
Abstract
Mutations in the ciliary gene TTC21B, NPHP4, and CRB2 cause familial focal and segmental glomerulosclerosis (FSGS). We report a girl with a mutation of the ciliary gene CC2D2A presenting with FSGS and nephronophthisis. The patient had mental retardation, postaxial polydactyly, and ataxic breathing, and was diagnosed as having compound heterozygous CC2D2A missense mutations at age 5. Retrospectively, azotemia at 1 year and proteinuria at 5 years were recorded but not investigated. At age 6, she was referred to the pediatric nephrology service because of hypertension, pretibial pitting edema, heavy proteinuria, and hematuria. eGFR was 66 ml/min/1.73 m2, total protein 5.3 g/dl, albumin 2.4 g/dl, and cholesterol 317 mg/dl. Ultrasonography showed normal-sized kidneys with a cyst in the right. Losartan was started. On renal biopsy, 8 out of 24 glomeruli were globally sclerosed, and three showed segmental sclerosis and/or hyalinosis with no immune deposits. Mild tubular dilatation, tubular atrophy, and interstitial fibrosis were observed. On electron microscopy, glomeruli showed focal foot process effacement with no electron dense deposits. Since losartan did not exert an obvious effect, treatment with prednisolone was tried. Urine protein decreased from 6.6 to 3.7 g/gCr. Prednisolone was discontinued after 10 days, however, because she developed duodenal ulcer perforation that necessitated omentoplasty. Subsequently, she was treated with losartan only. Her renal function deteriorated and peritoneal dialysis was initiated 8 months later. FSGS in this patient could be primary glomerular associated with CC2D2A mutation, rather than the consequences of tubulointerstitial fibrosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It has been reported that mutations in the ciliary gene TTC21B, NPHP4, and CRB2 cause familial focal segmental glomerulosclerosis (FSGS) [1,2,3]. We report a girl with a mutation of a ciliary gene CC2D2A (encoding coiled-coil and C2 domain-containing protein 2A) presenting with FSGS and nephronophthisis. This is another ciliary gene, the mutation of which is associated with FSGS.
Case report
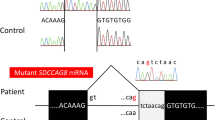
The patient is a 6-year-old girl with mental retardation, postaxial polydactyly, and ataxic breathing. She was diagnosed as having compound heterozygous CC2D2A missense mutations at age 5 years;
Chr4(GRCh37):g.15512892C>T, NM_001080522.2:c.563C>T, p.(Ala188Val)
Chr4(GRCh37):g.15591190C>G, NM_001080522.2:c.4202C>G, p.(Thr1401Ser)
inherited from each parent At age 6 years, she was referred to pediatric nephrology service because of proteinuria. In retrospect, azotemia at age 1 year {urea nitrogen (UN) 32.5 mg/dl} and proteinuria 3 + at age 5 years were recorded at local hospitals but no investigation was performed. Height was 106 cm (− 1.5 SD), weight was12.6 kg (− 2.3 SD), and blood pressure was 116/53 mmHg. The breathing was ataxic and there was pretibial pitting edema, but other physical examination was unremarkable. Urinalysis showed protein 3+, blood 2+, granular cast+, protein-to-creatinine ratio (pro/Cr) 6.6 g/g, ß2 micro-globulin 112 μg/l, N-acetyl-β-d-glucosaminidase (NAG)/Cr 33 U/g, and selectivity index 0.09. Blood test revealed hemoglobin 13.2 g/dl, UN 23.2 mg/dl, Cr 0.56 mg/dl (eGFR 66 ml/min/1.73 m2), cystatin C 1.34 mg/l, total protein 5.3 g/dl, albumin 2.4 g/dl, cholesterol 317 mg/dl, and uric acid 7.5 mg/dl. Serologic tests for glomerulonephritis were negative. Ultrasonography showed normal-sized kidneys with a cyst in the right. A diagnosis of nephrotic syndrome was made and losartan 12.5 mg/day was started for hypertension and proteinuria. One week later she was hospitalized for renal biopsy. Soon thereafter, she developed acute pyelonephritis due to E. coli. After antibiotic treatment, open renal biopsy was performed. On light microscopy, 8 out of 24 glomeruli were globally sclerosed, and three showed segmental sclerosis and/or hyalinosis (Fig. 1). Mild tubular dilatation, tubular atrophy, and interstitial fibrosis were observed. No immune deposits were seen. On electron microscopy, glomeruli showed focal foot process effacement with no electron dense deposits. Since losartan did not exert an obvious effect, treatment with prednisolone was tried. Urine pro/Cr decreased to 3.7 g/g, but prednisolone was discontinued after 10 days because she developed duodenal ulcer perforation that necessitated omentoplasty. CT scan performed then showed bilateral multiple cysts in the kidney. Since then she had been treated with losartan only. Voiding cystourethrogram showed right grade III and left grade II vesicoueteral reflux and residual urine. Her renal function deteriorated and peritoneal dialysis was initiated 8 months later.

