
Abstract
Immune checkpoint inhibitors (ICIs) have become the standard treatment for many types of cancer and have improved patient prognosis. However, ICIs upregulate the immune system against tumors, leading to immune-related adverse events (irAEs). Kidney irAEs are less common, and most of them are acute tubulointerstitial nephritis (ATIN). However, there has been a recent increase in recognition of glomerular disease related to ICI therapies. We report the case of a 65-year-old man with lung adenocarcinoma who was treated with pembrolizumab (a monoclonal antibody targeting programmed cell death protein-1 [PD-1]). Pembrolizumab was discontinued after seven cycles due to the development of destructive thyroiditis. Within three months of discontinuing the pembrolizumab treatment, the patient developed rapid progressive glomerulonephritis (RPGN), liver dysfunction, and dysgeusia. The patient underwent renal biopsy and was diagnosed with crescentic glomerulonephritis due to anti-glomerular basement membrane (GBM) antibodies complicated with membranous nephropathy (MN) and ATIN. Treatment with systemic corticosteroids resulted in a favorable clinical response. Various ICI-associated glomerular diseases have been described; however, this is the first reported case of anti-GBM glomerulonephritis associated with MN and ATIN following ICI treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Immune checkpoint inhibitor (ICI) therapies have dramatically changed cancer treatment and have improved the prognosis of many patients. However, the same mechanisms that activate antitumor immunity can lead to adverse effects, which are characterized as immune-related adverse events (irAEs). Several organs are affected by irAEs; however, adverse kidney effects are rare [1]. The incidence of kidney irAEs with monotherapy with ICIs is 2%, while that with combination therapy approaches 5% [1]. Although the most common pathology of kidney irAEs is acute kidney injury (AKI) due to acute tubulointerstitial nephritis (ATIN), there are increasing reports of glomerular diseases related to ICI therapies [2]. Kithlu et al. reviewed 45 cases of biopsy-confirmed ICI-related glomerular diseases and reported that the most frequent types were pauci-immune glomerulonephritis and renal vasculitis (27%), podocytopathies (24%), and complement 3 glomerulonephritis (11%) [3]. Concurrent ATIN has been reported in 41% of the cases [3].
Herein, we report the first case of a patient who developed anti-glomerular basement membrane (GBM) glomerulonephritis and membranous nephropathy (MN) with ATIN after treatment with pembrolizumab.
Case report
A 65-year-old Japanese man presented to our hospital with rapid progressive glomerulonephritis (RPGN) in May 2021. He was diagnosed with lung adenocarcinoma in August 2020 and was administered pembrolizumab (4 mg/kg) every three weeks until October 2020. At this time, his kidney function was normal (creatinine = 0.59 mg/dL). After two cycles, his urinalysis revealed hematuria. Pembrolizumab was discontinued after seven cycles due to the development of destructive thyroiditis in February 2021. Proteinuria was observed in March 2021. He noticed dysgeusia, and laboratory tests indicated an elevated C-reactive protein (CRP) level (8.93 mg/dL) in April 2021. Computed tomography (CT) performed then did not show any remarkable changes, which could explain the elevation in CRP levels. In May 2021, his loss of appetite progressed, and laboratory results indicated acute kidney injury and liver dysfunction. The patient was referred to the Nephrology Department. He had no personal or family history of chronic kidney disease. The patient denied exposure to nephrotoxic agents except for pembrolizumab. He was taking levothyroxine (75 μg) for destructive thyroiditis. His blood pressure was 146/87 mmHg. The results of the remaining physical examinations were normal. Laboratory findings indicated an elevated serum creatinine level (2.6 mg/dL) with a urinary protein-to-creatinine ratio of 2.22 g/g, an alkaline phosphatase level of 412 U/L, an aspartate aminotransferase level of 73 U/L, an alanine aminotransferase level of 125 U/L, and an elevated CRP level of 27.39 mg/dL. Urinalysis revealed the presence of dysmorphic erythrocytes and granular casts. Serologic testing was weakly positive for anti-GBM antibody (5 U/mL), positive for antinuclear antibody (homogeneous and speckled pattern 160 times) and lupus anticoagulant (1.4 s), but negative for specific antibodies for systemic lupus erythematosus, such as anti-double strand DNA antibody and anti-Smith antibody. The complementary levels were normal. The markers of other vasculitis types, such as myeloperoxidase antineutrophil cytoplasmic antibody (ANCA) and proteinase 3-ANCA, were negative. Tubular markers were slightly increased (urine N-acetyl-beta-D-glucosaminidase, 52.3 IU/L; urine neutrophil gelatinase-associated lipocalin, 85 μg/g Cre). CT scan of the abdomen revealed a slight swelling in the kidneys bilaterally. RPGN and liver dysfunction were suspected as irAEs, and the patient was treated with pulse intravenous methylprednisolone (500 mg daily for three days), followed by 1 mg/kg/day of prednisolone.
