
Abstract
TNF-α is a crucial cytokine in the process of inflammatory diseases. The adverse effect of TNF-α is mostly mediated by interaction of TNF-α with TNF-α receptor type I (TNFR1); therefore, discovery of molecules which can bind to TNFR1 preventing TNF-α-receptor complex formation would be of great interest. In the current study, using GRID/GOLPE program, a 3D-QSAR study was conducted on a series of synthetic TNFR1 binders, which resulted in a 3D-QSAR model with appropriate power of predictivity in internal (r2 = 0.94 and q2LOO = 0.74) and external (r2 = 0.66 and SDEP = 0.42) validations. The structural features of TNFR1 inhibitors essential for exerting activity were explored by analyzing the contour maps of the 3D-QSAR model showing that steric interactions and hydrogen bonds are responsible for exerting TNFR1 inhibitory activity. To propose potential chemical entities for TNFR1 inhibition, PubChem database was searched and the selected compounds were virtually tested for anti-TNFR1 activity using the generated model, resulting in two potential anti-TNFR1 compounds. Finally, the possible interactions of the compounds with TNFR1 were investigated using docking studies. The findings in the current work can pave the way for designing more potent anti-TNFR1 inhibitors.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Tumor necrosis factor-alpha (TNF-α, cachexin, cachectin) was first identified on the basis of its capability to induce in vitro shrinkage of tumor cells and hemorrhagic necrosis of transplantable tumors in mice [1]. Later studies demonstrated the powerful pro-inflammatory effects of TNF-α [2] and revealed its key role as a mediator of endotoxic shock [3]. TNF-α is mostly produced by activated monocytes and macrophages upon stimulation of its secretion either by exogenous or endogenous factors, and subsequently interacts with its two distinct receptors namely TNFR1 and TNFR2 [4]. At physiological levels, TNF-α regulates different biological processes such as body circadian rhythm, immune responses, remodeling and replacement of injured tissues and other important mechanism in the cell hemostasis [5]. However, overexpression of TNF-α has been implicated in the pathogenesis of different immune-dependent complications [2]. Inhibition of TNF-α activity is of great importance to alleviate the adverse effects originated from pathological amounts of TNF-α, which can be achieved by mechanistically different strategies. Most of the currently used biopharmaceuticals for controlling the effect of TNF-α in inflammatory diseases work based on direct binding to TNF-α and include etanercept (Enbrel, Amgen Incorporated, thousand Oaks, CA, Wyeth Pharmaceutical, Collegiville, PA), infliximab (Remicade, Centocor, Horsham, PA, Schering Plough, Kenilworth, NJ), adalimumab (Humera, Abbot Laboratories, Abbot Park, IL), certolizumab, and golimumab [6,7,8,9]. Since these biological therapeutics bind to TNF-α, the physiological processes regulated by TNF-α would be disrupted resulting in serious adverse effects such as increasing the risk of lymphoma and infections (such as tuberculosis and hepatitis B) [10, 11]. Moreover, immunogenic responses to these proteins as well as their high cost and time-consuming production processes necessitate identification of new small anti-TNF-α agents with high stability and less adverse effects [12,13,14].
As mentioned above, TNF-α exerts its biological effects through TNFR1 and TNFR2 receptors, among which, TNFR1 is expressed by most of the somatic cells, while TNFR2 is found on particular immune cells and contributes to T-cell survival [15]. Both of these receptors belong to the TNF receptor family. The members of this family are classified into three major groups, based on their cytoplasmic sequences and signaling diversity [16]. It has been demonstrated that the activation of TNFR1 contributes to the inflammation pathogenesis [17, 18], while, TNFR2 is implicated in cell-mediated immunity [19, 20]. Based on these differences, much attention has been dedicated to discriminate the mode of interaction of TNF-α with its two distinct receptors to promote specific binding to a particular type of TNF-α receptor [21].