Pathological findings. a Twenty-four glomeruli (eight global sclerosis, three segmental sclerosis and/or hyalinosis) were found in the wedged specimen. Tubular dilation, mild tubular atrophy and interstitial fibrosis were observed. b PAM staining. Segmental sclerosis and/or hyalinosis. Hyper-cellularity was absent. c Electron micrograph showing the presence of foot process effacement. Areas with intact foot processes were also observed (not shown). There was no electron dense deposit
No clinically significant variants in known FSGS genes, including ACTN4, ANLN, ARHGAP24, COL4A3, COL4A4, COL4A5, COL4A6, CHD1L, C3, CFI, CD46, FN1, GATA3, INF2, MYH9, PAX2, PODXL, RET, ROBO2, SALL1, SIX1, SRGAP1, TBX18, TNXB, TRPC6, UMOD, WNT4, and WT1 were detected in the patient [4].
Discussion
We reported a girl with compound heterozygous CC2D2A missense mutations who had nephrotic syndrome due to FSGS and nephronophthisis. Her FSGS was thought to be primary glomerular, based on the clinically overt nephrotic syndrome and on mild tubulointerstitial lesions.
Mutations in ciliary genes including CC2D2A cause a spectrum of overlapping and distinct clinical syndromes. Joubert syndrome, Meckel syndrome, their related disorders, and isolated rod-cone dystrophy have been reported in CC2D2A mutations [5]. Our patient does not fit the phenotype of any of the above, but cystic kidney disease has been associated with CC2D2A mutations. On the other hand, nephrotic syndrome or FSGS has not previously been reported. Méjécase et al. reported two brothers with compound heterozygous CC2D2A mutations with non-syndromic rod-cone dystrophy and nephrotic range proteinuria. However, the proteinuria was ascribed to the concurrent compound heterozygous variants in CUBN, a gene associated with steroid-resistant nephrotic syndrome [6]. The renal biopsy was reported as not compatible with nephronophthisis or glomerulonephritis.
CC2D2A is expressed in brain, prostate, pancreas, kidney, lung, liver, and retina. It is a centrosome–cilia protein essential for the assembly of subdistal appendages, anchoring cytoplasmic microtubules and priming the mother centriole for axoneme biogenesis [7]. In zebrafish, the role of CC2D2A is suggested to be ciliary trafficking [8]. In mammals, CC2D2A is thought to be necessary for primary cilia biogenesis. In case of another ciliary gene TTC21B mutation, podocyte cytoskeletal alteration and the subsequent loss of microtubular maintenance are suggested to be the causes of FSGS [1]. Thus TTC21B-deficient podocytes differentiate normally in vitro, suggesting that the cytoskeletal alterations are independent of the primary cilia defect. In a patient with CRB2 mutation, the expression of CRB2 was reported to be decreased [9]. In zebrafish, gene knock-down of CRB2 grossly disorganized the podocyte foot process architecture. The expression of CC2D2A in podocytes has not been described. A recent study, however, reported a role of CC2D2A as a downstream target of WT1, a master regulator of gene expression in podocytes [10]. Thus CC2D2A is one of WT1-bound genes in podocytes and its binding to WT1 changed dynamically during an adriamycin-induced reparative injury response in a mouse model of nephrotic syndrome.