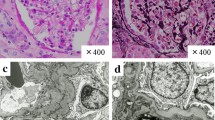
A renal biopsy from his left kidney performed five days after admission showed diffuse interstitial infiltration of lymphocytes at lower magnification (Fig. 1A) and diffuse crescent formation (14 of 29 glomeruli with cellular crescents and 8 of 29 with fibrocellular crescents) (Fig. 1B). Tubular atrophy and severe interstitial inflammation were seen around a normal glomerulus (Fig. 1C). Membrane alterations (spikes and thickening) were not visible by light microscopy. Immunofluorescence revealed linear and partially granular staining of the glomerular capillary loops for IgG (2 +), IgA (1 +), and C3 (1 +) (Fig. 2A). IgG1 and IgG3 were predominantly stained in glomerular capillary loops (Fig. 2B). Immunofluorescence results were negative for glomerular phospholipase A2 receptor (PLA2R) expression (data not shown). Electron microscopy revealed electron-dense deposits in the subepithelial and mesangial regions, thickening and irregularity of the GBM, and extensive podocyte foot process effacement (Fig. 3A, B). Based on the above findings, the patient was diagnosed with crescentic glomerulonephritis due to anti-GBM antibodies complicated with MN and ATIN.

Pathological findings of renal biopsy A The view at lower magnification shows diffuse interstitial infiltration of lymphocytes. (original magnification: 40x; Masson’s trichrome staining). B The representative glomerulus shows the entire circumference of cellular crescents that disrupt the Bowman capsule. (periodic acid-Schiff (PAS), original magnification: 200x). C Tubular atrophy and severe interstitial inflammation are shown around a normal glomerulus. (PAS, original magnification: 200x)

Immunofluorescence microscopy findings of renal biopsy A Immunofluorescence microscopy demonstrates that IgG is linear and partially granular stained, and IgA and C3 are granular stained in glomerular capillary loops. (original magnification: 400x) B Immunofluorescence microscopy demonstrates that IgG1 and IgG3 are dominantly stained in glomerular capillary loops. (original magnification: 400x)

Electron microscopy findings of renal biopsy (A, B). Electron microscopy reveals electron-dense deposits at the subepithelial and mesangial regions (arrow), thickened and irregular GBM, and extensive podocyte foot process effacement. (original magnification: 6000x)
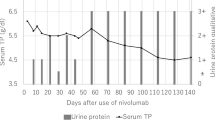
The clinical course of the patient is shown in Fig. 4. Although his creatinine levels eventually increased to a peak of 3 mg/dL, after nine months it improved to 1.54 mg/dL, and active urinary sediment resolved with a reduced protein-to-creatinine ratio of 0.45 g/g. The prednisolone dose was successfully tapered to 2.5 mg/day. The levels of CRP and hepatobiliary enzymes immediately decreased after corticosteroid initiation, and he gained appetite. In October 2021, CT scans showed that his primary lung cancer had enlarged; thus, carboplatin and paclitaxel were initiated.

