
Abstract
Polymeric nanoparticles, based on amphiphilic copolymers, have been extensively evaluated as a potential material for drug delivery systems (DDS), because they present a clear capability of forming micelles under an aqueous medium. Besides the amphiphilic properties, the thermosensitivity of polymers under aqueous media has attracted great attention for DDS. In this work, a well-defined amphiphilic poly(ε-caprolactone)-b-poly(N-isopropylacrylamide) (PCL-b-PNIPAAm) was used to prepare thermosensitive micelles as potential candidates for applications in drug delivery systems. First, hydroxyl-terminated PCL (PCL-OH) was synthesized by ROP, and then, the PCL-OH was converted to PCL-Br through reaction with 2-bromopropionyl bromide, followed by a chemical modification to ethyl xanthate in PCL-end chains through substitution reaction. The PCL-b-PNIPAAm block copolymers were obtained by RAFT polymerization of N-isopropylacrylamide (NIPAAm) monomer from the PCL-EX macroagent. Different size chains of PCL and PNIPAAm were evaluated as also their influence on the capacity of micelles formation. The polymers and their synthesis efficacy were characterized by chemical composition, molecular weight, and thermal and crystallinity properties. The CMC of the copolymers and the LCST of the micelles increased with the increase in the segmental length of PNIPAAm and decreased with the increase in PCL segmental length. The DLS demonstrated an increase in the micelles size with the increase of the proportion of both hydrophobic and hydrophilic segments. Finally, the morphology observed by AFM demonstrated that the micelles are of spherical shape.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Among different drug delivery systems (DDS), polymeric nanoparticles, formed by amphiphilic block copolymers, have attracted great interest in this field. Some reasons for their interest can be highlighted such as the fact that protein, DNA, and hydrophobic drugs can be easily incorporated into the hydrophobic core, and transported at higher concentrations than their intrinsic water solubility [1, 2]. The presence of hydrophilic blocks in the amphiphilic polymer’s structure can result in an interaction with the aqueous media, forming a shell surrounding the core and protecting the drug from the interaction with plasma proteins, such as opsonins, which enhance phagocytosis by macrophages in the liver and spleen [3, 4]. This provides a prolonged blood circulation time for enhanced permeability and retention (EPR) effect, which is based on tumor disposition [3, 5]. Thus, the carrier can accumulate within tumors, due to its small hydrodynamic diameter and its high stability in an aqueous medium at low concentrations [6, 7]. Besides these properties, in general, polymeric nanoparticles can present distinctive advantages over other formulations including easy control of biodegradability by adjusting the composition of the employed block copolymer. Moreover, the appropriate design of the diblock copolymer provides a rigid core structure leading to stable incorporation of drugs, giving them long pharmacological activity, without the need for further surface modification to prolong the blood circulation time of nanoparticles, which is commonly required for other nanoparticles such as liposomes and emulsions [8].
In this context, drug carriers formed by stimuli-responsive block copolymers have received special attention. Stimuli-responsive materials can be sensitive to specific factors, such as pH, temperature, light, ionic strength, electric field, chemicals, etc. The responses to these stimuli may be manifested as changes in their properties, such as chain dimensions, solubility, secondary structure, and degree of intermolecular association that can be detected, for example, as changes in dissolution/precipitation, swelling/collapsing, conformation changes, hydrophilic/hydrophobic changes, and micellization [9, 10]. The sensibility to external stimuli of copolymers can be influenced by different parameters: the functional group present in the block copolymer, the molecular weight, and the architecture structure. Therefore, during the synthesis of the block copolymer, these parameters can be manipulated to find a suitable polymer for biomedical applications [9,10,11], such as controlled and triggered drug delivery, diagnostics, tissue engineering, ‘smart’ optical systems, sensors and biosensors, bio-separation, immobilized biocatalysis, microelectromechanical systems, coatings, and textiles [9, 12, 13].
Among all smart polymers, those thermosensitive types are frequently applied, since temperature is an important physiological factor in the body. Because thermosensitive polymers can drastically and reversibly change the solubility under heating or cooling, the manifestation of some diseases, or even with the use of hyperthermia techniques can improve the targeting ability of thermoresponsive carriers [14, 15]. These polymers can be classified into two categories, based on their thermosensitive behavior in solution, such as lower critical solution temperature (LCST) and upper critical solution temperature (UCST) [16], which means that heating above the LCST or cooling under the UCST temperature the polymer water-solubility changes from hydrophilic to hydrophobic.
Poly(N-isopropylacrylamide) (PNIPAAm) is a widely studied thermosensitive polymer, which has been used in bioactive applications, such as cell culture, controlled protein adsorption, and drug delivery [17]. PNIPAAm is water soluble and undergoes its volume phase transition in water at around 32 °C, close to human body temperature [16]. Several efforts have been made to understand its phase behavior in water. Amphiphilic block copolymers containing a smart hydrophilic PNIPAAm block are interesting due to their thermal stimuli-responsive properties [18]. Besides that, poly(ε-caprolactone) (PCL) has attracted great interest in pharmaceutical and biomedical applications due to its biocompatibility and biodegradability properties [14]. Moreover, its miscibility with several polymers and easy processability have led to a wide range of applications, especially in food packaging, tissue engineering, and drug delivery [19, 20]. Therefore, amphiphilic and thermosensitive block copolymers containing a hydrophobic PCL segment and a hydrophilic and thermosensitive PNIPAAm segment are very useful in the delivery of active ingredients.
