
Abstract
The research involves synthesizing 1:1 salts of Pipemedic acid (PMA) with oxalic acid (OA), salicylic acid (SA), and p-Toluene sulfonic monohydrate (BS) using a slow evaporation method. Many characterization techniques, including FT-IR, DSC, Single XRD, and DFT calculations, were employed to analyze the salts’ structural and physicochemical properties. The proton is transferred from oxalic, salicylic acid, and p-toluene sulfonic monohydrate to pyridine nitrogen of PMA. The salt 1OA, crystallizes in the monoclinic space group P 21/c, with a = 9.923(3) Å a = 90°, b = 9.443(3) Å b = 92.470(10)°and c = 18.248(5) Å g = 90° and volume = 1708.3(9) Å3and Z = 4. The salt 2SA crystallizes in the monoclinic space group P 21/c, with a = 6.8877(3) Å a = 90°.b = 13.9149(6) Å b = 98.092(2)° and c = 21.5313(10) Å g = 90° with volume = 2043.05(16) Å3 and Z = 4.The salt 3BS crystallizes in the monoclinic space group P 21/c, with, a = 9.3352(4) Å a = 90°, b = 12.7754(5) Å b = 97.722(2)°, c = 19.5462(8) Å g = 90°,with volume = 2309.96(16) Å3 and Z = 4. Supramolecular centrosymmetric ring motifs are formed by N–H···O hydrogen bonds between protonated nitrogen of the pyridone ring and the carboxylic O atom of the oxalate ion, in both 1OA and 2SA. The dihedral angles of 1OA, 2SA, and 3BS are found to be 43.63°, 88.19°, and 53.89° respectively. The Hirshfeld surfaces and the related 2D fingerprint plots were explored which uncovered that more than two-thirds of close contacts were related to H⋯H, C–H, N–H, and C–C bonding interactions whereas in 3BS, the structure is stabilized by N–H···O and N–H···S hydrogen bonding interactions. These weak associations assume a significant role in crystal packing as revealed by the Hirshfeld surfaces and the related 2D fingerprint plots.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cocrystallization has emerged as a promising strategy [1] in pharmaceutical research for the enhancement of active pharmaceutical ingredients (APIs) through the formation of unique solid-state structures [2]. By combining an API with a suitable coformer, cocrystals can be generated [3]. By selecting coformers with complementary characteristics, researchers can modify the crystal lattice and influence the physicochemical properties of the resulting cocrystal [4]. This offers a promising solution to enhance drug solubility [5], and bioavailability [6], and optimize drug delivery systems [7]. It has the potential to revolutionize pharmaceutical development [8] by addressing critical challenges and improving patient outcomes [9]. Various strategies, such as crystal engineering [10], solvent evaporation, grinding [11], and co-crystallization in solution, have been employed to facilitate the formation of cocrystals [12]. The resulting cocrystals can be characterized using techniques like X-ray diffraction, thermal analysis, spectroscopy, microscopy, and computational modeling, which provide valuable insights into their crystal structure, stability, and behavior [13].
The formation of cocrystals involves the non-covalent bonding interactions between the API and the coformer, resulting in the formation of a new crystalline lattice with distinct properties [14]. These interactions can include hydrogen bonding, π-π stacking, halogen bonding, and other weak interactions [15], which play a crucial role in determining the structure and properties of the cocrystal [16]. Cocrystals can exhibit altered solubility, dissolution rate [17], melting point, stability, and mechanical properties compared to pure API, [18] offering numerous advantages for pharmaceutical development.
In this study, our focus lies on the utilization of Pipemidic acid (PMA) as the API for cocrystal synthesis. Pipemidic acid (PMA) is a promising active pharmaceutical ingredient (API) that belongs to the class of antibacterial agents known as fluoroquinolones [19]. It exhibits broad-spectrum activity against various bacterial strains, making it an effective treatment option for a range of infections [20].PMA exerts its antimicrobial action by inhibiting the bacterial DNA gyrase and topoisomerase IV enzymes, crucial for DNA replication and repair [21]. This mechanism of action helps to halt the growth and proliferation of bacteria, aiding in the resolution of infections [22].
Although PMA demonstrates potent antibacterial activity, it faces challenges related to its solubility and bioavailability [23]. These limitations can hinder its optimal therapeutic use, leading to suboptimal drug delivery and efficacy [24]. By forming cocrystals through non-covalent interactions with suitable coformers, it becomes possible to modify the drug’s characteristics, including its solubility, dissolution rate, and stability [25]. By carefully selecting and designing coformers with complementary functional groups and physicochemical characteristics, we strive to achieve cocrystals with optimized properties [26].
The novelty in my research work lies in the preparation of three distinct salts derived from pipimedic acid Three unique salts 1OA,2SA, and 3BS of PMA were successfully synthesized using the cocrystallization approach. These salts have not been previously reported or documented, making them unique contributions to the field. By employing a novel synthetic methodology and utilizing specific reaction conditions, successfully synthesized these salts. The characterization data and physicochemical properties further support the novelty of these salts, offering new opportunities for applications in various fields. Besides, we performed SCXRD, Hirshfeld analysis, and DFT void analysis to characterize these cocrystals. These comprehensive analyses provide valuable insights into the structural properties and intermolecular interactions within the PMA cocrystals.