The patient’s clinical course We set admission day at zero. mPSL methylprednisolone, PSL prednisolone, CBDCA + PTX carboplatin and paclitaxel
Discussion
Glomerular diseases with several pathologies related to kidney irAEs have been reported in the last few years. There are only three cases of anti-GBM glomerulonephritis and only two cases of MN associated with ICIs [4,5,6,7,8]; however, none of the cases of anti-GBM glomerulonephritis are accompanied by MN. To the best of our knowledge, this is the first report with such a condition. Anti-GBM glomerulonephritis typically presents with RPGN with or without pulmonary hemorrhage. Its histopathological features are linear GBM staining for IgG by immunofluorescence and diffuse crescentic glomerulonephritis affecting more than half of the glomeruli by light microscopy. The target antigen has been identified as the non-collagenous domain of the α3 chain of type IV collagen in GBM. In our case, the serum anti-GBM antibody was weakly positive; however, we diagnosed anti-GBM glomerulonephritis with diffuse crescentic formation and linear IgG staining. Nasr et al. reported 20 cases of atypical anti-GBM glomerulonephritis typified by linear GBM staining for immunoglobulins but without a diffuse crescentic formation and serum anti-GBM antibody [9]. The clinical features of this type of glomerulonephritis include hematuria, proteinuria, and mild renal insufficiency (mean serum creatinine, 2.2 mg/dL), without pulmonary hemorrhage. In histopathological aspects, all cases showed mesangial and/or endocapillary hypercellularity, and focal crescents and/or fibrinoid necrosis were seen in 40% of cases. One of the three reports of anti-GBM glomerulonephritis related to irAEs was an atypical type [6]. They reported that after half a year of anti-CTLA-4 blockage therapy, the patient developed focal crescentic, proliferative, and sclerosing glomerulonephritis with linear IgG deposition without serum anti–GBM antibody. Oral cyclophosphamide and corticosteroids were initiated, which stabilized his renal function. Our case and Kyriazis’s case [6] are similar in many aspects, such as low titer of anti-GBM antibody and good renal prognosis after ICI therapy, but differ in the formation of diffuse and focal crescents. Our case is unique in having a good renal prognosis in spite of diffuse crescent formations. A systemic review of glomerular disease related to irAE showed that only two out of 12 patients with pauci-immune glomerulonephritis had positive ANCA serology [3]. According to previous reports, glomerulonephritis related to irAEs is often serologically negative for ANCA or anti-GBM antibodies. One possible reason is that the autoantibodies acquired by ICI administration may have a different epitope than the typical ones.
The staining of IgG subclasses was useful for our diagnosis because of the combination of MN and anti-GBM glomerulonephritis. Human IgG is categorized into four subclasses (IgG1, IgG2, IgG3, and IgG4). Qu Z et al. reported the distribution of IgG subclass deposition in renal biopsy specimens from 46 patients with anti-GBM glomerulonephritis [10]. Anti-GBM IgG3 was detected in all patients (100%), IgG2 in 47.8%, IgG1 in 19.6%, and IgG4 in 15.2%. They also found that patients with strong linear staining of IgG3 along the GBM presented with lower creatinine levels than those with weak IgG3 staining. Although not statistically significant, the levels of anti-GBM antibodies also appeared to be lower in patients with strong IgG3 deposits than in those with weak IgG3 deposits. These features apply to our case, which predominantly showed IgG1 and IgG3 deposition along the GBM, low levels of serum creatinine, and weakly positive anti-GBM antibodies. Zhao J et al. demonstrated that subclasses of anti-GBM IgG were not restricted to specific types in the sera of anti-GBM patients, but the frequency of IgG1 and IgG3 increased while renal function decreased [11]. These results suggest that IgG1 and IgG3 subclasses are associated with disease severity. In particular, IgG3 binds complement 1q and mediates complement-associated inflammatory response [12]. The antibodies detected on primary MN, such as PLA2R and thrombospondin type 1 domain–containing 7A, belong to the IgG4 subclass [13]. The predominant deposition of IgG1 and IgG3 also indicates that MN concurrent with anti-GBM glomerulonephritis is secondary MN. The negative staining of the anti-PLA2R antibody in our case was consistent with these findings.