Some reports about the synthesis of linear block copolymers containing hydrophobic PCL block and hydrophilic PNIPAAm block can be found in the literature. Choi et al. [21], Duan et al. [22], and Lee et al. [23] showed the synthesis of PNIPAAm-b-PCL diblock copolymers by first synthesizing the hydroxyl-terminated PNIPAAm (PNIPAAm–OH) through radical polymerization of NIPAAm monomer using either 2-mercaptoethanol [21, 23] as the chain-transfer agent (CTA) or bifunctional initiator, 4,4′-azobis (4-cyano-1-pentanol) [22], in which both reactions were followed by ring-opening polymerization (ROP) of ε-CL using the PNIPAAm-OH as the initiator. Patil and Wadgaonkar [24] reported the synthesis of acetal containing PCL-b-PNIPAAm block copolymer by alkyne-azide click reaction of azido-terminated PNIPAAm (PNIPAAm-N3) with propargyl-terminated PCL (PCL-propargyl). The PNIPAAm-N3 was obtained by ATRP using a new ATRP atom initiator, namely 2-(1-(2-azidoethoxy)ethoxy)ethyl-2-bromo-2-methylpropanoate, which contains both “cleavable” acetal linkage and “clickable” azido groups. Finally, using a combination of three techniques (enzyme-catalyzed anionic ROP, RAFT polymerization, and triazolinedionediene click chemistry), the PCL-b-PNIPAM copolymers were prepared by Vandewalle et al. [25]. In the context of triblock copolymers, Chang et al. [26] reported the synthesis of PCL-b-PNIPAAm-b-PCL using the RAFT polymerization of NIPAAm mediated by the trithioncarbamate-based PCL macro-chain-transfer agent. Xu et al. [27], Loh et al. [28], and Li et al. [29] reported the synthesis of PNIPAAm-b-PCL-b-PNIPAAm prepared through CuBr/HMTETA (1,1,4,7,10,10-hexamethyltriethylenetetramine)-mediated atom transfer radical polymerization (ATRP) using Br-PCL-Br as the macroinitiator. Later, Mishra et al. [18] showed the controlled synthesis of PNIPAAm-b-PCL-b-PNIPAAm triblock copolymers by combining the controlled ROP of ε-CL and the xanthate-mediated RAFT polymerization of NIPAAm using a bifunctional-based PCL xanthate as RAFT agent.
Based on the above literature reports, in this work, we report the synthesis of well-defined linear AB-type amphiphilic, thermosensitive diblock copolymers, PCL-b-PNIPAAm with different blocks lengths, by combining the controlled ring-opening polymerization (ROP) followed by reversible addition–fragmentation chain-transfer (RAFT) polymerization. First, PCL containing hydroxyl end-group (PCL-OH) was synthesized by ROP using benzylic alcohol as an initiator. The OH end-group was then converted to the corresponding Br end-group (PCL-Br) through reaction with 2-bromopropionyl bromide. Then, this Br end-group was converted to the corresponding ethyl xanthate end-group (poly(ε-caprolactone)-ethyl xanthate, PCL-EX) through an ionic substitution reaction with potassium ethyl xanthate. PCL-EX was used as a macro-chain-transfer agent (macro-CTA) for the controlled/living radical polymerization of NIPAAm to obtain PCL-b-PNIPAAm block copolymers. All (co)polymers were characterized by SEC, FTIR, 1H NMR, DSC, and XRD measurements. Then, the effect of PCL and PNIPAAm chain lengths on the amphiphilicity of the copolymers, and their ability of micelles formation were evaluated. Finally, aiming for future application in drug delivery systems, PCL-b-PNIPAAm-based micelles were prepared by solvent evaporation method, and their physicochemical characteristics and thermosensitive behavior were investigated as well.
Experimental
Materials
ε-Caprolactone (ε-CL, Aldrich, 97%) was dried over CaH2 and distilled under reduced pressure before use. N-isopropylacrylamide (NIPAAm, Kohjin Co. Ltd.) was purified by recrystallization with toluene/hexane (6/4 v/v). 2,2′-Azobis(isobutyronitrile) (AIBN) was recrystallized in ethanol and stored at 4 °C. Benzyl alcohol (Synth, 99%) was dried over CaO and distilled under reduced pressure. Toluene (Synth, 99.5%), tetrahydrofuran (THF, Synth, 99%), 1.4-dioxane (Synth, 99%), triethylamine (TEA, Aldrich, 99%), and pyridine (Synth, 99%) were dried and fractionally distilled from sodium. Stannous octoate (Sn(Oct)2, Aldrich, 92.5–100%), 2-bromopropionyl bromide (BPB, Aldrich 97%), potassium ethyl xanthate (KEX, Aldrich, 96%), 1,3,5-trioxane (≥ 99%, Aldrich), sodium hydrogen carbonate (NaHCO3, Synth, 99.7–100%), ammonium chloride (NH4Cl, Synth, 100%), anhydrous magnesium sulfate (MgSO4 XH2O, Synth, 98%), pyrene (Aldrich, 98%), deuterated chloroform (CDCl3, Sigma, 99.8%) acetone (Synth, 99.5%), and the solvents used in the polymer purification steps, such as dichloromethane (DCM, Synth, 99.5%), diethylether (Synth, 98%), and methanol (Synth, 99.5%), were used as received. The water used in the entire process was deionized.
Synthesis of PCL macro-chain-transfer agent (PCL-EX)
The macro-chain-transfer agent (macro-CTA), poly(ε-caprolactone)-ethyl xanthate (PCL-EX), was synthesized in three chemical steps, as shown in Scheme 1.

Synthetic route favored in the synthesis of poly(ε-caprolactone)-ethyl xanthate (PCL-EX) chain-transfer agent
In the first step, PCL-OH was synthesized by ROP of ε-CL using benzyl alcohol as initiator, Sn(Oct)2 as the catalyst, and toluene as solvent (Scheme 1). Two different ε-CL molar concentrations were used, proportional to the molar amount of benzyl alcohol ([ε-CL]0:[benzyl alcohol]0 molar ratio = 66.67 and 133.33) aiming to obtain PCL homopolymers with two different lengths chain. In a typical experiment, 20 g ε-CL, 284.2 mg benzyl alcohol, 354.9 mg Sn(Oct)2, and 20 mL toluene were introduced in a 100 mL dry round-bottom flask equipped with a magnetic stirrer under a dry nitrogen atmosphere. Then, the flask was immersed in an oil bath preheated at 100 °C and kept for 8 h. The polymerization was stopped by cooling the reaction mixture in the ice bath. The polymer was purified in cold diethyl ether, and dried under vacuum at room temperature for 24 h.