From structural point of view, both TNF-α receptor types contain six cysteine residues, which constitute four cysteine-rich domains (CRDs) [13]. In TNFR2, the turn motif of CRD3 (Ser107 to Cys112) fits to CRD2 by a disulfide bond between Cys104 and Cys112, while in TNFR1 there is a space between the turn motif of the CRD3 and CRD2 resulting in generating of a binding pocket on TNFR1. One more difference between the structures of TNFR1 and TNFR2 arises from the longer loop structure in TNFR1 (highlighted by dashed oval in Fig. 1) compared to TNFR2, which is suspected for the formation of van der Waals interactions with TNF-α [21]. TNF-α initiates its most pathogenic effects through interaction with TNFR1. It has been shown that specific inhibitors of TNFR1 provide satisfactory inhibition of TNF-α-induced pathological effects, without interfering with the interaction of TNF-α with TNFR2 [22, 23]. In this context, the identification of TNFR1-blocking agents seems a promising strategy for achieving desired therapeutic effects due to inhibition of TNFR1-mediated effects of TNF-α, while leaving TNFR2 pathway unaffected. ATROSAB [24] and mouse TNFR1 antagonist (DMS5540) [25] are two anti-TNFR1 antibodies which have been recognized with antagonizing effect on TNF-α-mediated cytotoxicity. ATROSAB is a humanized full antibody with a strong inhibitory activity on TNFR1-mediated signaling pathways [24], whereas DMS5540 is an anti-TNFR1 antibody resulted from the fusion of a mouse anti-TNFR1 domain antibody (dA) with an albumin-binding domain antibody [25]. Another class of anti-TNFR1 agents belongs to low-molecular weight inhibitors reported by Carter and co-workers from Bristol-Myers Squibb company [26, 27].

Comparison of TNFR1 and TNFR2 structures. a TNFR1, b TNFR2. An extra loop is noticeable in TNFR1 structure
The aim of this study was to gain insight into the structural features of the small synthetic anti-TNFR1 inhibitors by generating a 3D-QSAR model for a series of TNFR1 inhibitors [26, 27]. To this end, alignment-dependent 3D-QSAR study was performed [28, 29] in which after initial superpositioning of the compounds on a TNFR1-bound ligand (IV703) [26], the aligned structures were introduced into GRID program to calculate molecular interaction fields (MIFs) [30,31,32,33]. Subsequently, the generated MIFs were transferred into GOLPE program [34] to perform the multivariate statistical analysis and model building. The produced 3D-QSAR model was statistically validated by both internal and external validation methods. Using the most potent compound, PubChem database was searched for the structurally similar compounds and the selected structures were evaluated for their anti-TNFR1 activity based on the generated model. Moreover, using the developed model, the important structural features affecting the biological activities of TNFR1 inhibitors were identified, which can be used for designing new drug candidates with TNFR1 inhibitory activities.
Methods
Dataset collection
A set of 47 5-arylidene-2-thioxodihydropyrimidine-4,6(1H,5H)-diones and 3-thioxo-2,3-dihydro-1H-imidazo[1,5-α] indol-1-ones derivatives was collected from the literature [26, 27] (Table 1). The binding affinity data for all compounds were experimentally determined as IC50 (µM) through TNF-α–TNFR1 binding assay. These values estimate the concentration of ligands that inhibit 50% of TNF-α binding to a soluble form of monomeric TNFR1. All inhibitory activities expressed as IC50 values in molar scale were converted to pIC50 and used as the dependent variable in 3D-QSAR study. The 3D structures of the ligands were generated using the Built Optimum option of HyperChem software [35] (HyperChem™ version 8.0.8, Hypercube, Inc. Gainesville, FL, USA), followed by energy minimization using MM+ force field based on Polak–Ribiere algorithm [36]. Then the structures were fully optimized by semi-empirical AM1 algorithm [37].
Using MOE program [38] (Molecular Operating Environment, Chemical Computing Group Inc. Montreal, QC, Canada) all the compounds were aligned on a co-crystallized ligand bound to TNFR1, namely IV703 (PDB ID code: 1FT4) [26]. To do so, the matching part of the compounds to the 5-Thioxo-5,6-dihydro-7H-thieno [2′, 3′:4, 5] pyrrolo[1,2-c]imidazol-7-one core scaffold in IV703 (Fig. 2) was paired up using its conformation found in the crystallographic coordinates and then letting the other parts of the molecules be aligned flexibly.

IV703 chemical structure. The core center for superpositioning of all the compounds have been highlighted
Grid calculations
The aligned compounds were introduced into GRID program [39] (version 1.2.2, Molecular Discovery Ltd, UK) in which the C3, OH2 and N1 (a neutral flat NH2) probes were used to mimic possible steric hindrance, hydrogen bond donor and hydrogen bond acceptor interactions of compounds with receptor. GRID analysis was performed using a grid box of 31, 29 and 23 Å in X, Y and Z dimensions, respectively, with a grid spacing of 1 Å (NPLA = 1). The calculations were carried out on a Linux-based Cluster consisting eight nodes running CentOS (ver. 5.6) operating system.
