
Abstract
A combination of candesartan cilexetil and hydrochlorothiazide in the tablet dosage form is developed by Takeda and recently approved by the United States Food and Drug Administration (USFDA) for the treatment of hypertension. The main objective of the current work was to optimize and develop a novel and robust stability-indicating reversed-phase ultra-performance liquid chromatography (RP-UPLC) method for simultaneous quantification of candesartan cilexetil and hydrochlorothiazide and their impurities in various tablet dosage forms and application to content uniformity and dissolution studies. The chromatographic condition is accomplished using a mobile-phase gradient system consisting of purified water–acetonitrile–glacial acetic acid (95:5:0.1, v/v) as mobile phase A and purified water–acetonitrile–glacial acetic acid (5:95:0.1, v/v) as mobile phase B at flow rate 0.4 ml/minute, wavelength 265 nm, injection volume 1.0 µL, column oven temperature 30 °C, sample cooler temperature 15 °C, and ZORBAX Eclipse Plus RRHD C18 column (5 cm × 2.6 mm, 1.8 µm). Calibration curves are achieved in the linearity range (1–240 µg/mL) and (3–225 µg/mL) with a correlation coefficient ≥ 0.9999 and a mean recovery percentage of 100.07 ± 0.89 and 100.62 ± 0.70 for candesartan cilexetil and hydrochlorothiazide, respectively. The suggested method is found to be selective as no overlapping showed from either solvent or placebo with the studied drugs and also purity threshold is greater than the purity angle under all forced degradation conditions. The current research of the developed UPLC method is validated as per the International Council for Harmonisation (ICH) guidelines and can be facilely applied in quality control or bioequivalence studies.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
USFDA approved Atacand Plus and Blopress Plus drugs which are manufactured by AstraZeneca and Takeda pharmaceutical companies, respectively, and these drugs contain a combination of candesartan cilexetil and hydrochlorothiazide for treating hypertension. Candesartan cilexetil is an “angiotensin II receptor,” which means that it prevents the function of a hormone in the body called angiotensin II. Angiotensin II is a strong vascular strait. By inhibiting receptors with which angiotensin II is usually associated, candesartan stops the hormone effect, allowing blood vessels to expand and lower blood pressure. Another sort of hypertension medication is hydrochlorothiazide, which is a diuretic. It works by boosting urine production, lowering blood pressure, and decreasing fluid in the blood. The combination of both drugs works together to reduce blood pressure more effectively than either drug alone [1].
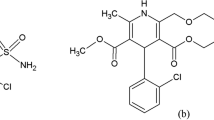
Candesartan cilexetil (CNC), angiotensin II (AT1) receptor antagonist, is chemically characterized as (1RS)-1-[[(cyclohexyloxy)carbonyl] oxy] ethyl 2-ethoxy-1-[[2′-(1H-tetrazol-5-yl) biphenyl-4-yl] methyl]-1H-benzimidazole-7-carboxylate with physical properties, white or almost white powder, practically insoluble in water, freely soluble in methylene chloride, and slightly soluble in anhydrous ethanol. Its chemical formula is C33H34N6O6, and its structural formula is shown in Fig. 1a [2].

Chemical structures of a CNC, b HCT
Hydrochlorothiazide (HCT) (Fig. 1b), thiazide diuretic, is chemically characterized as 6-chloro-3,4-dihydro-2H-1,2,4-benzothiadiazine-7-sulfonamide 1,1-dioxide with physical properties: white or almost white crystalline powder, low water solubility, soluble in acetone, and low ethanol solubility (96%). It dissolves in diluted solutions of alkaline hydroxides. Its chemical formula is C7H8ClN3O4S2 [2].
CNC and HCT are officially reported in British Pharmacopeia (BP) and the United States Pharmacopeia (USP) [2, 3]. The literature review showed various analytical methods have been published for the identification and estimation of CNC and HCT drugs by high-performance liquid chromatography (HPLC) methods [4,5,6,7,8,9,10,11,12,13,14,15,16], HPTLC methods [17, 18], LC–MS methods [19,20,21], spectroscopic methods [22, 23], and UPLC methods [24, 25].
Stability-indicating UPLC method is in high demand to assure the purity and efficacy of the pharmaceutical formulation through all stages and their long term. Furthermore, the separation of the degraded products is required to guarantee the safety and quality of the product and to estimate the degradation pathways [26,28,29,30,31,32,33,34,35,36,37,38,39].