In the second step, PCL-Br was prepared by a reaction between hydroxyl-terminated PCL (PCL-OH) and 2-bromopropionyl bromide (BPB) in the presence of triethylamine (TEA) ([PCL-OH]0:[BPB]0:[TEA]0 molar ratio = 1:7.5:10) (Scheme 1). In brief, 17 g (2.32 mMol, Mn NMR = 7328 g mol−1) PCL-OH and 2.35 g (23.2 mMol) TEA were dissolved in 150 mL THF in a nitrogen purged round-bottom flask. The reaction system was kept in an ice bath and a BPB solution (3.87 g in 20 mL THF, 17.94 mMol) was added dropwise over 1 h. Then, the temperature was increased to room temperature (RT), and the mixture was stirred for 72 h. The precipitate formed during the reaction (white precipitate corresponding to triethylammonium chloride) was removed by filtration, and THF was evaporated in a rotary evaporator. The reaction product was dissolved in 150 mL dichloromethane (DCM) and was washed first with 5% sodium bicarbonate solution (5 × 100 mL) and then with water (5 × 150 mL). After, the organic layer was dried over anhydrous MgSO4 and filtered. Finally, the filtrate was concentrated in a rotary evaporator, and the polymer was precipitated in cold methanol/ether (2:8 v/v) and dried under vacuum at room temperature for 24 h.
Finally, PCL-Br was reacted with potassium ethyl xanthate (KEX) (Scheme 1) using the following molar ratio [PCL-Br]0:[KEX]0:[pyridine]0 = 1:5:50. In a nitrogen purged round-bottom flask, 15.5 g (2.06 mMol, Mn NMR = 7538 g/mol) of PCL-Br, 1.72 g (10.71 mMol) of KEX and 125 mL of THF were mixed. A pyridine solution (8.13 g in 30 mL THF, 102.81 mMol) was then added to the mixture and stirred under a nitrogen atmosphere. The reaction mixture was stirred at room temperature (RT) for 48 h. After the removal of the precipitate by filtration, the THF was eliminated by rotary evaporation. The crude polymer was then dissolved in 150 mL DCM and washed with saturated NH4Cl solution (5 × 150 mL), saturated NaHCO3 solution (5 × 150 mL), and water (5 × 150 mL). After, the organic layer was dried over MgSO4 and filtered. The filtrate was concentrated by rotary evaporation, and the polymer was then precipitated in cold methanol/ether (2:8 v/v) and dried under vacuum at room temperature for 24 h.
Synthesis of PCL-b-PNIPAAm block copolymers
PCL-b-PNIPAAm block copolymers were synthesized through RAFT polymerization of NIPAAm monomer from PCL-EX macroagent, using AIBN as initiator and 1,4-dioxane as solvent at 70 °C (Scheme 2). Different NIPAAm molar concentrations relative to PCL-EX were used to obtain block copolymers with different PCL and PNIPAAM block lengths (Table 1). In a typical experiment, using the following molar ratio, [NIPAAm]0:[PCL-EX]0:[AIBN]0 = 62.5:1:0.2; for example, 1.9756 g PCL-EX (Mn NMR = 7763 g/mol), 1.8802 mg NIPAAm, and 8.36 mg AIBN were dissolved in 10.8 mL 1.4-dioxane. A homogeneous solution was obtained after stirring and degassing under nitrogen for 45 min. Next, the flask was immersed in an oil bath preheated at 70 °C for 6 or 8 h. The reaction was stopped by cooling the reaction mixture in the ice bath. The copolymer was purified in cold diethyl ether and dried under vacuum at room temperature for 24 h.

Synthetic route favored in the synthesis of poly(ε-caprolactone)-b-poly(N-isopropylacrylamide) (PCL-b-PNIPAAm) block copolymers
Polymeric particles’ preparation
The particles were prepared by nanoprecipitation technique (also known as the solvent displacement method). An amount of 12.5 mg block copolymer was dissolved in 5 mL THF; thereafter, 1 mL of this organic polymer solution was added dropwise into 45 mL of ultrapure water and stirred at 25 °C. The solvent THF was then removed using a rotary evaporator, and the obtained dispersion was transferred into a 50 mL volumetric flask, followed by dilution to the calibration mark with water.
Characterization
Polymer characterization
Number average molar masses (Mn) and dispersity (Đ) were determined by size exclusion chromatography (SEC) (Waters 1515, USA), using a triethylamine solution in THF (0.3% v/v) as eluent. The characterizations were performed at 35 °C with a flow rate of 1.0 mL min−1 on three Phenogel columns (103, 104, and 106 Å) connected in series to a 2414 differential refractive index detector. The columns were calibrated against polystyrene (PS) standard samples. The IR spectra were collected in an IR Prestige-21 spectrometer (Shimadzu, Japan) using KBr disks method. The 1H NMR spectra were obtained in a Mercury-300 NMR spectrometer (Varian, USA) operating at 300 MHz. These analyses were performed at room temperature using CDCl3 as the solvent and are reported in parts per million (δ) from internal standard tetramethylsilane (TMS). The absorbance measurements of the polymer organic solutions in THF were performed on a UV-1800 spectrophotometer (Shimadzu, Japan) in a range of 190–1100 nm. The (co)polymers’ thermal properties were evaluated using a differential scanning calorimeter (DSC) (TA Instruments, USA) under a nitrogen atmosphere. The instrument was calibrated with indium before use. The samples were submitted in two heaters running consecutively at a heating rate of 10 °C/min, first from − 80 to 180 °C, and followed by quenching to − 80 °C. Then, the samples were re-heated to 220 °C. X-ray diffraction patterns were obtained by a PAN-analytical Empyrean ACMS 101 (Malvern, UK) diffractometer at room temperature. The diffractometer was used with a monochromatic radiation beam of the CuKα. Experiments were conducted with a scan range from 10 to 90° (2θ), a step size of 0.01° (2θ), and a counting time of 10 s per step.