Experimental
Materials
The initial substances, namely PMA (CAS No. 51940-44-4) with a purity of at least 99%, and the conformers OA, SA, and (BS), along with solvents such as methanol, propanol 2, and acetonitrile, all of which have HPLC grade purity, were procured from Yarrow Chem Products in Mumbai and Lab Reagents and Allied Products in Bengaluru (India), respectively.
Synthesis and growth of molecular salts 1OA, 2SA, and 3BS
A mixture containing PMA (0.050 mg) and OA (0.020 mg) in a 1:1 ratio was dissolved in a 12 mL equimolar combination of methanol and water. The solution was then subjected to slow evaporation at a temperature below 80 °C. After 72 h, small, transparent, cubic crystals with a light blue had formed.
A 1:1 stoichiometric ratio of PMA (0.050 mg) and SA (0.027 mg) was thoroughly ground via liquid-assisted grinding using an equimolar volume of propanol-2 and acetonitrile for 20 min. The mixture was then heated to 50 °C until complete dissolution occurred, and afterward, slow evaporation was carried out at room temperature. Within 24 h, needle-shaped, transparent crystals had formed.
A solution was prepared by dissolving PMA (0.050 mg) and p-Toluene sulfonic monohydrate (0.020 mg) in an equimolar combination of 12 mL ethanol and water in a 1:1 ratio. The solution was then subjected to slow evaporation at a temperature below 70 °C. After 72 h, small, yellow-colored rod-shaped crystals had formed.
Fourier transform-infrared spectroscopy (FT-IR)
Infrared spectra of pure PMA and its binary solids were recorded on FTIR spectrometer (Thermo Nicolet 6700, Thermo Scientific, Waltham, MA) in which the samples were spread in KBr pellet, in the spectral range 4000–400 cm−1. Data were analysed using spectrum software version 2(JASCO, Easton, Maryland, USA).
Differential scanning calorimetry (DSC)
Thermal analysis of PMA and all three of its salts formed was recorded individually on Mettler Toledo model no. DSC00f3MAYA, company: Netzsch Samples were placed in crimped but vented aluminum sample pans with nitrogen purging.
Elemental analysis (EA)
Elemental analyses (C, H, N, and S) were conducted on a LECO Truspec Micro Analyser.
Single crystal X-ray diffraction (SCXRD)
Single X-ray diffraction measurements of the 1:1 salts of PMA with OA, SA, and BS were carried out on a Bruker AXS KAPPA APEX-II. CCD Diffractometer with Mo Kα radiation at 293 K. Structure refinement was done by using the SHELX program [27]. Besides the structures of 1OA, 2SA, and 3BS were drawn by using the Olex2 program [28]. Both primary and secondary atom site locations were found from the structure invariant direct method and difference Fourier map respectively. Hydrogen atom site areas were induced at neighbouring ring sites. The crystal data and data processing are given in Table 1. The final fractional atomic coordinates are listed in Table 2 whereas Table 3 gives information about bond lengths and bond angles. The entire details of structural parameters in the CIF format are accessible from the Cambridge Crystallographic Database Centre under No. CCDC 2114663, CCDC 2114662, and CCDC 2144823 respectively for 1OA, 2SA, and 3BS.
Hirshfeld surface analysis
For all three crystals (1OA,2SA, and 3BS), Hirshfeld surface calculations were performed using CrystalExplorer17. All the surfaces (including 2D fingerprints) were generated at a high standard surface resolution. The crystallographic information file (CIF) of 1OA 2SA and 3BS was read directly into the CrystalExplorer 17 program [29]. The Hirshfeld surface maps are that of normalized contact distance, dnorm, which is defined in terms of the distance to the nearest atoms outside (de), the distance to the nearest atoms inside (di), and the van der Waals radii of the two atoms external and internal to the surface. The dnorm ranges used to map the Hirshfeld surfaces were − 0.6504 to 1.3026 (1OA), − 0.5619 to 1.2844 (2SA), and − 0.5956 to 1.4123 (3BS). Besides, the enrichment ratio [30], ER(X, Y), for a pair of elements (X, Y) is calculated by comparing the actual proportion of contacts observed in the crystal to the theoretical proportion of random contacts. The ER provides information about the tendency of a pair of chemical species to form specific interactions within a crystal (Fig. 1).

DSC thermograms showing melting behaviour of PMA, 1OA, 2A, and 3BS
Theoretical calculations
For theoretical calculations, a crystal unit was selected as the initial structure from crystals 1OA, 2SA, and 3BS. DFT-B3LYP/6-311G + + (d,p) [31] methods in Gaussian 09 [32] were used to optimize the structure of the title compounds at a default temperature of 298.15 K. Vibration analysis showed that the optimized structure represented a minimum on the potential energy surface (no negative eigenvalues).Molecular parameters as estimated from XRD are very much upheld by the DFT computations. Crystal Explorer 17.5 software was used to generate molecular Hirshfeld surfaces and visualize intermolecular interactions in molecular crystals. The program accepts a structured input file in the CIF format [33].