At the tissue level, cancer cells avoid an immune response through the expression of ligands such as programmed cell death protein ligand-1 (PD-L1) and PD-L2, which deactivate T-cells by binding to PD-1 receptors expressed on their surface. The PD-1 inhibitors, which interrupt the interaction between PD-L1 and the T-cell PD-1 receptor, allow activated T-cells to identify and destroy cancer cells [2]. Several studies have revealed that T-cell-mediated cellular immune responses may be the most crucial mediators inducing anti-GBM glomerulonephritis in humans and mice [14,15,16,17,18]. T-cell activation caused by PD-1 inhibitors is likely to trigger anti-GBM glomerulonephritis.
Our case is the first report of combined anti-GBM glomerulonephritis and MN associated with ICI therapy, however, the combination of these two diseases has already been documented. Jia X et al. reported that eight patients with combined anti-GBM glomerulonephritis and MN presented significantly lower levels of serum creatinine at diagnosis, a lower level of oliguria/anuria and gross hematuria, and a higher level of urinary protein excretion compared with anti-GBM glomerulonephritis without MN [19]. Although the difference was not significant, biopsies from anti-GBM glomerulonephritis patients with MN showed fewer crescents in the glomeruli. This could be related to the fact that patients with MN had significantly better renal outcomes than those without. They reported that 62.5% of patients with MN recovered their renal function after treatment, compared to 86.7% of patients with anti-GBM glomerulonephritis without MN progressing to end-stage kidney disease. In 2021, Ahamad et al. reported that only two of the 12 anti-GBM glomerulonephritis patients with MN recovered their renal function, seven remained dialysis-dependent, and three required kidney transplantation [20]. The reasons why the renal outcome was different between the two reports were that the patients reported by Jia et al. were younger than those reported by Ahamad et al. (32 vs. 55 years old) and their creatinine levels on diagnosis were lower (5.2 vs. 9.8 mg/dL in the Ahamad et al. group). Two reports commonly documented that the positive ratio of PLA2R staining in renal biopsies was low (1/4 in the Jia et al. group and 1/5 in the Ahamad et al. group). Our patient was also negative for PLA2R staining. This feature could indicate that MN combined with anti-GBM glomerulonephritis occurs secondary to GBM damage, leading to epitope exposure and subsequent formation of antibodies. However, in previous report [20] and in our case the stages of MN were 1–2 which suggests that MN preceded anti-GBM glomerulonephritis. In our case, it is possible that secondary MN was caused by lung cancer. Constant follow-up after ICI therapy and early corticosteroid induction against irAE accompanied by liver dysfunction and dysgeusia may have led to good renal outcomes in our patient.
In our case, whether anti-GBM glomerulonephritis was related to ICI therapy remains inconclusive because it occurred three months after the completion of ICI therapy. In a study, Kichlu et al. showed that the median time to initiation of ICI therapies to diagnose the secondary glomerular disease as an irAE was three months; however, in a few cases, it was even more than three months after the therapy had been discontinued [3]. Nevertheless, whether ICI therapies create long-term risks for irAEs is unknown.
Here, we report a case of anti-GBM glomerulonephritis accompanied by MN and ATIN that occurred three months after ICI therapy. Staining of the IgG subclass and the findings of electron microscopy were useful for diagnosis. Early initiation of corticosteroids results in favorable renal outcomes.
References
Cortazar FB, Marrone KA, Troxell ML, et al. Clinicopathological features of acute kidney injury associated with immune checkpoint inhibitors. Kidney Int. 2016;90:638–47.
Perazella MA, Shirali AC. Immune checkpoint inhibitor nephrotoxicity: what do we know and what should we do? Kidney Int. 2020;97:62–74.
Kitchlu A, Jhaveri KD, Wadhwani S, et al. A systematic review of immune checkpoint inhibitor-associated glomerular disease. Kidney Int Rep. 2021;6:66–77.