Micelles characterization
Critical micelle concentrations (CMC) of the block copolymers were determined by fluorescence measurements at 390 nm emission wavelength (Varian Cary Eclipse fluorescence spectrophotometer) using pyrene as a probe. A pyrene solution in acetone was added into a series of volumetric flasks in such an amount that the final concentration of pyrene in each solution was 6 × 10–7 mol/L. The acetone was then allowed to completely evaporate. The polymer solution was added into the volumetric flasks and diluted to the calibration mark using deionized water to obtain different copolymer concentrations ranging from 5 × 10–5 to 0.05 mg/mL. The samples were stored at room temperature overnight to equilibrate micelles and pyrene. The size distribution of micelles was determined by dynamic light scattering (DLS) using a Malvern Nano ZS instrument. A 0.45 μm filter was used to remove the dust particles from the aqueous suspension solutions before the measurements. The LCST results were investigated on a Thermo Scientific Genesys 10UV spectrophotometer with a Thermo Fisher Scientific air-cooled temperature controller. The transmittance of the polymeric micelles aqueous solutions at ʎ = 500 nm was recorded in a 1.0 cm path length quartz cell. Atomic force microscope (AFM) images of the micelles were obtained using a Park Systems XE7 microscope. The samples were dropped onto a mica slide and dried at -4 °C.
Results and discussion
In this work, PCL-b-PNIPAAm block copolymers were synthesized by a combination of ROP and RAFT polymerization techniques, as shown in Scheme 1, and used to prepare self-assembly micelles in aqueous media. The copolymer synthesis can be divided into two steps: (i) synthesis of macro-chain-transfer-agent (PCL-EX) through ROP of ɛ-CL using two different proportions of benzyl alcohol followed by conversion of PCL-OH to PCL-Br through reaction with 2-bromopropionyl bromide and the insertion of EX-end chains through substitution reaction with potassium ethyl xanthate and (ii) polymerization of NIPAAm monomer from the PCL-EX macroagent. Finally, the block copolymer PCL-b-PNIPAAm was used to prepare self-assembly micelles in aqueous media.
Synthesis of PCL-EX
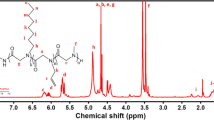
Figure 1a shows the 1H NMR spectrum of the PCL-OH homopolymer obtained through the PCL-OH [1] reaction. As expected, the sample gave the typical resonance signals of PCL-OH at δ (ppm) = 4.05 (–CH3C(=O)O–, f), 3.64 (–CH2OH–, f’), 2.30 (–C(=O)CH2–, c), 1.64 and 1.37 (–CH2–, d and e, respectively). The characteristic resonance signals at 5.11 ppm (–CH2, b) and 7.35 ppm (–CH– of the aromatic ring, a) refer to the protons of the methylene and methane groups, respectively, from the benzylic alcohol. Through the 1H NMR spectrum of PCL-OH (Fig. 1a), it was possible to determine the degree of monomer conversion in the polymerization reaction, resulting in 94.3% for PCL-OH[1] and 90.8% for PCL-OH[2], as observed in Table S1 of Supporting Information. Then, to evaluate the conversion of the hydroxyl end-group of the PCL-OH into bromopropionyl group and, subsequently, this last one into the corresponding ethyl xanthate group, PCL-Br and PCL-EX were analyzed by 1H NMR. Figure 1b and c shows the typical 1H NMR spectra of PCL-Br and PCL-EX polymers obtained in their corresponding PCL-Br[1] and PCL-EX[1] reactions. The incorporation of the 2-bromopropionyl group in the PCL segment was confirmed from the displacement of methylene (f’) protons at 3.64 ppm to 4.15 ppm, and from the appearance of methine (1H, –CH(CH3)Br, g) and methyl (3H, –CH(CH3)Br, h) protons of 2-bromopropionyl end-group of the PCL-Br at 4.30 and 1.80 ppm, respectively. The conversions (%) of PCL-OH into PCL-Br were estimated at 100% for PCL-Br[1] and PCL-Br[2] reactions by comparing the resonance area of methyl protons “h” of 2-bromopropionyl end-group with the resonance area of methylene protons “b” of the benzyl end-group.

1H NMR spectra of the a PCL-OH[1], b PCL-Br[1] and (c) PCL-EX[1]
The conversion of bromo end-group into xanthate end-group was confirmed by the appearance of a new characteristic resonance signal relative to the methylene protons of the xanthate end-group at 4.62 ppm (2H, –OCH2CH3, i) (Fig. 1c). The methyl protons’ resonance signals characteristics of the propionyl group of PCL-Br at 1.80 ppm (3H, h) had shifted to 1.6 ppm. In addition, the resonance signal characteristics of the methyl protons “j” (3H, –OCH2CH3) from of xanthate end-group were overlapped by the resonance signal of the methylene protons “c” from PCL backbone chain. The estimated conversion (%) of bromo end-group into xanthate end-group was 100% for both PCL-EX[1] and PCL-EX[2] reactions. This conversion value was obtained by comparing the resonance of methine protons “g” from the propionyl group with that of the methylene protons “i” from the xanthate end-group. UV/Vis technique was used as a complementary approach to confirm the presence of the xanthate group in PCL chain (PCL-EX) and the spectrum showed in Fig. S1 of Supporting Information and revealed an intense absorption band at around 250–320 nm due to the S–(C=S)– functional group that provided high absorption at λ∼280 nm [30,31,32].