Few UPLC methods had been published for concurrent quantification of CNC and HCT in tablet dosage form, and the comparison of the suggested method and the reported one is displayed in Table 1. The uniqueness of the proposed method is to establish fully validated stability-indicating UPLC method with high selectivity and sensitivity after exposing the studied drugs in pure and tablet dosage form to a wide range of exertion conditions, but the published UPLC methods were appropriated only for estimation of the examined drugs without broaching the forced degradation conditions. Moreover, the second goal of the current research work is the application of content uniformity and dissolution studies.
Materials and methods
Chemicals and reagents
UPLC grade acetonitrile and methanol and analytical grade polysorbate 20, hydrochloric acid, sodium hydroxide, glacial acetic acid, and hydrogen peroxide were purchased from Scharlau, Spain. Ultra-purified water was prepared with a Milli-Q water purification system (Millipore, USA). Working standard of CNC and HCT and finish products of Blopress® Plus 8/12.5 and 16/12.5 mg tablet were provided by Hikma Pharmaceutical Company (Amman, Jordan).
UPLC instrumentation
ACQUITY UPLC H-Class (Waters, Milford, MA, USA) complies with the Code of Federal Regulations (CFR) part 11 for data integrity and is equipped with the photodiode array (PDA) detector, binary solvent, sample, and column manager. Data analysis and processing supported with Empower 3 software.
Chromatographic conditions
Mobile phase contained purified water–acetonitrile–glacial acetic acid (95:5:0.1, v/v) as solution A and purified water–acetonitrile–glacial acetic acid (5:95:0.1, v/v) as solution B with gradient scheme (i) 0–1 min: 100% (A), 0% (B); (ii) 1–2.7 min: 5% (A), 95% (B), (iii) 2.7–2.8 min: 100% (A), 0% (B), and (iv) 2.8–4.5 min: 100% (A), 0% (B). Inject 1.0 µL of each solvent, standard, and sample using the following conditions: ZORBAX Eclipse Plus RRHD C18 column (5 cm × 2.6 mm, 1.8 µm), flow rate 0.4 ml/minute, wavelength 265 nm, column oven temperature 30 °C, and sample cooler temperature 15 °C. Perform the washing method before and after running the sequence about 15 min with mobile phase, 40 min with weak wash (10–20% organic:80–90% H2O), and 30 min with strong wash (70% organic:30% H2O).
Standard solution preparation (for LC 8/12.5 and 16/12.5)
Weigh accurately about the equivalent to 9.6 mg of CNC and 15 mg of HCT working standard, transfer completely into a 100-mL volumetric flask with the aid of 75 ml of solvent, sonicate for about 15 min, complete up to volume using solvent and mix well, and then filter through a syringe membrane filter.
Sample solution preparation for assay and content uniformity (for LC 8/12.5 and 16/12.5)
Weigh 20 tablets and calculate the average weight per tablet, weigh accurately about the equivalent to 9.6 mg of CNC and 15 mg of HCT from the sample being examined, transfer completely into a 100-mL volumetric flask with the aid of 75 ml of solvent, sonicate for about 20 min with handshaking (shake for 10 s every 2 min), complete up to volume using solvent and mix well, and then filter through a syringe membrane filter. HPLC chromatograms are displayed in Fig. 2.

a Standard solution of CNC and HCT, b assay test of Blopress Plus tablet, and c placebo
Solvent for assay, content uniformity, and impurity tests
Ultra-purified water and methanol (10:90) were used as solvents.
Standard solution for dissolution test
Weigh accurately about the equivalent of 8 mg of CNC and 12.5 mg of HCT working standard, transfer completely into a 100-mL volumetric flask with the aid of 50 ml of acetonitrile, sonicate for about 5 min, and complete up to volume with acetonitrile and mix well. Dilute 2 ml of the resulting solution into a 200-mL volumetric flask, complete up to volume with dissolution medium and mix well, and then filter through a syringe membrane filter.
Dissolution conditions
Operate the system at the following parameters: dissolution medium 1.0% (w/v) polysorbate 20 adjusted with 1.0 N NaOH at pH 6.8, volume 900 mL, apparatus: USP type II (paddle), speed 50 rpm, time interval 45 min, and temperature 37ºC ± 0.5 °C.