Variable selection using GOLPE analysis
The calculated MIFs for the molecules and the corresponding activity values were imported to GOLPE program [40] (version 4.6.0, Multivariate Infometric Analysis, Barcelona, Spain) running on a Silicon Graphics O2 workstation, in which interaction values (X values) less than 1.0E-7 were removed automatically. The studied compounds were randomly divided into train (37 compounds) and test (10 compounds) sets using SPSS (version 16.0) based on their activites. The additional pretreatment performed on train set compounds were as follows: X variables less than 0.1 were set to zero for all probes, and those with variance lower than 0.1 were rejected (minimum S.D. cut-off was regarded as 0.1). This pretreatment procedure reduced the number of variables to about 20% of that found in the previous step without considerably changing the model statistics. Based on the selected variables, primary PLS model was built to proceed with smart region definition (SRD) method with default number of seeds selected on the PLS weight space [41]. In SRD the variables were grouped within the distance of 2 Å to the seeds followed by collapsing the neighboring groups including the same information within the distances of 4 Å. On the generated groups, the fractional factorial design (FFD) procedure was performed with 20% dummy variables. Finally, remained variables were used to generate PLS model [42]. FFD selection was applied several times until no further improvement in statistical parameters (q2 and SDEP) was observed. The applicability domain analysis on the train and test sets for the generated 3D model was carried out using the method developed by Roy et al. named “AD using standardization approach (version 1.0)” [43], where the PCA scores obtained from the 3D-QSAR were used as the X variables and the biological activities of the studied compounds were considered as Y variable [44]. The modeling process was repeated after excluding the identified outlier to develop the final 3D model. In an effort to evaluate the predictive power of the generated 3D-QSAR model, internal validation methods such as leave-one-out (LOO), leave-two-out (LTO) and random-groups-out (RGO) as well as external validation were used. Standard deviation of the error of prediction (SDEP) value was also used to assess the predictive power of the model.
Database search for identification of anti-TNFR1 candidates
PubChem database of chemical molecules was searched based on the 90% structural similarity to the most potent compound (i.e., compound 45) in our data set. The search resulted in 52 compounds. The 3D structures of these compounds were retrieved and after superimpositioning on IV703, their activities were predicted using the proposed 3D-QSAR model.
Molecular docking studies
To investigate the possible interaction mode of the inhibitors with TNFR1, compounds IV703 and 45, and the two compounds with the highest predicted activities from the similarity search of PubChem database were docked into TNFR1 protein (PDB code: 1FT4) using GOLD docking program (version 5.0, CCDC, Cambridge, UK) running under LINUX operating system [45, 46]. LigPlot+ program [47] was used to extract the possible interactions between the best docking pose for each compound and TNFR1.
Results and discussion
Assessment of the model predictivity
TNF-α is an important cytokine which plays a key role in inflammatory processes. It has been shown that TNF-α-associated pathogenesis is mostly mediated upon activation of tumor necrosis factor-alpha receptor type 1 (TNFR1). Therefore, inhibition of TNF-α interaction with TNFR1 can be a promising approach to neutralize TNF-α induced adverse pro-inflammatory effect. In this regard, identification and development of new anti-TNFR1 candidates based on computational methods is of great importance. Ligand-based drug design is a strategy for lead identification and optimization using ligand–receptor three-dimensional structural information. On contrary to the structure-based drug design, this method uses structural features of the molecules that participate in receptor binding [48, 49]. In the current study, based on the crystal structure of a complex formed between a small molecule (i.e., IV703) and TNFR1 (PDB code: 1FT4), ligand-based drug design approach was employed to build 3D-QSAR model for a series of TNFR1 inhibitors. The receptor-bound mode of the studied molecules was attained by aligning them on IV703 as the reference structure (Fig. 3) and the obtained conformations were introduced to GRID program in which C3, OH2 and N1 probes were used for the calculation of MIFs. The obtained MIFs were in turn transferred into GOLPE program for model building and statistical analyses. In general, most of the variables obtained from GRID calculation are irrelevant in correlating the chemical structures to the biological activities, and therefore, must be removed to improve the quality of the 3D-QSAR model. Initially, the number of variables was decreased by 20% in pretreatment process. Then using SRD algorithm, the spatially close variables were grouped and the produced groups comprising the same energy information were collapsed to the regions. On the generated regions, the fractional factorial design (FFD) procedure was performed to weigh up the effects of individual regions. In each FFD, procedure only group of variables which had positive effect on the model more than the average dummy variable were retained. The selected variables were used for generating a PLS model based on three latent variables.