Figure 2 shows the SEC chromatogram obtained for the PCL-OH prepared using two different [ɛ-CL]0:[benzyl alcohol]0 molar ratios. The number average molar mass (Mn) and dispersity (Ð) values for these two homopolymers are presented in Table S1 of Supporting Information. The PCL-OH[2] prepared with higher ratio of [ε-CL]0:[benzyl alcohol]0 showed higher molar mass than PCL-OH[1], as expected. The theoretical molar masses (Mn theor) calculated on the basis of monomer conversion are close to the Mn determined using 1H NMR technique (Mn NMR). The difference in Mn NMR and Mn SEC can be attributed to PS standards used for calibration in the analysis of the SEC method. In addition, both materials have shown low dispersity values (Ð ≤ 1.28). However, the SEC chromatogram of the PCL-OH[2] showed a shoulder at a higher molar mass (Fig. 2), which was probably due to an intermolecular transesterification reaction during the ROP.

SEC chromatograms of the a PCL-OH[1], PCL-Br[1], and PCL-EX[1] and b PCL-OH[2], PCL-Br[2] and PCL-EX[2]
Table S2 and Table S3 in Supporting Information show the molar masses and dispersity data of PCL-Br and PCL-EX homopolymers, respectively. As expected, the Mn NMR, Mn SEC, and Đ values of the converted PCL-Br were close to the values observed for the corresponding PCL-OH, and PCL-EX values were close to the values observed for the corresponding PCL-Br. Moreover, after the conversion of the hydroxyl end-group into bromo, and then xanthate, there was no significant change observed in the chromatograms, which could demonstrate only the chemical modification of the end-group without secondary reactions or degradation of the polymers.
Therefore, based on SEC, 1H NMR, and UV/Vis results, it was possible to confirm that PCL-EX was successfully prepared.
Synthesis of PCL-b-PNIPAAm block copolymers
Aiming to evaluate the livingness of the RAFT polymerization of the NIPAAm mediated by PCL-EX macro-CTA, and choose the appropriate reaction time, a study on polymerization kinetics was carried out using a [NIPAAm]:[Macro-CTA]:[AIBN] molar ratio of 62.5:1:0.2. Figure S2a in Supporting Information shows the plot of the % monomer conversion and ln([M0]/[M]) as a function of the reaction time. A good linear correlation for ln[([M0]/[M])] against time was observed as high as 95.5% conversion, indicating the pseudo-first-order monomer conversion kinetics. A plot of Mn and Ð of the block copolymer versus monomer conversion showed a linear increase of Mn as a function of NIPAAm conversion, while Ð remained almost constant (1.28–1.35) (Fig. S2b).
After the kinetic estimation, copolymerization was carried out to obtain PCL-b-PNIPAAm block copolymers synthesized from different lengths of hydrophobic PCL and hydrophilic PNIPAAm segments. The results obtained for conversion, molar masses, and dispersity are presented in Table 1.
The molar mass of the resultant block copolymers increases with an increase in NIPAAm monomer loading, which can be evidenced by the displacement of the SEC curve to lower values of retention time (Fig. 3), as expected. The Mn theo values of the PCL-b-PNIPAAm block copolymers were close to their Mn NMR values. However, in general, the Mn SEC of the block copolymers were quite different from their Mn theo and Mn NMR, mainly for the material formed with the highest NIPAAm segment length. These results may be: (i) due to the change in the hydrodynamic volume owing to the incorporation of PNIPAAm in the block copolymer, or (ii) the use of PS polymers as standard in the SEC analyses, or (iii) the formation of PNIPAAm homopolymers, or even (iv) to the sum of the three hypotheses. Nonetheless, the SEC chromatograms of the block copolymers (Fig. 3) were monomodal without the tailing caused by residual macro-CTA. In addition, the five polymerization products showed dispersity values (Ð) below 1.5 (Table 1).

SEC chromatograms of the a1 PCL-EX[1], a2 PCL7479-b-PNIPAAm5813and a3 PCL7479-b-PNIPAAm13822, and b1 PCL-EX[2], b2 PCL14031-b-PNIPAAm5743, b3 PCL14031-b-PNIPAAm13310 and b4 PCL14031-b-PNIPAAm29755
The IR and 1H NMR spectra of the block copolymer PCL7479-b-PNIPAAm5813 are shown in Figs. 4 and 5, respectively. As expected, the IR spectrum of the block copolymer (Fig. 4b) showed the characteristic vibration bands of both PCL and PNIPAAm. The PCL absorption bands highlighted in the spectrum showed in Fig. 4a are the band of characteristic methylene (CH2) groups at 2944 cm−1 (C–H asymmetric axial deformation), 2895 cm−1 (C–H symmetric axial deformation) and 1472 cm−1 (C–H symmetrical angular deformation in the plane), two C–O axial deformation bands at 1180 cm−1 and 1050 cm−1, C=O stretching of esters at 1730 cm−1, and C–C axial deformation at 960 cm−1. The characteristic bands of PNIPAAm, such as C=O absorption of amides at 1643 cm−1, NH stretching vibration at 3600–3000 cm−1, and NH-deformation vibration of amides at 1541 cm−1, are observed IR spectrum shown in Fig. 4b.

IR spectra of the a PNVCL-EX[1] and b PCL7479-b-PNIPAAm5813

1H NMR spectra of the a PNVCL-EX[1] and b PCL7479-b-PNIPAAm5813
The 1H NMR spectrum of the block copolymer showed the typical resonance signals of both PCL and PNIPAAm segments. In Fig. 5a, it is possible to observe the resonance signal characteristics of PCL, previously discussed, and after the copolymerization reaction, there is the appearance of characteristic resonance signals of the PNIPAAm backbone, such as methane (–CH–, j, 2, 2–2, 4 ppm) and methylene (–CH2–, i, 1, 8–2, 3 ppm) protons, and the methyl (–CH(CH3)2, m, 1.13 ppm), imine (–NH–, k, ∼6.45 ppm), and methane (–CH(CH3)2, l, 3.80–4.20 ppm) protons of the isopropyl amide group are present in the spectrum shown in Fig. 5b.