Result and discussion
FT-IR spectral analysis
This is generally used in the characterization of cocrystal interactions and through which we can distinguish the salts from cocrystals. The hydrogen bonding patterns of a molecule will change after the formation of cocrystal which results in the change of vibration modes of functional group and IR frequencies [34].
PMA shows an IR absorption frequency at 1640 cm−1 for CO (of the carboxylic group), CO of (pyridone) ketone at 1616 cm−1, and for OH, the absorption frequency was found to at3383cm−1 as a medium peak [35]. However, we can observe the changes in the CO stretching of (the carboxylic group) from 1640 to 1732 cm−1 which indicates the formation of salt rather than a cocrystal for 1OA, whereas in the case of 2SA the shift is found in the region of 1729 cm−1 as shown in Fig. 2. In the FT-IR spectrum of PMA, the peak at 1627 cm−1 was assigned to the N–H bending vibration of the quinolones. In the spectrum of the 3BS the –NH stretching region of PMA (3366 cm − 1) is shifted at 3446 cm−1.

a Basic unit structure of cocrystal Fig. 1b Hydrogen-bonded tecton in 1OA c 1OA salt molecules are connected through N−H∙∙∙O, O−H∙∙∙O, and C −H∙∙∙O interactions. d crystal packing due to weak intermolecular C−O··· O and C−O···N contacts
Thermal analysis
The specific melting point of the PM, OA, SA, and BS and salt formed were obtained by DSC investigation Table 2. The difference in the melting point of the salt when compared to that of pure PMA indicates interaction between the drug and conformers which proves that first-order transformation that is meting endotherm has disappeared.
Elemental analysis
Single-crystal X-ray analysis
Description of the crystal structure of molecular salt 1OA
The molecular salt crystallizes as light blue color cubic-shaped crystals. The structural determination shows that the PMA forms a 1:1 molecular salt in the monoclinic P 21/c space group with Z = 4 as shown in Fig. 2a.
In 1OA, the OA is connected to PMA through the two intermolecular hydrogen bonds i.e. N(1)-H(1C)…O(6) and N(1)-H(1C)…O(4) resulting in the formation of the R12(5) and R12 (5) ring motifs.The anion forms an S(6) graph-set motif through an O—H…O hydrogen bond (Fig. 2a and b). In the crystal lattice, adjacent anions and cations are interconnected by N—H…O and C—H…O hydrogen bonds, forming a two-dimensional network that extends parallel to the ac plane. This network encompasses ring motifs Fig. 2c, including R2 2(10) [36] and R1 2(5) R12 (5) [37] R22(7) [38]and R22(13) [39] Table 4. The proton transfer of OA on the N1 site has also resulted in the change in the small ∆C–O value. The torsion angles N1–C2–C2–N2 and N1–C4–C3–N2 is 58.54(11)°, and − 51.81(12) ° respectively, which implies that both the amine groups are nearly in the same plane as the benzene ring of the anion. Table1 presents the measured bond distances corresponding to these interactions. The crystallographic program MERCURY 4.2.0 software [40]facilitated the analysis of crystal structure and packing arrangements.
The molecular conformation is stabilized by weak intermolecular C−O··· O and C−O···N contacts C12−O1··· N1 = 2.984 Å, C12−O1··· O6 = 3.038 Å, C15−O4··· N1 = 2.874 Å. Figure 2d.
Description of the crystal structure of molecular salt 2SA
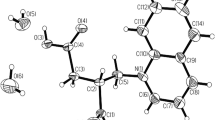
The molecular salt 2SA crystallizes as colorless needle-shaped crystals. The structural determination shows 2 forms of 1:1 2SA molecular salt in the monoclinic P21/n space group with Z = 4, with the asymmetric unit (Fig. 3a).

a Molecular structure of pipimedic acid co-crystals with salicylic acid, showing the atom-labeling scheme and H atoms are shown as small spheres of arbitrary radius; hydrogen bonds are represented by dashed lines. b The crystal packing of (2SA) viewed down the b-axis, showing 1-D extended chains along the c-axis through N—H···O and C—H ∙∙∙O hydrogen bonding. c box-shaped chain through C—H…O hydrogen bond d Molecular packing stabilized by intermolecular interactions that are weaker than hydrogen bond
Hydrogen-bonding interactions, involving N—H…O and C—H…O, hold the molecules of the title compound together in its crystal structure Fig. 3b. These interactions play a vital role in stabilizing the structure, contributing to its overall stability and properties [41].PMA molecules form a box-shaped chain through C—H…O hydrogen bond with the R24(10) [42] ring motif Fig. 3, Table 5 (c). Proton transfer is evidenced by the difference between the C–O bond distances C21–O5 = 0.260(16) Å, and C21–O4 = 1.251(15) Å of the carboxylate group with the ∆DC–O value of 0.009 Å a relatively small value, which is expected for the carboxylate group [13]. The protonation of SA on the N1 site has resulted in, a change in the bond angle. The angle at the unprotonated ring N atom is 118.56(13)° while for protonated ring N atom, the angle (C3–N1–C2) is 111.59(14) The torsion angles O6–C20–C19–C18 and O4–C21–C15–C16 are 178.9(2)°, and − 11.8(4)°, respectively. This shows that both the amine groups are nearly in the same plane as the benzene ring of the anion. Further, the molecular conformation is stabilized by C−O·· N hydrogen bond C21−O5·· N1 = 2.762 Å C21−O4·· N1 = 2.856 Å forming layers parallel to the ab plane Fig. 3d.
