinence worsened. Seizures episodes became more frequent in one patient, from about one every six months to one a month and the two other patients with active seizures developed dependance in anti-epileptic drugs. Two patients seen in our clinic died, a female from unknown cause and a male from acute nephrotic syndrome.
Discussion
HSPs are heterogeneous neurodegenerative disorders that have been clinically well characterized in other populations, including Europe, Asia, and North America [1, 10, 23]. Cases have also been reported in North Africa [11,12,13,14,15]. Although reports in sub-Saharan Africa are rare, novel clinical and genetic entities have been identified, particularly in West Africa [16]. The rarity of reported HSP cases in this region may be due to the predominance of other causes including infections and traumatic spine injury that overshadow genetic causes and the limited resources that prevent a thorough evaluation of suspected HSP cases. In many sub-Saharan African countries, there is limited access to MRI and CT-scans. For example, during our study, only one MRI and eight CT-scan machines were available, and all were in the capital city. In addition, testing for other causes of spasticity and rigidity such as HTLV-1 and Wilson’s disease are not available locally. The HTLV-1 testing reported here was done in a private laboratory in France for serology and at the National Institutes of Health for the PCR. Lastly, the expertise and infrastructure to establish the molecular basis of these diseases is scarce. Few laboratories in North and South Africa offer genetic testing, and generally on a research basis, limiting the number of patients who have access to it. In additional to these difficulties, access for patients from rural zone to the capital city where is located the study site was another challenge particularly for large family with several affected individuals. Exclusion of common or non-genetic causes of spastic paraplegia is critical to consolidate HSP clinical diagnosis. Availability of more powerful IRM and CT-scans with higher tesla and equipped laboratories may help to rule out acquired causes of spastic paraplegia. Hence, implementation of genetic testing facilities in this region of the world could enhance the diagnosis of genetic diseases in general and enrich the literature data. These challenges are common to almost all African countries. Therefore, there is no reliable national epidemiological data on spastic paraplegia in general and with genetic causes in sub-Saharan Africa, in particular. In this study, the frequency of HSP is estimated to be about 9% among hereditary neurological disorders (HND) seen in our neurogenetics clinic, and 0.3% all pooled neurological diseases seen during the time of the study in the Neurology Department of the Teaching Hospital of Point “G”, Bamako, Mali. This is like previous reports, where HSP represented the third most common HND worldwide [24]. Although autosomal dominant cases are more common elsewhere [6], a recessive inheritance pattern was predominant in our study. This is likely due to the high consanguinity rate in Mali [25, 26], which has been shown to increase the prevalence of recessive diseases in other populations [8].
In HSPs, symptoms can begin at any age, from early childhood up to the fourth decade [27]. The mean age at disease onset and at first visit in our cohort was 14.7 years and 25.8 years, respectively. These ages are lower than reported elsewhere [6, 12, 28]. These differences are likely due to the high prevalence of recessive cases (50%) that tend start earlier than dominant cases. However, the mode of inheritances is based only on family history, which could be biased. The longer time between disease onset and the first visit is likely due to the fact that most families are seen first by a traditional healers known to be more accessible and unexpensive. Secondary, the delay may be related to the lack of specialists in most of the country. In fact, 95% of the neurologists in Mali are operating in the capital city. Generalists are more prone to link these diseases to more common causes such as cerebral palsy and spine injuries. Sporadic cases are common in clinical practice [28, 29], and represented 31% in this study. However, this proportion may decrease as we establish the molecular basis of the disease in these families and additional family members are diagnosed.
Clinically, spasticity and brisk reflexes were present in all our patients. These features were shown to be present in > 95% HSPs patients in the literature [9]. We report a predominance of complex forms, which were found in 21 patients (84%). This is because complex forms are more common in autosomal recessive cases [1], which were more prevalent in our cohort. The clinical variability reported in the literature [8] was found in this study. In fact, in one family (Family #1 in Fig. 3A) with four affected siblings, both pure and complex clinical forms were seen. In six patients, peripheral neuropathy was part of the clinical presentation. Peripheral neuropathy is one of the most common associated findings in HSPs. Although the neuropathy was variable in six cases, three of them from three different families had lower motor neuron involvement. In four patients, bulbar symptoms were often associated with peripheral neuropathy including problems with choking on food or liquid, swallowing, and phonation.