A comparative study of the crystallinity and thermal properties of the homopolymer PCL-EX and the block copolymer PCL-b-PNIPAAm was performed through differential exploratory calorimetry (DSC). The DSC thermograms of these materials are shown in Fig. S3 of Supporting Information, and their main transition temperature data are shown in Table 2.
PNIPAAm is an amorphous polymer, with a glass transition temperature (Tg) of around 140 °C [34, 35]. The determined Tg and Tm values of both PCL-EX[1] and PCL-EX[2] were about -57.2 and 55.2 °C, respectively, which are close to the Tg and Tm values of PCL homopolymer reported in the literature [36]. Comparing the Tm and the crystallinity degree (Xc) of both macro-CTAs (PCL-EX[1] and PCL-EX[2]), the second one showed the highest Tm and crystallinity degree (Xc) values due to its higher molar mass, as expected. On the other hand, by comparing the Tm and Xc of the PCL-EX with its corresponding block copolymers, these values were inversely proportional to the length of the PNIPAAm segment incorporated in the PCL. Therefore, the copolymers with the same length of semicrystalline block (PCL) presented a lower crystallinity and a smaller Tm value and thermal stability when a larger amorphous polymer block (PNIPAAm) was applied. This trend agrees with the literature [18], and it may be due to the PNIPAAm segments chemically linked to the PCL which restrict the crystallization of the PCL, so the enthalpy associated with this phase transition is smaller and the fusion is shifted to lower temperature values. In addition, the thermograms of the PCL7479-b-PNIPAAm13822, PCL14031-b-PNIPAAm13310, and PCL14031-b-PNIPAAm29755 did not show any event that could be attributed to Tg of the PCL segment. This is due to the longer length of the PNIPAAm segment. However, a Tg at 134 °C was observed for the block copolymers PCL7479-b-PNIPAAm13822, PCL14031-b-PNIPAAm13310, and PCL14031-b-PNIPAAm29755, which can be attributed to the Tg of PNIPAAm block, and a shift toward a lower temperature due to the plasticizing effect of the PCL block [37]. Nevertheless, this event was not observed for PCL7479-b-PNIPAAm5813 and PCL14031-b-PNIPAAm5743, probably due to the PCL block being the dominant phase and/or the plasticizing effect of the PCL block being more pronounced due to the longer PCL segment.
Typical X-ray diffractograms (XRD) of the PCL-EX and PCL-b-PNIPAAm block copolymers are shown in Fig. 6. The X-ray diffractogram of the PCL-EX showed intense diffraction peaks located at 2θ = 21.3°, 22°, and 23.8° that may correspond to the (110), (111), and (200) PCL reflection planes (Fig. 6a). Comparing the diffractograms of the PCL-EX, it was observed that the PCL-EX[2] showed higher intensity peaks than PCL-EX[1], confirming that the longer size of the chain resulted in a higher crystallinity degree. In addition, comparing the macro-CTA diffractograms with those of their block copolymers (Fig. 6b and c), it was observed that the intensity of characteristic peaks of the PCL crystalline structure was inversely proportional to the length of PNIPAAm segment chemically linked to PCL, which suggests that an increase in the PNIPAAm segment length leads to a decrease in the crystallinity of the copolymer. Therefore, these results determined by XRD agree with the data obtained by DSC analyses.

a X-ray diffractograms of the a1 PCL-EX[1] and a2 PCL-EX[2]. b X-ray diffractograms of the b1 PCL-EX[1], b2 PCL7479-b-PNIPAAm5813 and b3 PCL7479-b-PNIPAAm13822. c X-ray diffractograms of the c1 PCL-EX[2], c2 PCL14031-b-PNIPAAm5743, c3 PCL14031-b-PNIPAAm13310 and c4 PCL14031-b-PNIPAAm29755
Preparation of thermoresponsive micelles based on PCL-b-PNIPAAm block copolymers
Determination of the critical micelle concentration (CMC)
The onset of micellization and the critical micelle concentration (CMC) were obtained using a fluorescence technique with pyrene as the fluorescence probe, and the CMC values of the PCL-b-PNIPAAm block copolymers are shown in Table 3. As expected, the results demonstrated lower CMC values for the copolymers containing a larger hydrophobic PCL segment, and higher CMC values as the proportion of the hydrophilic PNIPAAm segment were increased. These results agree with those previously reported in the literature for amphiphilic block copolymers [18, 28, 38]. This trend is strictly related to the hydrophilicity of the copolymer. For amphiphilic block copolymers, the longer the hydrophilic block, the higher their CMC, as the solubility in water of such copolymers shall increase with the increase in its hydrophilic block length. On the other side, the CMC will decrease with the increase in the hydrophobic block length due to the decrease in the solubility of the copolymers in water [18].
Effect of hydrophobic and hydrophilic block lengths and temperature on the diameter of polymeric micelles
To evaluate the hydrodynamic diameter (Zav.) of the polymeric micelles composed of these block copolymers, the micellar solutions were analyzed by DLS. The measurements were taken at two different temperatures (25 and 37 °C). Then, from the data presented in Table 3, it is possible to assert that the length of both hydrophobic and hydrophilic segments and the temperature affect the Zav of the micelles. The micellar diameter ranged from 97.50 to 175.10 nm at 25 °C, since the larger the PNIPAAm and/or PCL block length, the higher the Zav. values of the polymer micelles. In addition, as expected, a decrease in Zav. values could be observed as soon as the temperature of the micellar solution was increased from 25 to 37 °C. This last-mentioned behavior is due to a change in the solubility of the PNIPAAm hydrophilic segment caused by an increase in solution temperature, which led to the dissolution of this segment, and the consequent reduction of the hydrodynamic diameter of the micelles, without micellar aggregation.