Calibration curves construction
Different concentrations of CNC and HCT were obtained by diluting aliquots of standard solution in a series of 10-ml volumetric flasks to get concentrations equivalent to (1–240 µg/mL) and (3–225 µg/mL) of CNC and HCT, respectively. The sequence of injection was performed as follows: one injection of standard solution # 01 (system setup), five replicate injections of standard solution # 01 (system suitability), two replicate injections of standard solution # 02 (standard check recovery), one injection of diluent (wash), one injection of diluent (blank), two replicate injections of standard solution #02 (two-point calibration #01), two replicate injections of test solution # 01, two replicate injections of test solution # 02, and two replicate injections of standard solution #02 (two-point calibration #02). The maximum number of sample injections between two consecutive two-point calibration sets was 12 (for all type of testing). In the sequence, the two-point calibrations must be injected from the same standard vial all over the sequence.
Results and discussion
Methods development and optimization
Many items were addressed and investigated during the optimization a new RP-UPLC method for simultaneous determination of CNC and HCT in their tablet, and the obtained results are displayed in Table S1 for mobile phase, flow rate, columns, and wavelengths. Initially, more than one method was tried with different compositions of the mobile phase, such as methanol/water (50:50, v/v), methanol/water (70:30, v/v), methanol:/water (80:20, v/v), and methanol/water (95:5, v/v), but no peak was found; 0.05 M phosphate buffer, pH 3.0/acetonitrile (50:50, v/v), 0.05 M phosphate buffer, pH 5.5/acetonitrile (70:30, v/v), but only peak of HCT was observed; and acetonitrile/water (50:50, v/v), acetonitrile/water (55:45, v/v), acetonitrile/water/glacial acetic acid (55:45:1, v/v), the peak of CNC was observed, and so, gradient system was established to achieve the best separation for CNC and HCT. Mobile phase A consists of purified water–acetonitrile–glacial acetic acid (95:5:0.1, v/v), and mobile phase B including purified water–acetonitrile–glacial acetic acid (5:95:0.1, v/v) with gradient profile (i) 0–1 min: 100% (A), 0% (B); (ii) 1–2.7 min: 5% (A), 95% (B), (iii) 2.7–2.8 min: 100% (A), 0% (B), and (iv) 2.8–4.5 min: 100% (A), 0% (B), showed the best development system for the separation of both drugs CNC and HCT, respectively. Scanning of the wavelengths in the range (200–400 nm) shows the best detector with a high resolution at 265 nm. Flow rates were tried from 0.1 to 1.0 mL/min, which showed the best one was 0.4 mL/min. Different types of stationary phases with different suppliers were addressed as Hypersil Gold C18 column (5 cm × 2.1 mm, 1.9 µm), ZORBAX Eclipse Plus RRHD C18 column (5 cm × 2.6 mm, 1.8 µm), and Ace Excel 2 C18 (5 cm × 2.1 mm, 2 µm), showing ZORBAX Eclipse Plus RRHD C18 column (5 cm × 2.6 mm, 1.8 µm) the higher-definition column with a rapid resolution. Also, the column oven temperature was maintained at 30 °C with a sampler cooler temperature of 15 °C. The final optimization system consists of a mobile phase containing purified water–acetonitrile–glacial acetic acid (95:5:0.1, v/v) as solution A and purified water–acetonitrile–glacial acetic acid (5:95:0.1, v/v) as solution B with gradient program (i) 0–1 min: 100% (A), 0% (B); (ii) 1–2.7 min: 5% (A), 95% (B), (iii) 2.7–2.8 min: 100% (A), 0% (B), and (iv) 2.8–4.5 min: 100% (A), 0% (B), injection volume 1.0 µL, ZORBAX Eclipse Plus RRHD C18 column (5 cm × 2.6 mm, 1.8 µm), flow rate 0.4 ml/minute, wavelength 265 nm, column oven temperature 30 °C, and sample cooler temperature 15 °C.
Method validation
The current UPLC method was developed and adjusted following the ICH Q2(R1) guidelines concerning the validation of the analytical method [40].
Linearity and range
A series of different concentrations were prepared for 1–240 and 3–225 µg/mL for CNC and HCT, respectively, to evaluate the linearity of the UPLC method procedure. After chromatographing each preparation in triplicate, a linear regression analysis was performed on the average peak areas versus the concentration of the level studied. The displayed result showed the method is linear over the concentration ranges with a correlation coefficient equal to 0.9999 as demonstrated in Table 2.