The pathogenicity of the c.563C>T and c.4202C>G variants was predicted as follows: the combined annotation-dependent depletion (CADD) score corresponded to deleterious results (29.5 and 24.8) [11] and Mutation Taster (both scores with disease causing (prob: 0.999 and 1.000) [12]. Overall, both the allele c.563C>T and c.4202C>G were scored as likely pathogenic (PM1, PM2, PP3, and PP4) according to the standards and guidelines for the interpretation of sequence variants by the American College of Medical Genetics and Genomics [13].
Patients with FSGS caused by genetic mutations are less likely to present with clinically overt nephrotic syndrome compared with those with primary FSGS [14]. Foot process effacement is also helpful to differentiate primary and secondary FSGS. It is non-existent or focal in secondary FSGS including genetic FSGS as in our patient [15]. Secondary or genetic FSGS is usually steroid-resistant. Our patient responded partially, but because of the serious side effect, glucocorticoid was withdrawn. In this regard, there is a report that FSGS secondary to a genetic mutation may respond to calcineurin inhibitors [16]. Beyond their immunosuppressive activity, calcineurin inhibitors exhibit a protective effect on podocyte injury [17]. We discussed their use, but her renal function rapidly deteriorated before we made the decision. Taken together, our patient’s FSGS was thought to be primary glomerular, because her tubulointerstitial lesions were mild. Her FSGS is probably due to podocytopathy associated with CC2D2A mutation in consideration of the literature and the genetic databases.
Patients with ciliopathy, such as autosomal dominant polycystic kidney disease, Bardet–Biedl syndrome, and Joubert syndrome with CSPP1 mutation, are known to be associated with vesicoureteral reflux [18,19,20]. Although not described in patients with CC2D2A mutation, or Meckel syndrome, VUR seen in our patient may be related to cilia dysfunction.
In conclusion, we report a girl with CC2D2A mutation who showed FSGS and nephronophthisis. This is another example of ciliary gene involved in glomerulopathy.
References
Huynh Cong E, Bizet AA, Boyer O, Woerner S, Gribouval O, Filhol E, et al. A homozygous missense mutation in the ciliary gene TTC21B causes familial FSGS. J Am Soc Nephrol. 2014;25(11):2435–43. https://doi.org/10.1681/ASN.2013101126 (Epub 2014 May 29).
Mistry K, Ireland JH, Ng RC, Henderson JM, Pollak MR. Novel mutations in NPHP4 in a consanguineous family with histological findings of focal segmental glomerulosclerosis. Am J Kidney Dis. 2007;50(5):855–64. https://doi.org/10.1053/j.ajkd.2007.08.009.
Lamont RE, Tan WH, Innes AM, Parboosingh JS, Schneidman-Duhovny D, Rajkovic A, et al. Expansion of phenotype and genotypic data in CRB2-related syndrome. Eur J Hum Genet. 2016;24(10):1436–44. https://doi.org/10.1038/ejhg.2016.24. (Epub Mar 23).
Wang M, Chun J, Genovese G, Knob AU, Benjamin A, Wilkins MS, et al. Contributions of rare gene variants to familial and sporadic FSGS. J Am Soc Nephrol. 2019;30(9):1625–40. https://doi.org/10.1681/ASN.2019020152. (Epub 2019 Jul 15).
Barroso-Gil M, Olinger E, Ramsbottom SA, Molinari E, Miles CG, Sayer JA. Update of genetic variants in CEP120 and CC2D2A-With an emphasis on genotype-phenotype correlations, tissue specific transcripts and exploring mutation specific exon skipping therapies. Mol Genet Genomic Med. 2021;00:e1603. https://doi.org/10.1002/mgg3.1603.
Méjécase C, Hummel A, Mohand-Saïd S, Andrieu C, El Shamieh S, Antonio A, et al. Whole exome sequencing resolves complex phenotype and identifies CC2D2A mutations underlying non-syndromic rod-cone dystrophy. Clin Genet. 2019;95(2):329–33. https://doi.org/10.1111/cge.13453 (Epub 2018 Nov 4).
Veleri S, Manjunath SH, Fariss RN, May-Simera H, Brooks M, Foskett TA, et al. Ciliopathy-associated gene Cc2d2a promotes assembly of subdistal appendages on the mother centriole during cilia biogenesis. Nat Commun. 2014;5:4207. https://doi.org/10.1038/ncomms5207.