Morphology of the polymeric micelles
As visualized by AFM, the morphology of the micelles formed by PCL14031-b-PNIPAAm13310 block copolymers is shown in Fig. 7. It appears that most micelles showed a spherical-shaped morphology. However, the wide particle-size distribution and the presence of aggregates made it difficult to estimate the average diameter size of such nanoparticles from the AFM images.

Images of the micelles formed by PCL14031-b-PNIPAAm13310 block copolymers
Effect of hydrophobic and hydrophilic block lengths on the LCST of polymeric micelles
To evaluate the effect of hydrophobic and hydrophilic block lengths on the LCST of PCL-b-PNIPAAm block copolymers, the optical transmittance of micellar solutions (0.1 mg/mL) obtained by the nanoprecipitation process was measured as a function of temperature (Fig. 8). LCST of the block copolymers was estimated to be the temperature value relative to the inflection point of the second derivative of the curves shown in Fig. 8. Thus, the LCST values of the PCL7479-b-PNIPAAm5813, PCL7479-b-PNIPAAm13822, PCL14031-b-PNIPAAm13310, and PCL14031-b-PNIPAAm29755 block copolymers were estimated in 26.8, 28.2, 27.4, and 28.6 °C, respectively. As expected, these results confirmed that the LCST decreases with the increase in PCL block length, and increases with the increase in PNIPAAm block length due to the overall copolymer hydrophilicity. Therefore, these results agree with the literature [39, 40], and show that the phase transition of the copolymers can be controlled within a temperature range by tuning the hydrophobicity of the (co)polymers.

Effect of hydrophobic and hydrophilic block lengths on the LCST of the a PCL7479-b-PNIPAAm5813, b PCL7479-b-PNIPAAm13822, c PCL14031-b-PNIPAAm13310, and d PCL14031-b-PNIPAAm29755 block copolymers micelles
Conclusion
Poly(ε-caprolactone)-b-poly(N-isopropylacrylamide) (PCL-b-PNIPAAm) block copolymers with different PCL and PNIPAAm block lengths were successfully obtained by combining ring-opening polymerization (ROP) and subsequent reversible addition-fragmentation chain-transfer (RAFT) polymerization as evidenced by SEC, FTIR, and NMR analyses. The DSC and XRD analysis showed that the PCL-b-PNIPAAm block copolymers are less crystalline than PCL-based macro-chain-transfer agent. The CMC of the block copolymers was affected by their composition, since the CMC values decreased with the enlargement of PCL segment and/or with the decrease of the length of PNIPAAm segment. The copolymers showed the formation of micelles, and the size and the thermosensitivity properties of these nanostructures were investigated as functional attributes of the block copolymers’ composition. The average micelles sizes, determined by DLS, increased with the increase in the extension of both PCL and/or PNIPAAm segments. On the other hand, the LCST decreased with the increase in PCL chain length, and nevertheless, it increased with the increase in PNIPAAm chain length. Polymeric micelles with the micellar diameter and phase transition, easily adjusted as a function of chemical composition of amphiphilic block copolymer, is of great interest for several fields of application including controlled drug delivery.
Data Availability
The authors confirm that the data supporting the findings of this study are available within the article (and/or) its supplementary materials. If necessary, the extra data that support the findings of this study are available on request from the corresponding author.
References
Xiong XB, Falamarzian A, Garg SM, Lavasanifar A (2011) Engineering of amphiphilic block copolymers for polymeric micellar drug and gene delivery. J ControlRelease 155:248–261. https://doi.org/10.1016/j.jconrel.2011.04.028
Nicolas J, Mura S, Brambilla D, Mackiewicz N, Couvreur P (2013) Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chem Soc Rev 42:1147–1235. https://doi.org/10.1039/C2CS35265F
Di J, Gao X, Du Y, Zhang H, Gao J, Zheng A (2021) Size, shape, charge and “stealthy” surface: carrier properties affect the drug circulation time in vivo. Asian J Pharm Sci 16:444–458. https://doi.org/10.1016/j.ajps.2020.07.005
Yorulmaz Avsar S, Kyropoulou M, Di Leone S, Schoenenberger CA, Meier WP, Palivan CG (2019) Biomolecules turn self-assembling amphiphilic block co-polymer platforms into biomimetic interfaces. Front Chem 6:645. https://doi.org/10.3389/fchem.2018.00645
Leong J, Teo JY, Aakalu VK, Yang YY, Kong H (2018) Engineering polymersomes for diagnostics and therapy. Adv Healthc Mater 7:e1701276. https://doi.org/10.1002/adhm.201701276
Mei H, Cai S, Huang D, Gao H, Cao J, He B (2022) Carrier-free nanodrugs with efficient drug delivery and release for cancer therapy: from intrinsic physicochemical properties to external modification. Bioactive Mater 8:220–224. https://doi.org/10.1016/j.bioactmat.2021.06.035
Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R (2021) Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov 20:101–124. https://doi.org/10.1038/s41573-020-0090-8