Sammartino C, Goodman D, Flanagan G, Hill P. Anti-GBM disease following CTLA4 blockade in a patient with metastatic melanoma. Clin Kidney J. 2010;3:135–7.
Takahashi N, Tsuji K, Tamiya H, et al. Goodpasture’s disease in a patient with advanced lung cancer treated with nivolumab: an autopsy case report. Lung Cancer. 2018;122:22–4.
Kyriazis P, Tiwary A, Freeman J, et al. Atypical anti-glomerular basement membrane glomerulonephritis in a patient with metastatic melanoma treated with mitogen-activated protein kinase and immune checkpoint inhibitors: a case report. J Med Case Rep. 2021;15:186.
Chen M, Zhang L, Zhong W, et al. Case report: THSD7A-positive membranous nephropathy caused by Tislelizumab in a lung cancer patient. Front Immunol. 2021;12:619147.
Lin JS, Wang DY, Mamlouk O, et al. Immune checkpoint inhibitor associated reactivation of primary membranous nephropathy responsive to rituximab. J Immunother Cancer. 2020;8:e001287. https://doi.org/10.1136/jitc-2020-001287.
Nasr SH, Collins AB, Alexander MP, et al. The clinicopathologic characteristics and outcome of atypical anti-glomerular basement membrane nephritis. Kidney Int. 2016;89:897–908.
Qu Z, Cui Z, Liu G, Zhao MH. The distribution of IgG subclass deposition on renal tissues from patients with anti-glomerular basement membrane disease. BMC Immunol. 2013;14:19.
Zhao J, Yan Y, Cui Z, et al. The immunoglobulin G subclass distribution of anti-GBM autoantibodies against rHalpha3(IV)NC1 is associated with disease severity. Hum Immunol. 2009;70:425–9.
Hamilton RG. Human IgG subclass measurements in the clinical laboratory. Clin Chem. 1987;33:1707–25.
Cattran DC, Brenchley PE. Membranous nephropathy: integrating basic science into improved clinical management. Kidney Int. 2017;91:566–74.
Robertson J, Wu J, Arends J, et al. Characterization of the T-cell epitope that causes anti-GBM glomerulonephritis. Kidney Int. 2005;68:1061–70.
Huang XR, Tipping PG, Apostolopoulos J, et al. Mechanisms of T cell-induced glomerular injury in anti-glomerular basement membrane (GBM) glomerulonephritis in rats. Clin Exp Immunol. 1997;109:134–42.
Kitching AR, Huang XR, Ruth AJ, et al. Effects of CTLA4-Fc on glomerular injury in humorally-mediated glomerulonephritis in BALB/c mice. Clin Exp Immunol. 2002;128:429–35.
Reynolds J, Tam FWK, Chandraker A, et al. CD28-B7 blockade prevents the development of experimental autoimmune glomerulonephritis. J Clin Invest. 2000;105:643–51.
Salama AD, Chaudhry AN, Holthaus KA, et al. Regulation by CD25+ lymphocytes of autoantigen-specific T-cell responses in Goodpasture’s (anti-GBM) disease. Kidney Int. 2003;64:1685–94.
Jia XY, Hu SY, Chen JL, et al. The clinical and immunological features of patients with combined anti-glomerular basement membrane disease and membranous nephropathy. Kidney Int. 2014;85:945–52.
Ahmad SB, Santoriello D, Canetta P, et al. Concurrent anti-glomerular basement membrane antibody disease and membranous nephropathy: a case series. Am J Kidney Dis. 2021;78:219-225.e1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared that no conflicts of interest exist.
Human and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Hoshina, A., Endo, S. Anti-glomerular basement membrane glomerulonephritis concurrent with membranous nephropathy and acute tubular interstitial nephritis in a lung cancer patient treated with pembrolizumab. CEN Case Rep 12, 230–236 (2023). https://doi.org/10.1007/s13730-022-00750-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13730-022-00750-x