An overlap of HSPs and Charcot-Marie-Tooth disease symptoms is common, and in some cases, only genetic testing may differentiate these diseases. A family (Family #2 in Fig. 3B) with dominant HSP features also had peripheral neuropathy and bulbar symptoms. SPG panel and C9ORF72 repeat expansion testing was negative. Although the clinical presentation could suggest another form of familial ALS, the disease progression was slower in this family than is usually reported in ALS. These two families (#1 and #2) highlight the intra- and inter-familial clinical variability reported in the literature.
The majority of the families were from Bambara ethnic group, which could be explain by the fact, Bambara is the largest ethnic group in Mali about 33,3% of the Malian population followed by Fulani and Sarakolé according (CIA, The World Factbook, April 2022, https://www.cia.gov/the-world-factbook/countries/mali/).
Mental retardation was another most common associated symptom in these families. This symptom is primarily reported in complex cases [7], which represent the majority in our cohort. Four patients from two families (16%) presented with bladder disturbance, which has been reported in 40% of cases in the literature [30]. The relative rarity of this symptom in our study can be explained by the young age of the patients and cultural factors that could limit its reporting. The cerebellar involvement seen in some patients could suggest spinocerebellar ataxia, however, spastic paraplegia dominated the clinical picture. This finding has been reported in several types of HSPs including SPG11, which was present in one of the families studied here [18]. Bilateral sensorineural hearing impairment was found in four patients (16%) from three families. There is a possibility that this was caused by environment factors such as infection or drug toxicity (chloroquine), which are frequent in Mali [31]. However, this phenotype has also been reported in previous studies of HSPs [32, 33]. Other findings including cognitive decline and epilepsy were also present in our cohort. All three patients who presented with seizures or abnormal EEG had cognitive impairment with varying degrees. The two patients with active epilepsy had worse cognitive decline compared to the one who presented slow background at EEG. These features have been reported previously in HSP studies [34, 35].
SPG gene panel testing identified a variant in only 30%. This could be due the previously reported genetic diversity in Africa. The African population may harbor genetic variants that are not present in these panels. In keeping with this, C19orf12, a gene found to mutated in SPG43, which was first identified in a Malian family [16], is not included in the gene panel used for this study. Therefore, the use of such panels in the African population may not be the best approach but a necessary as cost and accessibility are more important factors than the coverage of mutations in this population, and until African data are inserted in those panels. All variants reported in this study are classified as pathogenic according (ACMG) and were not present in SNP databases. Taken all together, using the WES or WGS approach should be considered for HSP diagnosis, especially for genetically unexplored population.
Many of the patients were not ambulatory. Some never walked, and others lost motility at an earlier age than reported elsewhere. This may be due to the underlying disease progression. However, the relative lack of appropriate treatment and follow-up could also explain this rapid decline. Although HSP is untreatable, symptomatic treatment and restorative surgery can improve patient function [36].
Conclusion
This study helps to establish the epidemiological and clinical spectrum of HSPs in a resource-limited setting. It has delineated the clinical heterogeneity of HSPs in Mali and confirmed the intrafamilial and inter-familial variability and the high frequency of complex forms. Larger cohort studies in neighboring sub-Saharan countries could help to confirm and extend our findings. With molecular diagnosis becoming less expensive and more accessible, there is a potential to identify novel genetic entities that may then be studied in other populations.
Data availability
Data are available upon request from the corresponding author.
Abbreviations
- HSP:
-
Hereditary spastic paraplegia
- HND:
-
Hereditary neurological disorders
- HTLV-1:
-