The superposition of all studied molecule on crystal structure of IV703
According to the rule of Organization for Economic Co-operation and Development (OECD) [43, 50], the applicability domain (AD) for the developed PLS model must be defined. The calculated PC values and the biological activities of the train and test set compounds were used for outliers identification as described previously [44]. This was done by Applicability Domain (using standardization approach) tool version 1.0 developed by Roy and co-workers [43]. The results of this analysis were identified compounds 15, 43, and 46 as the outliers in both data sets. It seems that the presence of two bulky phenyl groups on the dihydropyrimidine moiety in compound 15 may interfere with the appropriate three-dimensional arrangement of this compound compared to the rest of the data set. In the case of compounds 43 and 46, the boat conformation of piperazine and the presence of a bulky substituent at the meta position of the phenyl group may be responsible for their structural differences to the rest of data set. By excluding compounds 15, 43, and 46, the QSAR model was rebuilt as outlined above. Initially, 58,500 variables were generated from MIF calculation in GRID, which was subsequently decreased to 10,156 during pretreatment process. Further variable selection was performed using SRD method along with several rounds of FFD resulting in 1359 variables. The obtained model with three latent variables has a correlation coefficient value (r2) of 0.94 (Table 2). The internal predictive power of the model was evaluated by LOO, LTO and RGO methods. The results show that the q2 of all internal cross-validation analyses are greater than 0.7 indicating good predictive power of the model in predicting the anti-TNFR1 activity of compounds (Table 2). Since relying on the internal cross-validation is not enough for model validation [51], in the current study external validation of the developed QSAR model was performed. The results for experimental versus predicted values for the train and test compounds are depicted in Fig. 4. Table 3 shows the r2 and SDEP values for test set compounds, which are 0.66 and 0.42, respectively, indicating its suitability. Further assessment of the 3D-QSAR model was performed by analyzing the errors observed for the predictive values. The mean absolute percentage error of the predictions (MAPE) for test and training set data are 5.63 and 4.07%, respectively, showing acceptable level of errors in the prediction. On the other hand, the mean absolute error of the predictions for the test set was calculated to be 0.33 which is more than 0.25 (i.e., 10% of the training set activity range) but still smaller than 0.49 (i.e., 20% of the training set activity range), indicating moderate prediction power of the built 3D-QSAR model based on MAE measure [52].

Experimental versus predicted pIC50 for compounds. Open squares indicate training set and filled circles show the test set compounds
Analysis of structural features of anti-TNFR1 compounds
In addition to predicting the biological activity, 3D-QSAR studies aim to identify the important structural molecular features contributing to the activity. In the current study, PLS coefficient plots generated in GOLPE program were analyzed to recognize the structural features of compounds whose interactions with the given probes influence the activity significantly. To simulate the virtual receptor C3, OH2 and N1 probes in GRID program were used to calculate the molecular interaction fields for the studied compounds. Subsequently, the calculated fields were used by GOLPE program to generate a 3D-QSAR model to predict the biological activity of TNFR1 inhibitors. The contour maps corresponding to the interaction of the compounds with different probes were generated and illustrated using the PLS coefficients (Fig. 5). Totally five regions were identified as the key structural features having impact on the binding ability of the inhibitors. These regions are indicated with R1, R2, R3, R4, and R5 in Fig. 5, in which the most active compound (i.e., compound 45) was inserted in the maps for depicting the important structural features needed for inhibitory activity of the studied compounds.

Contour maps for the PLS-coefficients (3LVs) illustrated for the most potent compound in the data set. a C3 probe; b N1 probe and c OH2 probe. Blue areas indicate negative coefficients and yellow areas indicate positive coefficients
R1 region
Based on the analysis of C3 contour map (Fig. 5a), it seems there is a pocket on the TNFR1 receptor which can accommodate a substituent on the inhibitor at the meta position of an aromatic ring (R1 region) distal to the core center of the molecule. Based on the substituents in different inhibitors, it seems that their interactions could be of van der Waals nature resulting in the increased biological activity. In some cases such as compounds 14, 23, 44, and 47, the nature of the substituent in terms of inductive effect on the aromatic ring affects the binding ability. The optimum biological effect is achieved where the electron-donating substituent is introduced on the aromatic ring. This explanation is supported by the data achieved from the PLS coefficients for OH2 and N1 probes. Figure 5b, c shows the contour maps of the PLS coefficients for the N1 and OH2 probes, respectively, in which R1 region represents favorable interactions with positive PLS coefficients (positive interaction energy with probes), but leading to decreased inhibitory activities suggesting that the existence of electrostatic interactions at R1 region between the inhibitors and TNFR1 is unfavorable.