Description of the crystal structure of molecular salt 3 (PMA-3BS)
The molecular salt 3BS crystallizes as yellow rod-shaped crystals. The structural determination shows that salt 3, forms a 1:1 (PMA-SA) molecular salt in the monoclinic P21/n space group with Z = 4, with the asymmetric unit. The crystal was stabilized by inter-molecular N–H···O and N–H···S hydrogen bonding interactions (Fig. 4a). There is a complete transfer of a proton from the sulphonic group to the N of the pyrimidine group of PMA, which is evidenced by an increase in the bond length of S(1)–O(4), from 1.451(2) [43] to 1.459(11) and the new bond formed N(5)-H(5B) = 0.888(14). Besides, the torsion angles, C(18)-C(19)-C (20) = 121.8(2) and C(15)-C(16)-C(17) = 119.91(19) of the p-toluene sulfonic monohydrate and the torsion angles, of PMA i.e., N(5)-C(10)-C(9) = 110.72(11) and N(4)-C(11)-C(12) = 109.82(11) shows that they are in the same plane. The crystal is also stabilized by intermolecular interactions viz, N(5)-H(5B)···S1, N(5)-H(5B)···O(4) and intramolecular O(1)-H(1)…O(3). In the crystal structure of 3BS, N5–H5B∙∙∙O4, C5–H5∙∙∙O4 and, C11–H11B∙∙∙O3 bonds generate R23(11) ring motif whereas C11–H11B∙∙∙O1 and C13–H13B∙∙∙O3 bonds forming R23(12) [44] ring motif,Fig. 4b. Table 6 presents the metrics related to hydrogen bonding. The molecules are linked via weak C−H···O and N−H···S hydrogen bonds, forming chains propagating along the c-axis direction Fig. 4c. N5−H5A···O1W = 2.69(2) Å C10−H10B···O5 = 2.519 Å, C12−H12A···O6 = 2.638 Å, C8−H8···O1W = 2.480 Å, N5−H5B···S1 = 2.84(2) Å.

a Intermolecular N–H···S and N–H···O and intramolecular O–H···O– hydrogen bonding patterns in the basic supramolecular packing unit of 3BS. H atoms are shown as small spheres of arbitrary radius; hydrogen bonds are represented by dashed lines. b Crystal packing stabilised by R23(11) and R23(12) ring motifs. c The molecular packing linked via weak C−H···O and N−H·· S hydrogen bonds
The crystallographic analysis of compound 1OA, 2SA and 3BS reveals that the heterocyclic ring adopts a chair conformation, as determined by calculations of torsion angles, asymmetry parameters, and ring puckering parameters. According to the Cremer & Pople and Nardelli methods, the total puckering amplitude, QT, is determined to be 0.5875 (8) Å. Additionally, the phase angle θ is measured to be 0.94 (8)°, and phi is found to be 34 (4)°. Similarly, the torsion angles of N2—C5—N3—C8 in 1OA and 2SAand C11—C5—N3—C8 in 3BS are 178.44, 179.08(8)and − 176.8 (1) respectively. The torsion angle values also clearly confirm the chair conformation.
Salt or cocrystal by ΔpKa
ΔpKa is one of the tools used to determine whether the formed substance is a co-crystal or salt ΔpKa < 0 indicates the formation of a co-crystal whereas, salt is formed if ΔpKa > 3 and ΔpKa value is found to be in between, then it is considered partial proton transfer. The pKa values of PMA, OA, SA, and BS are 8.57, 1.23, 4.19, and − 2.1 respectively. So, the corresponding ΔpKa value of the complex formed is 7.34, 4.38, and 10.67 indicating the formation of salts, not cocrystals (Fig. 5).

dnorm mapped on Hirshfeld surfaces for 1OA, 2SA, and 3BS
Hirshfeld surface analysis
The detailed proximity of intermolecular interactions in crystal packing was studied by Hirshfeld surface analysis. Figure 6 shows the Hirshfeld surface mapped over dnorm based on van der Waals in 1OA, 2SA, and 3BS. Dark red indicates negative norm values indicating contacts shorter than the sum of van der Waals radii. Dark blue indicates positive dnorm values indicating contacts longer than the sum of van der Waals radii while white depicts zero dnorm values (contact distances close to van der Waals).