Detection and quantitation limit
The limit of detection and quantitation of the proposed UPLC method was computed using the validation of an excel sheet for the linearity range, using the equation formula (3.3δ/slope) and (10δ/slope), respectively, as mentioned in Table 2.
Accuracy and recovery
The objective of this study was to determine whether any inert ingredient in the finished product interferes with CNC and HCT peaks, so the placebo was prepared and spiked with the three different concentrations (50%, 100%, and 150%) to evaluate the recovery study. The results met the acceptance limit (98–102%), as given in Table S2.
Assay of dosage form
Test preparation was performed to evaluate the assay of CNC and HCT in dosage form with different strengths. Each sample was injected in duplicate, and the results were calculated using the average responses of two injections. The assay of the dosage form meets the acceptance limit (90- 110%) of the labeled amount as reported in Table 3.
Content uniformity test
Calculation of the acceptance value was done for the assay of 10 samples of dosage unit individually using the equation formula: acceptance value = ┃M-X ┃ + KS, showing the acceptance value within the limit of stage I in USP NMT 15% as listed in Table S3.
Precision
System precision
The objective of this item was to find the degree of agreement between the separate test results when the method is utilized repeatedly for multiple injections of the same homogeneous samples as listed in Table 2.
Intermediate precision (Ruggedness)
The ruggedness of the analytical method may be shown by running composite samples on each of the two days by the second analysts. The first analyst analyzed the test preparations of the composite sample on the first day. A second analyst used different equipment and repeated the analysis on a second day. The assay method is rugged, and RSD is less than 2.0% between the assay results through all change stages as shown in Table 4.
Robustness
The robustness of the UPLC method is addressed to evaluate the minor changes in the proposed method as pH of mobile phase, flow rate, wavelength, and column oven temperature changes. The obtained result showed UPLC method is a robust with relative standard deviation less than 2.0% between the assay results through all change stages as displayed in Table 4.
Stability of the analytical solution
This study intends to check the stability of active ingredients by analyzing the standard and sample preparations after two days in the fridge and at room temperature. The assay results showed within ± 2.0% of the actual amount, as given in Table S4.
Filter compatibility
The purpose of this study was to determine the effect of the filtration procedure on the standard and sample solution using two types of filters (Micro Solv cellulose and PTFE syringe membrane filter 25 mm and 0.2 µm, respectively). Standard and sample solutions were filtered and two portions of filtrate of each first four milliliters and first eight milliliters and analyzed in duplicate corresponding to unfiltered standard and sample. The assay results showed within the limit (98–102%), and the two types of filters are suitable after discarding the first three milliliters, as given in Table S4.
Application of dissolution test
Operate the system at the parameters specified. At the specified time, withdraw 10 ml from each dissolution vessel. Filter through a syringe membrane filter and inject the test preparation after transferring vials to the HPLC system to evaluate the dissolution rate of CNC and HCT in dosage form with different strengths. The dissolution results of dosage form meet the acceptance limit NLT 80% of the labeled amount of CNC and HCT dissolved with RSD NMT 10% as shown in Table S5.
Specificity
Selectivity
The objective of the selectivity test is to make sure that the proposed method is selective to the targeted dosage form. The selectivity test was established by injecting all materials than the active ingredient as solvent and placebo, and the figures show no interference from solvent and placebo in the CNC and HCT retention time.
Forced degradation
In order to establish stability-indicating capability method for CNC and HCT in Blopress Plus tablet, CNC raw material, HCT raw material, Placebo, and Blopress plus tablet were subjected separately to the following conditions: acidic ( 5 mL of 1 N HCl for 60 min at 95 °C), basic (5 mL of 1 N NaOH for 60 min at 95 °C), heat (30 min at 95 °C), photolysis (sun test 765 w/m2 at 35 °C for 3 h), and oxidation (5 mL of 30% H2O2 for 30 min at 95 °C). CNC and HCT peaks were found to be pure under all forced degradation conditions since the purity threshold is greater than the purity angle and the purity flag value is none for the active ingredients under all conditions, as shown in Figure S1, Figs. 3 and 4, and Table 5.