Chen HY, Kelley RA, Li T, Swaroop A. Primary cilia biogenesis and associated retinal ciliopathies. Semin Cell Dev Biol. 2021;110:70–88. https://doi.org/10.1016/j.semcdb.2020.07.013 (Epub Jul 31).
Udagawa T, Jo T, Yanagihara T, Shimizu A, Mitsui J, Tsuji S, et al. Altered expression of Crb2 in podocytes expands a variation of CRB2 mutations in steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2017;32(5):801–9. https://doi.org/10.1007/s00467-016-3549-4 (Epub 2016 Dec 10).
Ettou S, Jung YL, Miyoshi T, Jain D, Hiratsuka K, Schumacher V, et al. Epigenetic transcriptional reprogramming by WT1 mediates a repair response during podocyte injury. Sci Adv. 2020;6(30):5460. https://doi.org/10.1126/sciadv.abb5460 (eCollection 2020 Jul).
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5. https://doi.org/10.1038/ng.2892 (Epub 014 Feb 2).
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. https://doi.org/10.1038/nmeth.2890.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30 (Epub Mar 5).
Kiffel J, Rahimzada Y, Trachtman H. Focal segmental glomerulosclerosis and chronic kidney disease in pediatric patients. Adv Chronic Kidney Dis. 2011;18(5):332–8. https://doi.org/10.1053/j.ackd.2011.03.005.
Sethi S, Glassock RJ, Fervenza FC. Focal segmental glomerulosclerosis: towards a better understanding for the practicing nephrologist. Nephrol Dial Transpl. 2015;30(3):375–84. https://doi.org/10.1093/ndt/gfu035 (Epub 2014 Mar 2).
Sinha A, Sharma S, Gulati A, Sharma A, Agarwala S, Hari P, et al. Frasier syndrome: early gonadoblastoma and cyclosporine responsiveness. Pediatr Nephrol. 2010;25(10):2171–4. https://doi.org/10.1007/s00467-010-1518-x (Epub 2010 Apr 24).
Mallipattu SK, He JC. The podocyte as a direct target for treatment of glomerular disease? Am J Physiol Renal Physiol. 2016;311(1):F46-51. https://doi.org/10.1152/ajprenal.00184.2016 (Epub 2016 Apr 20).
Koslowe O, Frank R, Gauthier B, Vergara M, Trachtman H. Urinary tract infections, VUR, and autosomal dominant polycystic kidney disease. Pediatr Nephrol. 2003;18(8):823–5. https://doi.org/10.1007/s00467-003-1211-4 (Epub 2003 Jun 17).
Marchese E, Ruoppolo M, Perna A, Capasso G, Zacchia M. Exploring key challenges of understanding the pathogenesis of kidney disease in Bardet–Biedl syndrome. Kidney Int Rep. 2020;5(9):1403–15. https://doi.org/10.1016/j.ekir.2020.06.017. (eCollection 2020 Sep)
Akizu N, Silhavy JL, Rosti RO, Scott E, Fenstermaker AG, Schroth J, et al. Mutations in CSPP1 lead to classical Joubert syndrome. Am J Hum Genet. 2014;94(1):80–6. https://doi.org/10.1016/j.ajhg.2013.11.015 (Epub Dec 19).
Acknowledgements
We thank Ms. Chika Kanoe, Ms. Mineko Fujiwara, Ms. Yumi Obayashi, and Ms. Keiko Tsukue who have provided technical assistance.
Funding
This work was supported by Initiative on Rare and Undiagnosed Diseases (Grant number JP21ek0109549) from the Japan Agency for Medical Research and Development.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human and animal rights
This article does not describe any studies with human participants or animals performed by any of the authors.
Consent for publication
Informed consent was obtained from the parents.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Awazu, M., Yamada, M., Asada, N. et al. A girl with a mutation of the ciliary gene CC2D2A presenting with FSGS and nephronophthisis. CEN Case Rep 11, 116–119 (2022). https://doi.org/10.1007/s13730-021-00640-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13730-021-00640-8