Fingerprint plot for 1OA, 2SA, and 3BS depicting percentages for O–H, H–H, and C–H contacts in the total Hirshfeld surface area
The fingerprint plots for the O···H, H···H, and C···H interactions are depicted in Fig. 7. The central spike of the fingerprint plot corresponds to the percentage of H···H interactions for 1OA (26.9%), 2SA(40.3%), and 3BS (45.6%). The large spikes show the O···H interactions in the crystals which were 43.4%, 32.1%, and 31.8% for 1OA, 2SA, and 3BS, respectively.

Percentage contribution of each interaction between atoms in 1OA, 2SA, and 3BS
The contribution of all interactions is shown as a histogram in Fig. 8, for crystals (1OA, 2SA, and 3BS). The bond distances were found to be in good agreement with those of crystal data. The three major contributions were found to be from O–H, H–H, and C–H interactions.

Graphical representation of voids
The enrichment ratio, ER(X, Y), for a pair of elements (X, Y) is calculated by comparing the actual proportion of contacts observed in the crystal to the theoretical proportion of random contacts. If the ratio is greater than unity, it indicates that the pair of elements has a higher likelihood of forming contacts in the crystal. Conversely, if the ratio is less than one, it suggests that the pair of elements tend to avoid contact with each other in the crystal [45] Table 7.
Voids within the crystal lattice significantly influence the response of crystals to applied stress or force [46]. The size of these voids serves as an indicator of the strong interatomic packing within the crystal structure, which, in turn, determines the crystal’s ability to withstand substantial amounts of stress [47]. To investigate the presence of voids, we employed the Crystal Explorer software for comprehensive void analysis. In our calculations, we assumed that the electronic density surrounding each atom exhibits spherical symmetry [48]. This investigation reveals that the void volume and surface area in the crystal 1OA is measured to be 155.72 Å3 and 568.13 Å2, respectively. Additionally, the analysis shows that the globularity and asphericity values are determined to be 0.246 and 0.196, respectively. The determined percentage of void space (Fig. 8a) in the crystal packing is 9.11%.
The crystal voids for 2SA (Fig. 8b) have a calculated volume of 82.71 Å3. Additionally, the percentage of free space within the unit cell is determined to be 4.05% with the globularity and asphericity values 0.248 and 0.252 respectively. A similar calculation for crystal voids reveals for 3BS, that it has a void volume estimated at approximately 278.02 Å3 and a corresponding surface area of about 888.62 Å2. The calculated percentage of void space (Fig. 8c) is found to be 12.03%.
This observation uses a 0.002 a.u. isovalue, suggests that the molecules within the crystal are firmly held together by non-covalent interactions, indicating strong intermolecular forces at play [49].
Theoretical calculations
Theoretical calculations were performed using Gaussian09 in the gaseous phase [50]. Geometry optimization was performed using density functional theory (DFT) employing the B3LYP (Becke three parameters Lee–Yang–Parr exchange–correlation functional), which combines the hybrid exchange functional of Becke with the gradient-correlation functional of Lee, Yang, and Parr using 6–311G + + (d,p) basis set was performed in the gas-phase at 298.15 K [51]. Input geometries were taken from X-ray refined data. The optimized geometry results in the free molecule state were found to be consistent with those in the crystalline state (Fig. 9). No negative vibrational modes were obtained. The DFT calculated structure and geometric parameters (bond lengths and bond angles) agreed with each other. Optimized structures of 1OA and 2SA, both had a C1 point group. The presence of intermolecular H-bonding is evident from the proton transfer from oxalic acid to 1 in the case of 1OA and from salicylic acid to 2 in the case of 2SA.

Optimized structure of 1OA, 2SA, and 3BS
Frontier orbital energy calculations
Molecular Total Energy and Frontier Orbital energy levels were calculated using DFT (Fig. 10). The energy gap (ΔE) has been calculated for both crystals as shown in Table 8. The energy gap between HOMO and LUMO was calculated by the B3LYP method using the 6-311G + + (d,p) basis set. The compounds 1OA, 2SA, and 3BS showed an energy gap (ΔE) for HOMO → LUMOofs 4.43, 3.98, and 4.38 eV, respectively (Table 6). HOMO and LUMO are essential factors that affect bioactivity, chemical reactivity and electron affinity, and ionization potential. Thus, a study of frontier orbital energy can provide useful information about the biological and chemical reaction mechanism. The HOMO–LUMO gap in 2SA (3.98 eV) was smaller than 1OA (4.43 eV) and 3BS (4.38 eV), thereby indicating its high reactivity.

HOMO and LUMO molecular orbitals in 1OA, 2SA and 3BS with band gaps
Conclusions
To sum up, we have reported, Crystal growth and its X-ray analysis and Hirshfeld surface analysis of 1OA, 2SA, and 3BS. Proton transfer is evidenced by the change between CO bond length in OA (1.18A0) and PMA (1.60 A0). The ∆C–O was 0.42, with a newly formed bond which is further confirmed by the bathochromic shift in FT-IR analysis. Further, the DSC analysis also provided evidence for the formation of new compounds by showing the change in the melting points from that of initial compounds. The Hirshfield analysis reveals the percentage of contribution of O–H, H–H, and C–H interactions and other interactions in the atoms of 1OA,2SA and 3 BS. We calculated the band structure and energy gap (ΔE) by B3LYP method.