Human T-cell leukemia virus type 1
- HIV:
-
Human immunodeficiency virus
- EEG:
-
Electroencephalography
- NCS:
-
Nerve conduction studies
- PCR:
-
Polymerase chain reaction
- MRI:
-
Magnetic resonance imaging
- CT-scans:
-
Computed tomography scans
- AD:
-
Autosomal dominant
- AR:
-
Autosomal recessive
- ALS:
-
Amyotrophic lateral sclerosis
- ACMG:
-
American College of Medical Genetics and Genomics
- WES:
-
Whole Exome Sequencing
- SPRS:
-
Spastic Paraplegia Rating Scale
References
Blackstone C (2018) Hereditary spastic paraplegia. Handb Clin Neurol 148:633–652. https://doi.org/10.1016/B978-0-444-64076-5.00041-7
Finsterer J, Löscher W, Quasthoff S, Wanschitz J, Auer-Grumbach M, Stevan G (2012) Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci 318(1–2):1–18. https://doi.org/10.1016/j.jns.2012.03.025
Fink JK (2013) Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol 126(3):307–328. https://doi.org/10.1007/s00401-013-1115
Harding AE (1983) Classification of the hereditary ataxias and paraplegias. Lancet 1983:1151–1155. https://doi.org/10.1016/S0140-6736(83)92879-9
Ruano L, Melo C, Silva MC, Coutinho P (2014) The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 42:174–183. https://doi.org/10.1159/0003588101
Erichsen AK, Koht J, Stray-Pedersen A, Abdelnoor M, Tallaksen CME (2009) Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: a population-based study. Brain 132(6):1577–1588. https://doi.org/10.1093/brain/awp056
Coutinho P, Ruano L, Loureiro JL, Cruz VT, Barros J et al (2013) Hereditary ataxia and spastic paraplegia in Portugal: a population-based prevalence study. JAMA Neurol 70(6):746–755. https://doi.org/10.1001/jamaneurol.2013.1707
Boukhris A, Stevanin G, Feki I et al (2009) Tunisian hereditary spastic paraplegias: clinical variability supported by genetic heterogeneity. Clin Genet 75(6):527–536. https://doi.org/10.1111/j.1399-0004.2009.01176.x
Sireesha M, Elanagan N, Pradeep CB (2021) Hereditary spastic paraplegia. J Neurol Sci 42:883–894. https://doi.org/10.1007/s10072-020-04981-7
Bouty M, Morais Stevanin G (2019) Update on the genetics of spastic paraplegias. Curr Neurol Neurosci Rep 19(4):18. https://doi.org/10.1007/s11910-019-0930-2
Mahungu CA, Monnakgotla N, Nel M, Heckmann MJ (2022) A review of genetic spectrum of hereditary spastic paraplegias, inherited neuropathies and spinal muscular atrophies in Africans. Orphanet J Rare Dis 17:133. https://doi.org/10.1186/s13023-022-02280-2
Coutinho P, Barros J, Zemmouri R et al (1999) Clinical heterogeneity of autosomal recessive spastic paraplegias: analysis of 106 patients in 46 families. Arch Neurol 56(8):943–949. https://doi.org/10.1001/archneur.56.8.943
Lesca G, Eymard-Pierre E, Santorelli FM et al (2003) Infantile ascending hereditary spastic paralysis (IAHSP): clinical features in 11 families. Neurology 60(4):674–682. https://doi.org/10.1212/01.WNL.0000048207.2879.25
Boukhris A, Stevanin G, Feki I et al (2008) Hereditary spastic paraplegia with mental impairment and thin corpus callosum in Tunisia. Arch Neurol 65(3):393–402. https://doi.org/10.1001/archneur.65.3.393
Bouslam N, Benomar A, Azzedine H et al (2005) Mapping of new form of pure autosomal recessive spastic paraplegia (SPG28). Ann Neurol 57(4):567–571. https://doi.org/10.1002/ana.20416
Landouré G, Zhu P-P, Lourenco CM et al (2013) Hereditary spastic paraplegia type 43 (SPG43) is caused by mutation in C19orf12. Hum Mutat 34(10):1357–1360. https://doi.org/10.1002/humu.22378
Guinto CO, Diarra S, Diallo S et al (2017) A novel mutation in KIF5A in a Malian family with spastic paraplegia and sensory loss. Ann Clin Transl Neurol 4(4):272–275. https://doi.org/10.1002/acn3.402
Landouré G, Dembélé K, Cissé L et al (2019) Hereditary spastic paraplegia type 35 in a patient from Mali. Am J Med Genet A 179(7):1122–1125. https://doi.org/10.1002/ajmg.a.6117
Landouré G, Dembélé K, Diarra S, Cissé L, Samassékou O, Yalcouye A, Traoré M, Fischbeck HK, Guinto OC, The H3Africa Consortium (2020) A novel variant in the spatacsin gene causing SPG11 in a Malian family. J Neurol Scienc 411:116675. https://doi.org/10.1016/j.jns.2020.116675
Schüle R, Holland-Letz T, Klimpe S, Kassubek J, Klopstock T, Mall V et al (2006) The spastic paraplegia rating scale (SPRS): a reliable and valid measure of disease severity. Neurology 67:430–434. https://doi.org/10.1212/01.wnl.0000228242.53336.90
Do GJ, Kim JB, Kim N, Sung HD (2022) Hereditary spastic paraplegia in Koreans: Clinical characteristics and factors influencing the disease severity. J Clin Neur 18(3):343–350. https://doi.org/10.3988/jcn.2022.18.3.343