R2 region
In the case of R2 region in C3 probe, interactions between a substituent in the molecule and the C3 probe in the areas with both negative (shown in blue) and positive PLS coefficients (shown in yellow) are noticeable. According to the C3 PLS coefficient maps, it seems that positioning of a methoxy group at R2 region (compounds 28, 29, and 30) is favorable for ligand–receptor interaction; however, the presence of more bulky groups may interfere with such favorable contacts. Analysis of the OH2 contour maps indicates that ligand–receptor interaction at R2 region with electrostatic nature is not favored supporting the idea that the hydrophobic contact at R2 region was well tolerated. In N1 contour maps, both favorable and unfavorable interactions are seen for R2 region. The establishment of favorable interactions by N1 region at R2 region can be attributed to the steric nature of N1 probe [53] showing the importance of spatial arrangement for ligand–receptor interaction at this region.
R3 region
Comparing the contour maps of different probes in R3 region shows that a steric repulsion at this region may prevent appropriate accommodation of inhibitors at the binding site of TNFR1. On the other hand, OH2 and N1 contour maps suggest negligible interaction due to electrostatic forces for the inhibitors. As it can be observed in Table 1, the compounds with low activities in the data set (i.e., compounds 10, 19, 25, and 26) are those in which the five-membered ring at R3 region is replaced with a relatively more bulky six-membered ring posing steric repulsion and significant loss of activity.
R4 region
Close inspection of R4 region indicates its significant effect due to hydrogen bond interactions. This information was deduced from contour maps of OH2 and N1 probes implying the positive influence of electrostatic interactions for TNFR1-binding ability of studied compounds. Additionally, C3 contour map in R4 region shows that positioning hydrophobic substituents at this site can result in decreased activity indicating that this region is more likely to contribute to just electrostatic interactions rather than hydrophobic and steric contacts.
R5 region
R5 region is another important part of the molecule where the results show that the more bulky the substituents in this region, the more active the compounds (Fig. 5a). The effect of this structural feature is more pronounced when the methyl group in compound 10 is replaced with ethyl and toluoyl groups in the compounds 13 and 12, respectively, leading to the increased activity. It can be hypothesized that TNFR1 inhibitors with a bulky substituent at this region form more proper interaction with the receptor.
Identification of anti-TNFR1 candidates
To propose new compounds with potential inhibitory activity on TNFR1, the database of chemical molecules in PubChem was searched to identify the most structurally similar compounds to that of the most potent compound in our data set. Among the 52 selected structures, the predicted inhibitory activities of compounds 8a and 33a on TNFR1 with one, two, and three latent variables were comparable with the experimental activities of the highly potent compounds in the data set (Table 4). Based on these results, it seems that the compounds 8a and 33a can be considered as promising TNFR1 inhibitors.
Prediction of binding mode between TNFR1 and its inhibitors
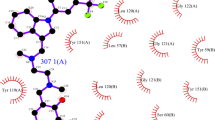
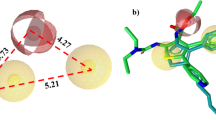
To predict the possible interactions between TNFR1 and its inhibitors, compounds IV703, 45, 8a and 33a were docked into TNFR1 (Fig. 6). The obtained best pose for each compound in complex with TNFR1 was analyzed using LigPlot + program (Fig. 7) in an effort to identify interacting moieties of the inhibitors and TNFR1. The 2D analyses of the complexes revealed that all of the docked inhibitors follow the same interaction pattern with TNFR1. Detailed analyses showed that these compounds interact with residues Ser74, Lys75 and Arg77 in the extra loop region of TNFR1 (Fig. 1) via R1 hydrophobic region identified by the grid analyses in our 3D QSAR study. The results are in agreement with previous studies suggesting this region of TNFR1 as part of the binding pocket for the inhibitors [21]. The other residue involved in the interaction between the docked molecules and the receptor is Ala62, which has been also suggested to be part of TNF-α-binding site based on homology modeling of TNF-α–TNFR1complex [26]. In the case of R2 region in PLS-contour maps, it seems that hydrophobic interactions are more favored at this part as (Fig. 5) the docking studies have proved such contacts between the inhibitors and Phe60, Asp91, Arg92, and Asp93 of TNFR1 (Fig. 7). Moreover, PLS-contour maps suggest that the formation of hydrogen bonds at R4 region between TNFR1 and inhibitors can improve the binding characteristics (Fig. 5), the idea which has been supported by the docking studies where compound 8a can form hydrogen bonds with Asp93 leading to an increased activity (Fig. 7c). Another point of agreement between 3D-QSAR and docking studies is that the hydrophobic-interacting R5 region in the counter map can be mimicked by the residues Ala62 and Ser63, which are in hydrophobic contact with functional groups responsible for the generation of R5 region (Fig. 7). Taking all these information into account, it can be concluded that the interaction mode presented in the current study may explain a logical way of ligand–protein complex formation. This binding mode is somewhat different from that shown in the crystallographic data [26], where the introduced binding pocket on TNFR1 possesses less than ten residues and, therefore, rarely can be considered as a binding site [54]. Moreover, the LigPlot+ analysis of the solved structure of IV703 bound to TNFR1 revealed no interaction suggesting that the binding mode of IV703 in crystal structure (PDB code: 1FT4) may be the artifact of crystallization not representing the true ligand-TNFR1 interaction.