References
S.N. Wong, Y.C.S. Chen, B. Xuan, C.C. Sun, S.F. Chow, Cocrystal engineering of pharmaceutical solids: therapeutic potential and challenges. CrystEngComm 23(40), 7005–7038 (2021)
S.L. Childs, K.I. Hardcastle, Cocrystals of piroxicam with carboxylic acids. Cryst. Growth Des. 7(7), 1291–1304 (2007)
N.P. Jaywant, D.A. Purnima, Development of efavirenz cocrystals from stoichiometric solutions by spray drying technology. Mater. Today Proc. 3(6), 1742–1751 (2016)
J.A.B. dos Santos, J.V. Chaves Júnior, R.S. de Araújo Batista, D.P. de Sousa, G.L.R. Ferreira, S.A. de Lima Neto, C.F.S. Aragão, Preparation, physicochemical characterization and solubility evaluation of pharmaceutical cocrystals of cinnamic acid. J. Therm. Anal. Calorim. 145, 379–390 (2021)
O.O. Abosede, A.T. Gordon, T.O. Dembaremba, C.M. Lorentino, H.F. Frota, A.L. Santos, A.S. Ogunlaja, Trimesic acid–theophylline and isopthalic acid–caffeine cocrystals: synthesis, characterization, solubility, molecular docking, and antimicrobial activity. Cryst. Growth Des. 20(5), 3510–3522 (2020)
R. Kumar Bandaru, S.R. Rout, G. Kenguva, B. Gorain, N.A. Alhakamy, P. Kesharwani, R. Dandela, Recent advances in pharmaceutical cocrystal. Front. Pharmacol. 12, 2954 (2021)
G.J. Fernandes, M. Rathnanand, Formulation optimization for gastroretentive drug delivery system of carvedilol cocrystals using design of experiment. J. Pharm. Innov. 15, 455–466 (2020)
A.N. Allam, V.F. Naggar, S.S. El Gamal, Pharmaceutical development and technology. Pharm. Dev. Technol. 18(4), 856–865 (2013)
Y.M. Yu, F.Z. Bu, L. Liu, C.W. Yan, Z.Y. Wu, Y.T. Li, A novel sustained-release formulation of 5-fluorouracil-phenylalanine cocrystal self-assembled by cocrystal-entrapped micelle strategy displays enhanced antitumor efficacy. J. Mol. Liq. 368, 120665 (2022)
C. Cappuccino, D. Cusack, J. Flanagan, C. Harrison, C. Holohan, M. Lestari, M. Lusi, How many cocrystals are we missing? Assessing two crystal engineering approaches to pharmaceutical cocrystal screening. Cryst. Growth Des. 22(2), 1390–1397 (2022)
M.C. Mannava, M.K.D.R. Bommaka, K.A. Solomon, A.K. Nangia, Fluorobenzoic acid coformers to improve the solubility and permeability of the BCS class IV drug naftopidil. Chem. Commun. 58(37), 5582–5585 (2022)
R. Thakuria, B. Sarma, Drug-drug and drug-nutraceutical cocrystal/salt as alternative medicine for combination therapy: a crystal engineering approach. Crystals 8(2), 101 (2018)
K.A. Solomon, O. Blacque, R. Venkatnarayan, Molecular salts of 2, 6-dihydroxybenzoic acid (2, 6-DHB) with N-heterocycles: crystal structures, spectral properties and Hirshfeld surface analysis. J. Mol. Struct. 1134, 190–198 (2017)
G.N. Karthammaiah, S. Rao Amaraneni, A.K. Solomon, Co-crystal of nadifloxacin with oxalic acid. Acta Crystallogr. Sec. E Crystallogr. Commun. 79(4), 319–322 (2023)
S.M.A. Mashhadi, D. Yufit, H. Liu, P. Hodgkinson, U. Yunus, Synthesis and structural characterization of cocrystals of isoniazid and cinnamic acid derivatives. J. Mol. Struct. 1219, 128621 (2020)
K.A. Solomon, Molecular modelling and drug design (MJP Publisher, Chennai, 2019)
J.V.C. Júnior, J.A.B. Dos Santos, T.B. Lins, R.S. de Araújo Batista, S.A. de Lima Neto, A. de Santana Oliveira, C.F.S. Aragão, A new ferulic acid–nicotinamide cocrystal with improved solubility and dissolution performance. J. Pharm. Sci. 109(3), 1330–1337 (2020)
S. Karki, T. Friščić, L. Fabian, P.R. Laity, G.M. Day, W. Jones, Improving mechanical properties of crystalline solids by cocrystal formation: new compressible forms of paracetamol. Adv. Mater. 21(38–39), 3905–3909 (2009)
M. Shimizu, Y. Takase, S. Nakamura, H. Katae, A. Minami, K. Nakata, Y. Kubo, Pipemidic acid, a new antibacterial agent active against Pseudomonas aeruginosa: in vitro properties. Antimicrob. Agents Chemother. 8(2), 132–138 (1975)