Chromatograms of raw materials for CNC and HCT under conditions: a normal condition, b heat, c acid, d base, e sunlight, f oxidation


Chromatograms of impurity test of CNC and HCT under conditions: a normal condition, b heat, c acid, d base, e sunlight, f oxidation
System suitability
It is generally desirable to ascertain the suitability and effectiveness of the operating system when employing chromatographic methods such as pressurized liquid chromatography. It should be noted that the specification of the effective parameter in the CNC and HCT assay procedures does not preclude the use of other suitable operating conditions. To obtain acceptable operations, chromatograms may be required to ascertain the effectiveness of the final operating system. The system suitability results for five replicates in the standard of CNC and HCT are given in Table 6 showing the system is suitable.
Conclusion
A novel and accurate stability-indicating RP-UPLC method is developed and verified for the estimation of the combination of CNC and HCT and their related substances in Blopress® Plus 8/12.5 and 16/12.5 mg tablet dosage. Application to content uniformity weight and dissolution studies showed the results matching with the acceptance criteria of stage I in USP. All items of test method validation matched with the acceptance criteria that set in ICH guidelines. Stress degradation and selectivity were performed, showing no interference between the placebo and the studied drugs, and the active ingredient peaks are pure under all stress degradation conditions. Relying on the accepted results, the proposed UPLC method is confirmed and capable of indicating stability and can be used in quality control for release and stability studies.
References
E.B. Melian, B. Jarvis, Drugs (2002). https://doi.org/10.2165/00003495-200262050-00006
British Pharmacopoeia Stationary Office, Medicines, and Healthcare Products Regulatory Agency. 2, 1248 (2022)
U.S. Pharmacopoeia, United States Pharmacopoeia Convention Inc. 43, 3761 (2022).
N. Erk, J. Liq. Chromatogr. Relat. Technol. (2003). https://doi.org/10.1081/JLC-120023802
K.S. Babu, N.D.A. Kumar, U. Gosada, N. Sharma, Pharm. Methods. (2012). https://doi.org/10.4103/2229-4708.97718
R. A. Mhaske, S. Sahasrabudhe, A. A. Mhaske, D. J. Garole, Int. J. Pharm. Sci. Res. (2012). https://doi.org/10.13040/IJPSR.0975-8232.3(3).793-01
S.M. Al-Adl, L.M. Abdel-Aziz, M.A. Mohamed, Anal. Chem. Lett. (2017). https://doi.org/10.1080/22297928.2017.1326840
M. de Diego, R. Godoy, S. Mennickent, C. Vergara, D. Miranda, P. Navarro, J. Chromatogr. Sci. (2018). https://doi.org/10.1093/chromsci/bmx068
N. Pappula, S. Ravichandra, S.L. Sindhura, P.A. Rani, V.S. Madhuri, V. Ajay, P.N. Reddy, Asian J. Pharm. Ana. (2019). https://doi.org/10.5958/2231-5675.2019.00038.3
I.A. Naguib, E.A. Abdelaleem, M.E. Draz, H.E. Zaazaa, Anal. Chem. Lett. (2015). https://doi.org/10.1080/22297928.2015.1026394
V.N.R. Ganipisetty, D. Jalandhar, G. Gnanadev, P. Manoj, R. Venkata Nadh, Sep. Sci. Technol. (2015). https://doi.org/10.1080/01496395.2016.1154873
R.M. Gaurkhede, A.V. Chandewar, Res. J. Pharm. Technol. (2018). https://doi.org/10.5958/0974-360X.2018.00084.7
P. Prajapati, A. Patel, S. Shah, J. Chromatogr. Sci. (2022). https://doi.org/10.1093/chromsci/bmab070
P.B. Prajapati, A. Patel, S.A. Shah, J. AOAC Int. (2021). https://doi.org/10.1093/jaoacint/qsab079