3D-represantation of the inhibitors docked into TNFR1. a IV703, b compound 45, c compound 8a, and d compound 33a. PyMol program (version 1.7.0.0) was used to generate the images

2D-representation of interactions of docked IV703 (a), compounds 45 (b), 8a (a), and 33a (a) into the binding site of TNFR1 generated by LigPlot+ program
Conclusion
In the current work, we aimed to build a 3D-QSAR predictive model for a series of TNFR1 inhibitors. The validity of the developed model was evaluated by the internal and external cross-validation methods. The generated model has reasonable statistical parameters in terms of predictivity. Structural similarity searching of PubChem chemical structure database using potent TNFR1 inhibitor as the query structure identified 52 compounds of which compounds 8a and 33a showed adequate predicted anti-TNFR1 inhibitory activities. Using 3D-QSAR PLS contour maps and docking studies, hydrophobic interactions and hydrogen bonds were identified as the key elements in ligand–receptor interactions. The potent compounds from the data set used for QSAR model development and those identified by database similarity search showed similar docking poses on the receptor adding to the validity of the proposed model. The result of this work can be useful for designing novel potent TNFR1 inhibitors where blocking of TNFR1 is desired.
References
E.A. Carswell, L.J. Old, R.L. Kassel, S. Green, N. Fiore, B. Williamson, An endotoxin induced serum factor that cuases necrosis of tumors. Proc. Natl. Acad. Sci. USA. 72(9), 3666–3670 (1975)
I.A. Clark. How TNF was recognized as a key mechanism of disease. Cytokine Growth Factor Rev. 18(3–4), 335–343 (2007)
A. Ceramil, B. Beutler, The role of cachectin/TNF in endotoxic shock and cachexia. Immunol. Today. 9(1), 28–31 (1988)
P. Vandenabeele, W. Declercq, R. Beyaert, W. Fiers, Two tumour necrosis factor receptors: structure and function. Trends Cell. Biol. 5(10), 392–399 (1995)
K.J. Tracey, A. Cerami, Metabolic responses to cachectin/TNF. A brief review. Ann. N. Y. Acad. Sci. 587, 325–331 (1990)
M.J. Elliott, R.N. Maini, M. Feldmann, J.R. Kalden, C. Antoni, J.S. Smolen et al., Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor α (cA2) versus placebo in rheumatoid arthritis. Lancet. 344(8930), 1105–1110 (1994)
M.A. Palladino, F.R. Bahjat, E.A. Theodorakis, L.L. Moldawer, Anti-TNF-α therapies: the next generation. Nat. Rev. Drug Discov. 2(9), 736–746 (2003)
W.J. Sandborn, S.B. Hanauer, S. Katz, M. Safdi, D.G. Wolf, R.D. Baerg et al., Etanercept for active Crohn’s disease: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 121(5), 1088–1094 (2001)
D. Tracey, L. Klareskog, E.H. Sasso, J.G. Salfeld, P.P. Tak, Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol. Therapeutics. 117(2), 244–279 (2008)
J.J. Gómez-Reino, L. Carmona, V. Rodríguez Valverde, E.M. Mola, M.D. Montero, Treatment of rheumatoid arthritis with tumor necrosis factor inhibitors may predispose to significant increase in tuberculosis risk: a multicenter active-surveillance report. Arthritis Rheum. 48(8), 2122–2127 (2003)
J.S. Lubel, A.G. Testro, P.W. Angus, Hepatitis B virus reactivation following immunosuppressive therapy: Guidelines for prevention and management. Int. Med. J. 37(10), 705–712 (2007)
C. Antoni, J. Braun, Side effects of anti-TNF therapy: current knowledge. Clin. Exp. Rheumatol. 20(6 SUPPL. 28), S-152-S-7 (2002)
S.A. Devos, N. Van Den Bossche, M. De Vos, J.M. Naeyaert, Adverse skin reactions to anti-TNF-alpha monoclonal antibody therapy. Dermatology. 206(4), 388–390 (2003)
R. Wolf, H. Matz, E. Orion, V. Ruocco, Anti-TNF therapies—the hope of tomorrow. Clin. Dermatol. 20(5), 522–530 (2002)
D. Faustman, M. Davis, TNF receptor 2 pathway: drug target for autoimmune diseases. Nat. Rev. Drug Discov. 9(6), 482–493 (2010)