A. Ito, K. Hirai, M. Inoue, H. Koga, S. Suzue, T. Irikura, S. Mitsuhashi, In vitro antibacterial activity of AM-715, a new nalidixic acid analog. Antimicrob. Agents Chemother. 17(2), 103–108 (1980)
P.B. Fernandes, Mode of action, and in vitro and in vivo activities of the fluoroquinolones. J. Clin. Pharmacol. 28(2), 156–168 (1988)
Y. Zhang, Y. Duan, J. Su, L. Liu, Y. Feng, L. Wu, Y. Liu, Inspiration for revival of old drugs: improving solubility and avoiding hygroscopicity of pipemidic acid by forming two pharmaceutical salts based on charge-assisted hydrogen bond recognitions. New J. Chem. 45(42), 19704–19713 (2021)
P.C. Alves, P. Rijo, C. Bravo, A.M.M. Antunes, V. André, Bioactivity of isostructural hydrogen bonding frameworks built from pipemidic acid metal complexe. Molecules 25(10), 2374 (2020)
A.S. Al-Wasidi, Synthesis and spectroscopic characterizations of some essential metal ion (MgII, CaII, ZnII, and FeIII) pipemidic acid chemotherapeutic agent complexes. Crystals 13(4), 596 (2023)
B.S. Chethan, N.K. Lokanath, Study of the crystal structure, H-bonding and noncovalent interactions of novel cocrystal by systematic computational search approach. J. Mol. Struct. 1251, 131936 (2022)
L.W.Y. Sun, F. Yang, X. Zhang, W. Hu, Cocrystal engineering: a collaborative strategy toward functional materials. Adv. Mater. 31(39), 1902328 (2019)
M.C.G.A. Mannava, M.K. Bommaka, K.A. Solomon, A.K. Nangia, Solubility and permeability enhancement of BCS class IV drug ribociclib through cocrystallization. CrystEngComm 24(45), 7915–7923 (2022)
O. Dolomanov, L. Bourhis, R. Gildea, J. Howard, H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Cryst 42, 339–341 (2009)
P.R. Spackman, M.J. Turner, J.J. McKinnon, S.K. Wolff, D.J. Grimwood, D. Jayatilaka, M.A. Spackman, CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 54(3), 1006–1011 (2021)
C. Jelsch, K. Ejsmont, L. Huder, The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 1(2), 119–128 (2014)
S.V. Puchkov, Y.V. Nepomnyashchikh, Evaluation of the reactivity of cyclohexanone CH bonds in reactions with tert-butylperoxy radical by quantum chemical methods. Kinet. Catal. 62, 479–487 (2021)
J.S. Singh, IR and Raman spectra with Gaussian-09 molecular analysis of some other parameters and vibrational spectra of 5-fluoro-uracil. Res. Chem. Intermed. 46(5), 2457–2479 (2020)
K.E. Srikanth, A. Veeraiah, T. Pooventhiran, R. Thomas, K.A. Solomon, C.J.S. Raju, J.N.L. Latha, Detailed molecular structure (XRD), conformational search, spectroscopic characterization (IR, Raman, UV, fluorescence), quantum mechanical properties and bioactivity prediction of a pyrrole analogue. Heliyon 6(6), e04106 (2020)
A.N. Manin, K.V. Drozd, A.V. Churakov, G.L. Perlovich, Hydrogen bond donor/acceptor ratios of the coformers: do they really matter for the prediction of molecular packing in cocrystals? The case of benzamide derivatives with dicarboxylic acids. Cryst. Growth Des. 18(9), 5254–5269 (2018)
V. Andre, P.C. Alves, M.T. Duarte, Exploring antibiotics as ligands in metal–organic and hydrogen bonding frameworks: Our novel approach towards enhanced antimicrobial activity (mini-review). Inorg. Chim. Acta 525, 120474 (2021)
I. Warad, A. Alruwaili, S.I. Al-Resayes, M.I. Choudhary, S. Yousuf, 5, 5-dimethyl-2, 2-bis (pyridin-2-yl)-1, 3-diazinane. Acta Crystallogr. Sect. E Struct. Rep. Online 68(6), o1786–o1786 (2012)
A. Saeed, Z. Ashraf, H. Nadeem, J. Simpson, H. Pérez, M.F. Erben, An investigation of supramolecular synthons in 1, 2, 4-triazole-3 (4H)-thione compounds. X-ray crystal structures, energetic and Hirshfeld surface analysis. J. Mol. Struct. 1195, 796–806 (2019)
O. Büyükgüngör, M. Odabaşoğlu, A.M. Vijesh, H.S. Yathirajan, (1Z)-Phthalazin-1 (2H)-one isopropylidenehydrazone. Acta Crystallogr. Sect. E Struct. Rep. Online 63(10), o4084–o4085 (2007)