P.B. Prajapati, A.S. Patel, S.A. Shah, J. AOAC Int. (2022). https://doi.org/10.1093/jaoacint/qsab097
P.B. Prajapati, M.A. Thakor, K.B. Bodiwala, S.A. Shah, J. AOAC Int. (2021). https://doi.org/10.1093/jaoacint/qsaa140
I.A. Naguib, E.A. Abdelaleem, F.F. Abdallah, A.A. Emam, J. AOAC Int. (2020). https://doi.org/10.1093/jaocint/qsz015
M.E. Abou Kull, I.A. Naguib, Curr. Pharm. Anal. (2017). https://doi.org/10.2174/1573412911666151020003509
A. Gumieniczek, J. Galeza, T. Mroczek, K. Wojtanowski, K. Lipska, R. Pietras, Chromatographia (2018). https://doi.org/10.1007/s10337-018-3555-8
A. Tan, X. Gui, M. Wong, H. Deng, G. Gu, C. Fanaras, J.C. Fanaras, Biomed. Chromatogr. (2020). https://doi.org/10.1002/bmc.4808
D.V. Bharathi, K.K. Hotha, P.K. Chatki, V. Satyanarayana, V. Venkateswarlu, Bioanalysis (2012). https://doi.org/10.4155/bio.12.83
M.A. Mukthinuthalapati, J.S.P. Kumar, Pharm. Methods 6, 148 (2015)
Y.A. Workie, A.E. Mohamed, A.A. Bekhit, A. Hymete, Am. J. Pharm. Pharmacol 4, 50 (2017)
Ö. Üstündağ, E. Dinç, Monatsh. Fur Chem. (2021). https://doi.org/10.1007/s00706-021-02822-7
B. Singh, R.S. Lokhandae, A. Dwivedi, S. Sharma, N. Dubey, J. Pharm. Anal. (2014). https://doi.org/10.1016/j.jpha.2013.05.003
N.Y. Hassan, E.M. Abdel-Moety, N.A. Elragehy, M.R. Rezk, Spectrochim. Acta, Part A (2009). https://doi.org/10.1016/j.saa.2008.12.025
M.R. Elghobashy, A.M. Mahmoud, M.R. Rezk, M.K. Abd El-Rahman, J. Electrochem. Soc. (2014). https://doi.org/10.1149/2.0161501jes
M.A. Hegazy, H.M. Lotfy, M.R. Rezk, Y.R. Omran, Spectrochim. Acta, Part A (2015). https://doi.org/10.1016/j.saa.2014.12.098
M.R. Rezk, M.A. Tantawy, M. Wadie, S.A. Weshahy, Spectrochim. Acta, Part A (2020). https://doi.org/10.1016/j.saa.2019.117547
M.R. Rezk, A.S. Fayed, H.M. Marzouk, S.S. Abbas, J. Electrochem. Soc. (2017). https://doi.org/10.1149/2.0921709jes
M.R. Elghobashy, M.R. Rezk, Anal. Bioanal. Electrochem 6, 461 (2014)
M.A. Tantawy, S.A. Weshahy, M. Wadie, M.R. Rezk, Anal. Methods. (2020). https://doi.org/10.1039/D0AY00822B
H.M. Marzouk, M.R. Rezk, A.S. Gouda, A.M. Abdel-Megied, Microchem. J. (2022). https://doi.org/10.1016/j.microc.2021.106917
M.A. Tantawy, S.A. Weshahy, M. Wadie, M.R. Rezk, Microchem. J. (2020). https://doi.org/10.1016/j.microc.2020.104905
M.R. Rezk, E.M. Abdel-Moety, M. Wadie, M.A. Tantawy, J. Sep. Sci. (2021). https://doi.org/10.1002/jssc.202000975
A.H. Nadim, A.R. Hussein, M.R. Rezk, F.A. Fathalla, Y.S. El-Saharty, Anal. Biochem. (2022). https://doi.org/10.1016/j.ab.2022.114790
K.M. Kelani, M.R. Rezk, A.S. Saad, M.S. ElSherbiny, H.H. Monir, J. Anal. Chem. (2022). https://doi.org/10.1134/S1061934822080032
S.K. Muchakayala, N.K. Katari, T. Dongala, V.M. Marisetti, G. Vyas, R.V. Vegesna, J Iran Chem. Soc. (2022). https://doi.org/10.1007/s13738-021-02388-5
M.A. Mohamed, Ann. Pharm. Fr. (2022). https://doi.org/10.1016/j.pharma.2022.09.001
P. Borman, D. Elder, Q2 (R1) validation of analytical procedures. ICH Qual. Guidel. 5, 127 (2017)
Acknowledgements
The authors acknowledge the facilities provided by Hikma Pharmaceutical Company during performing this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mohamed, M.A., Nassar, H.F. Stability-indicating RP-UPLC method for determination of antihypertensive drugs and their degradation products in tablets: application to content uniformity and dissolution studies. J IRAN CHEM SOC 20, 763–773 (2023). https://doi.org/10.1007/s13738-022-02725-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-022-02725-2