R.M. Locksley, N. Killeen, M.J. Lenardo, The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell. 104(4), 487–501 (2001)
M. Leist, F. Gantner, S. Jilg, A. Wendel, Activation of the 55 kDa TNF receptor is necessary and sufficient for TNF- induced liver failure, hepatocyte apoptosis, and nitrite release. J. Immunol. 154(3), 1307–1316 (1995)
L. Mori, S. Iselin, G. De Libero, W. Lesslauer, Attenuation of collagen-induced arthritis in 55-kDa TNF receptor type 1 (TNFR1)-IgG1-treated and TNFR1-deficient mice. J. Immunol. 157(7), 3178–3182 (1996)
M.L. Olleros, R. Guler, N. Corazza, D. Vesin, H.P. Eugster, G. Marchal et al., Transmembrane TNF induces an efficient cell-mediated immunity and resistance to Mycobacterium bovis bacillus Calmette-Guérin infection in the absence of secreted TNF and lymphotoxin-α. J. Immunol. 168(7), 3394–3401 (2002)
B.M. Saunders, S. Tran, S. Ruuls, J.D. Sedgwick, H. Britton, B.J. Warwick, Transmembrane TNF is sufficient to initiate cell migration and granuloma formation and provide acute, but not long-term, control of Mycobacterium tuberculosis infection. J. Immunol. 174(8), 4852–4859 (2005)
Y. Mukai, T. Nakamura, M. Yoshikawa, Y. Yoshioka, S.I. Tsunoda, S. Nakagawa et al., Solution of the structure of the TNF-TNFR2 complex. Sci. Signal. 3(148), ra83 (2010)
T. Banno, A. Gazel, M. Blumenberg, Effects of tumor necrosis factor-α (TNFα) in epidermal keratinocytes revealed using global transcriptional profiling. J. Biol. Chem. 279(31), 32633–32642 (2004)
M.F. Rodrigues, C. Alves, B. Figueiredo, A.B. Rezende, S. Wohlres-Viana, Silva VLd et al., Tumour necrosis factor receptors and apoptosis of alveolar macrophages during early infection with attenuated and virulent Mycobacterium bovis. Immunology. 139(4), 503–512 (2013)
K.A. Zettlitz, V. Lorenz, K. Landauer, S. Münkel, A. Herrmann, P. Scheurich et al., ATROSAB, a humanized antagonistic anti-tumor necrosis factor receptor one-specific antibody. mAbs. 2(6), 639–647 (2010)
F.E. McCann, D.P. Perocheau, G. Ruspi, K. Blazek, M.L. Davies, M. Feldmann et al., Selective tumor necrosis factor receptor i blockade is antiinflammatory and reveals immunoregulatory role of tumor necrosis factor receptor II in collagen-induced arthritis. Arthritis Rheumatol. 66(10), 2728–2738 (2014)
P.H. Carter, P.A. Scherle, J.A. Muckelbauer, M.E. Voss, R.Q. Liu, L.A. Thompson et al., Photochemically enhanced binding of small molecules to the tumor necrosis factor receptor-1 inhibits the binding of TNF-α. Proc. Natl. Acad. Sci. USA. 98(21), 11879–11884 (2001)
M.E. Voss, P.H. Carter, A.J. Tebben, P.A. Scherle, G.D. Brown, L.A. Thompson et al., Both 5-arylidene-2-thioxodihydropyrimidine-4,6(1H,5H)-diones and 3-thioxo-2,3-dihydro-1H-imidazo[1,5-a]indol-1-ones are light-dependent tumor necrosis factor-α antagonists. Bioorg. Med. Chem. Lett. 13(3), 533–538 (2003)
J. Tong, Y. Wu, M. Bai, P. Zhan, 3D-QSAR and molecular docking studies on HIV protease inhibitors. J. Mol. Struct. 1129, 17–22 (2017)
A. Ghaleb, A. Aouidate, M. Ghamali, A. Sbai, M. Bouachrine, T. Lakhlifi, 3D-QSAR modeling and molecular docking studies on a series of 2,5 disubstituted 1,3,4-oxadiazoles. J. Mol. Struct. 1145, 278–284 (2017)
R.D. Cramer Iii, D.E. Patterson, J.D. Bunce, Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 110(18), 5959–5967 (1988)
P.J. Goodford, A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 28(7), 849–857 (1985)
R.C. Wade, K.J. Clark, P.J. Goodford, Further development of hydrogen bond functions for use in determining energetically favorable binding sites on molecules of known structure. 1. Ligand probe groups with the ability to form two hydrogen bonds. J. Med. Chem. 36(1), 140–147 (1993)
R.C. Wade, P.J. Goodford, Further development of hydrogen bond functions for use in determining energetically favorable binding sites on molecules of known structure. 2. Ligand probe groups with the ability to form more than two hydrogen bonds. J. Med. Chem. 36(1), 148–156 (1993)