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Ortega RPM, Rosemberg S (2019) Hereditary spastic paraplegia: A clinical and epidemiological study of a Brazilian pediatric population. Arq Neuropsiquitar 77(1):10–18. https://doi.org/10.1590/0004-282X20180153
Meilleur KG, Coulibaly S, Traoré M et al (2012) Genetic testing and counseling for hereditary neurological diseases in Mali. J Community Genet 2(1):33–42. https://doi.org/10.1007/s12687-011-0038-0
Sangaré M, Hendrickson B, Sango HA et al (2014) Genetics of low spinal muscular atrophy carrier frequency in sub-Saharan Africa. Ann Neurol 75(4):525–532. https://doi.org/10.1002/ana.2414
Landouré G, Cissé L, Touré BA et al (2017) Neurological complications in subjects with sickle cell disease or trait: Genetic results from Mali. Glob Heart 12(2):77–80. https://doi.org/10.1016/j.gheart.2017.01.014
Fink JK, Hedera P (1999) Hereditary spastic paraplegia: genetic heterogeneity and genotype-phenotype correlation. Semin Neurol 19:301–309. https://doi.org/10.1055/s-2008-1040846
Racis L, Tessa A, Di Fabio R, Storti E, Agnetti V, Casali C et al (2014) The high prevalence of hereditary spastic paraplegia in Sardinia, insular Italy. J Neurol 261(1):52–59. https://doi.org/10.1007/s00415-013-7151-4
Faber I, Servelhere KR, Martinez AR, D’abreu A, Lopes-Cendes I, França MC Jr (2014) Clinical features and management of hereditary spastic paraplegia. Arq Neuropsiquiatr 72(3):219–226. https://doi.org/10.1590/0004-282X2013248
Schneider SA, Beckinger VE, Möller B, Knüpfer S, Haman M, Deuschl G (2019) Urinary symptoms, quality of life, and patient satisfaction in genetic and sporadic hereditary spastic paraplegia. J Neurol 266(1):207–211. https://doi.org/10.1007/s00415-018-9129-8
Sako AB (1990) Particularities of hearing disorders in inhabitants of Mali. Vestn Otorinolaringol 1:18–20 (PMID:2316107)
Donkervoort S, Bhaurucha-Goebel D, Yun P et al (2017) HSP and deafness neurocristopathy caused by a novel mosaic SOX10 mutation. Neurol Genet 3(3):e151. https://doi.org/10.1212/NXG.0000000000000151
Xiao XW, Du J, Jiao B et al (2019) Novel ATL1 mutation in a Chinese family with hereditary spastic paraplegia: a case report and review of literature. World J Clin Cases 7(11):1358–1366
Helbig KL, Hedrich UBS, Shinde DN et al (2016) A recurrent mutation in KCNA2 as a novel cause of hereditary spastic paraplegia and ataxia. Ann Neurol 80(4):479–637. https://doi.org/10.1002/ana.24762
Hardies K, May P, Djémié T et al (2015) Recessive loss-of-function mutations in AP4S1 cause mild fever-sensitive seizures, developmental delay and spastic paraplegia through loss of AP-4 complex assembly. Hum Mol Genet 24(8):2218–2227. https://doi.org/10.1093/hmg/ddu740
Bellofatto M, De Michele G, Iovino A, Filla A, Santorelli FM (2019) Management of hereditary spastic paraplegia: A systematic review of the literature. Front Neurol 10:3. https://doi.org/10.3389/fneur.2019.00003
Acknowledgements
The authors thank patients and families for their participation in this study.
Funding
This work was supported by grant number U01HG007044 funded by the National Institute of Neurological Disorders and Stroke (NINDS) and administered by the National Human Genome Research Institute under the H3Afria initiative, NINDS intramural research funds, and the Centre Hospitalier Universitaire du Point “G”, Bamako, Mali.
Author information
Authors and Affiliations
Consortia
Contributions
SD and GL: drafted the document; TC, SD, SHD, SD, KB, DC, KD, MO, OC, AS, KHF, GL, and COG: examined patients; SD, TC, SHD, SD, KB, DC, KD, MO, AS, HAS, MT, COG, GL, and KHF: collected and analyzed the data. All authors read, edited and approved the final version of the article.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest.
Ethical approval
The ethical committee of the University of Science, Technique and Technology of Bamako (USTTB) approved this study, and it was conducted according to the principles of the declaration of Helsinki. Patients gave their written consent to publish their data and pictures.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Diarra, S., Coulibaly, T., Dembélé, K. et al. Hereditary spastic paraplegia in Mali: epidemiological and clinical features. Acta Neurol Belg 123, 2155–2165 (2023). https://doi.org/10.1007/s13760-022-02113-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13760-022-02113-w