V. Sethuraman, N. Stanley, P.T. Muthiah, W.S. Sheldrick, M. Winter, P. Luger, M. Weber, Isomorphism and crystal engineering: organic ionic ladders formed by supramolecular motifs in pyrimethamine salts. Cryst. Growth Des. 3(5), 823–828 (2003)
I.S.C.F. Macrae, S.J. Cottrell, P.T.A. Galek, P. McCabe, E. Pidcock, M. Platings, G.P. Shields, J.S. Stevens, M. Towler, P.A. Wood, Mercury 4.0: from visualization to analysis, design and prediction. J. Appl. Cryst 53, 226–235 (2020)
B.A. Stenfors, R.C. Collins, J.R. Duran, R.J. Staples, S.M. Biros, F.N. Ngassa, Crystal structure of 4-methyl-N-propylbenzenesulfonamide. Acta Crystallogr. Sec. E Crystallogr. Commun. 76(7), 1070–1074 (2020)
O. Şahin, O. Büyükgüngör, S. Şaşmaz, N. Gümrükçüoğlu, C. Kantar, N—H⋯ N and C—H⋯ π interactions in 4-amino-3-methyl-5-(p-tolyl)-4H-1, 2, 4-triazole and 4-amino-3-methyl-5-phenyl-4H-1, 2, 4-triazole. Acta Crystallogr. Sec. C Cryst. Struct. Commun. 62(11), o643–o646 (2006)
K.K. Sarmah, T. Rajbongshi, S. Bhowmick, R. Thakuria, First-line antituberculosis drug, pyrazinamide, its pharmaceutically relevant cocrystals and a salt. Acta Crystallogr. Sec. B: Struct. Sci. Crys. Eng. Mater. 73(5), 1007–1016 (2017)
S. Kalaiyarasi, S.R. Devi, R. Akilan, R.M. Kumar, G. Chakkaravarthi, 4-methylanilinium 3-carboxy-2-hydroxypropanoate. IUCrData 1(9), x161525 (2016)
H. Amarne, W. Helal, D. Taher, M. Korb, A. Al-Hunaiti, Crystal structure, Hirshfeld surface analysis and contact enrichment ratios of 5, 5-dimethyl-2-(2, 4, 6-tris (trifluoromethyl) phenyl)-1, 3, 2-dioxaborinane. Mol. Cryst. Liq. Cryst. 743(1), 77–88 (2022)
H. Kargar, M. Fallah-Mehrjardi, R. Behjatmanesh-Ardakani, K.S. Munawar, M. Ashfaq, M.N. Tahir, Experimental and theoretical studies of new dioxomolybdenum complex: synthesis, characterization and application as an efficient homogeneous catalyst for the selective sulfoxidation. Inorg. Chimica Acta 527, 120568 (2021)
C.J. Wilson, T. Cervenka, P.A. Wood, S. Parsons, Behavior of occupied and void space in molecular crystal structures at high pressure. Cryst. Growth Design 22(4), 2328–2341 (2022)
H. Aziz, A. Saeed, C.J. McAdam, J. Simpson, T. Hökelek, E. Jabeen, H.R. El-Seedi, Synthesis, single crystal structure determinations, Hirshfeld surface analysis, crystal voids, interaction energies, and density functional theory studies of functionalized 1, 3-thiazoles. J. Mol. Struct. 1281, 135108 (2023)
E. Irrou, Y.A. Elmachkouri, A. Oubella, H. Ouchtak, S. Dalbouha, J.T. Mague, M.L. Taha, Crystal structure determination, Hirshfeld surface, crystal void, intermolecular interaction energy analyses, as well as DFT and energy framework calculations of 2-(4-oxo-4, 5-dihydro-1H-pyrazolo [3, 4-d] pyrimidin-1-yl) acetic acid. Acta Crystallogr. Sec. ECrystallogr. Commun. 78(9), 953–960 (2022)
M. Karimi-Jafari, L. Padrela, G.M. Walker, D.M. Croker, Creating cocrystals: a review of pharmaceutical cocrystal preparation routes and applications. Cryst. Growth Des. 18(10), 6370–6387 (2018)
P. Garbacz, D. Paukszta, A. Sikorski, M. Wesolowski, Structural characterization of co-crystals of chlordiazepoxide with p-aminobenzoic acid and lorazepam with nicotinamide by dsc, x-ray diffraction, ftir and raman spectroscopy. Pharmaceutics 12(7), 648 (2020)
Acknowledgements
SJC, thanks Sri Sathya Sai University for Human Excellence for all the support and encouragement. The authors acknowledge SAIF, IIT Madras for single-crystal X-ray data collection.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest in the manuscript.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shwetha, J.C., Sharma, A. & Solomon, K.A. Molecular salts of pipemedic acid and crystal structure, spectral properties, and Hirshfeld surface analysis. J IRAN CHEM SOC 20, 3161–3176 (2023). https://doi.org/10.1007/s13738-023-02905-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-023-02905-8