M. Baroni, G. Costantino, G. Cruciani, D. Riganelli, R. Valigi, S. Clementi, Generating Optimal Linear PLS Estimations (GOLPE): An Advanced Chemometric Tool for Handling 3D-QSAR Problems. Quant. Struct. Activ. Relationsh. 12(1), 9–20 (1993)
HyperChem(TM) Professional 8.0.8; Hypercube, Inc, 1115 NW 4th Street, Gainesville, Florida 32601, USA, (2010)
C.B. Xue, X.T. Chen, X. He, J. Roderick, R.L. Corbett, B. Ghavimi et al., Synthesis and structure-activity relationship of a novel sulfone series of TNF-α converting enzyme inhibitors. Bioorg. Med. Chem. Lett. 14(17), 4453–4459 (2004)
N.L. Allinger. Conformational, analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 99(25), 8127–8134 (1977)
Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group Inc, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7 (2016)
P. Goodford, GRID Molecular Discovery Ltd, Oxford, England (1995)
GOLPE 4.5, Multivariate Infometric Analysis; Srl., Viale dei Castagni 16 (Perugia, Italy, 1999)
M. Pastor, G. Cruciani, S. Clementi, Smart region definition: a new way to improve the predictive ability and interpretability of three-dimensional quantitative structure-activity relationships. J. Med. Chem. 40(10), 1455–1464 (1997)
G. Cruciani, K.A. Watson, Comparative molecular field analysis using GRID force-field and GOLPE variable selection methods in a study of inhibitors of glycogen phosphorylase b. J. Med. Chem. 37(16), 2589–2601 (1994)
K. Roy, S. Kar, P. Ambure, On a simple approach for determining applicability domain of QSAR models. Chemometr. Intell. Lab. Syst. 145, 22–29 (2015)
A.A. Alizadeh, M. Hamzeh-Mivehroud, B. Sokouti, S. Dastmalchi, An alignment-independent 3D-QSAR study on series of hydroxamic acid-based tumor necrosis factor-α converting enzyme inhibitors. J. Chemometr. 30(9), 537–547 (2016)
G. Jones, P. Willett, R.C. Glen, Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 245(1), 43–53 (1995)
G. Jones, P. Willett, R.C. Glen, A.R. Leach, R. Taylor, Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267(3), 727–748 (1997)
R.A. Laskowski, M.B. Swindells, LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inform. Model. 51(10), 2778–2786 (2011)
G.L. Wilson, M.A. Lill, Integrating structure-based and ligand-based approaches for computational drug design. Future Med. Chem. 3(6), 735–750 (2011)
C.H. Lee, H.C. Huang, H.F. Juan, Reviewing ligand-based rational drug design: the search for an ATP synthase inhibitor. Int. J. Mol. Sci. 12(8), 5304–5318 (2011)
O. Nicolotti, D. Gadaleta, G.F. Mangiatordi, M. Catto, A. Carotti, Applicability domain for QSAR models: where theory meets reality. Int. J. Quant. Struct. Prop. Relationsh. (IJQSPR). 1(1), 45–63 (2016) (2379–7487)
A. Golbraikh, A. Tropsha, Beware of q2! J. Mol. Graph. Model. 20(4), 269–276 (2002)
K. Roy, R.N. Das, P. Ambure, R.B. Aher, Be aware of error measures. Further studies on validation of predictive QSAR models. Chemometr. Intell. Lab. Syst. 152, 18–33 (2016)
C. Tintori, M. Magnani, S. Schenone, M. Botta, Docking, 3D-QSAR studies and in silico ADME prediction on c-Src tyrosine kinase inhibitors. Eur. J. Med. Chem. 44(3), 990–1000 (2009)
M. Gao, J. Skolnick, A comprehensive survey of small-molecule binding pockets in proteins. PLoS Comput. Biol. 9(10), e1003302 (2013)
Acknowledgements
The authors would like to thank the Research Office and Biotechnology Research Center of Tabriz University of Medical Sciences for providing financial and facility support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sharifi, M., Alizadeh, A.A., Hamzeh-Mivehroud, M. et al. Computational explorations to gain insight into the structural features of TNF-α receptor I inhibitors. J IRAN CHEM SOC 15, 2519–2531 (2018). https://doi.org/10.1007/s13738-018-1440-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-018-1440-x