Kawakami S, Opanasopit P, Yokoyama M, Chansri N, Yamamoto T, Okano T, Yamashita F, Hashida M (2005) Biodistribution characteristics of all-trans retinoic acid incorporated in liposomes and polymeric micelles following intravenous administration. J Pharm Sci 94:2606–2615. https://doi.org/10.1002/jps.20487
Bratek-Skicki A (2021) Towards a new class of stimuli-responsive polymer-based materials: recent advances and challenges. Appl Surf Sci Adv 4:100068. https://doi.org/10.1016/j.apsadv.2021.100068
Mertoglu M, Garnier S, Laschewsky A, Skrabania K, Storsberg J (2005) Stimuli responsive amphiphilic block copolymers for aqueous media synthesised via reversible addition fragmentation chain transfer polymerisation (RAFT). Polymer 46:7726–7740. https://doi.org/10.1016/j.polymer.2005.03.101
Kelley EG, Albert JNL, Sullivan MO, Epps TH (2013) Stimuli-responsive copolymer solution and surface assemblies for biomedical applications. Chem Soc Rev 42:7057–7071. https://doi.org/10.1039/c3cs35512h
Stuart MAC, Huck WTS, Genzer J, Muller M, Ober C, Stamm M, Sukhorukov GB, Szleifer I, Tsukruk VV, Urban M, Winnik F, Zauscher S, Luzinov I, Minko S (2010) Emerging applications of stimuli-responsive polymer materials. Nat Mater 9:101–113. https://doi.org/10.1038/nmat2614
Wei M, Gao Y, Li X, Serpe MJ (2017) Stimuli-responsive polymers and their applications. Polym Chem 8:127–143. https://doi.org/10.1039/C6PY01585A
Wu Q, Wang L, Fu X, Song X, Yang Q, Zhang G (2014) Synthesis and self-assembly of a new amphiphilic thermosensitive poly(N-vinylcaprolactam)/poly(ε-caprolactone) block copolymer. Polym Bull 71:1–18. https://doi.org/10.1007/s00289-013-1041-x
Topuzogullari M, Bulmus V, Dalgakiran E, Dincer S (2014) pH- and temperature-responsive amphiphilic diblock copolymers of 4-vinylpyridine and oligoethyleneglycol methacrylate synthesized by RAFT polymerization. Polymer 55:525–534. https://doi.org/10.1016/j.polymer.2013.12.040
Sponchioni M, Capasso Palmiero U, Moscatelli D (2019) Thermo-responsive polymers: applications of smart materials in drug delivery and tissue engineering. Mater Sci Eng C 102:589–605. https://doi.org/10.1016/j.msec.2019.04.069
Rosenthal A, Rauch S, Eichhorn K-J, Stamm M, Uhlmann P (2018) Enzyme immobilization on protein-resistant PNIPAAm brushes: impact of biotin linker length on enzyme amount and catalytic activity. Colloid Surf B171:351–357. https://doi.org/10.1016/j.colsurfb.2018.07.047
Mishra AK, Vishwakarma NK, Patel VK, Biswas CS, Paira TK, Mandal TK, Maiti P, Ray B (2014) Synthesis, characterization, and solution behavior of well-defined double hydrophilic linear amphiphilic poly (N-isopropylacrylamide)-b-poly (ε-caprolactone)-b-poly (N-isopropylacrylamide) triblock copolymers. Colloid Polym Sci 292:1405–1418. https://doi.org/10.1007/s00396-014-3201-4
Labet M, Thielemans W (2009) Synthesis of polycaprolactone: a review. Chem Soc Rev 38:3484–3504. https://doi.org/10.1039/B820162P
Mondal D, Griffith M, Venkatraman SS (2016) Polycaprolactone-based biomaterials for tissue engineering and drug delivery: current scenario and challenges. IntJ Polym Mater Polym Biomater 65:255–265. https://doi.org/10.1080/00914037.2015.1103241
Choi C, Chae SY, Nah JW (2006) Thermosensitive poly(N-isopropylacrylamide)-b-poly(ε-caprolactone) nanoparticles for efficient drug delivery system. Polymer 47:4571–4580. https://doi.org/10.1016/j.polymer.2006.05.011
Duan Z, Zhang L, Wang H, Han B, Liu B, Kim LI (2014) Synthesis of poly(N-isopropylacrylamide)-b-poly(ɛ-caprolactone) and its inclusion compound of β-cyclodextrin. React Funct Polym 82:47–51. https://doi.org/10.1016/j.reactfunctpolym.2014.05.004
Lee RS, Lin CH, Aljuffali IA, Hu KY, Fang JY (2015) Passive targeting of thermosensitive diblock copolymer micelles to the lungs: synthesis and characterization of poly(N-isopropylacrylamide)-block-poly(ε-caprolactone). J Nanobiotechnol 13:42. https://doi.org/10.1186/s12951-015-0103-7
Patil SS, Wadgaonkar PP (2017) Temperature and pH dual stimuli responsive PCL-b-PNIPAAm block copolymer assemblies and the cargo release studies. J Polym Sci A 55:1383–1396. https://doi.org/10.1002/pola.28508
Vandewalle S, Van De Walle M, Chattopadhyay S, Du Prez FE (2018) Polycaprolactone-b-poly(N-isopropylacrylamide) nanoparticles: synthesis and temperature induced coacervation behavior. Eur Polym J 98:468–474. https://doi.org/10.1016/j.eurpolymj.2017.11.027
Chang C, Wei H, Quan CY, Li YY, Liu J, Wang ZC, Cheng SX, Zhang XZ, Zhuo RX (2008) Fabrication of thermosensitive PCL-PNIPAAm-PCL triblock copolymeric micelles for drug delivery. J Polym Sci A 46:3048–3057. https://doi.org/10.1002/pola.22645
Xu FJ, Li J, Yuan SJ, Zhang ZX, Kang ET, Neoh KG (2008) Thermo-responsive porous membranes of controllable porous morphology from triblock copolymers of polycaprolactone and poly(N-isopropylacrylamide) prepared by atom transfer radical polymerization. Biomacromoleclues 9:331–339. https://doi.org/10.1021/bm7008922
Loh XJ, Wu YL, Seow WTJ, Norimzan MNI, Zhang ZX, Xu FJ, Kang ET, Neoh KG, Li J (2008) Micellization and phase transition behavior of thermosensitive poly(N-isopropylacrylamide)-poly(ɛ-caprolactone)-poly(N-isopropylacrylamide) triblock copolymers. Polymer 49:5084–5094. https://doi.org/10.1016/j.polymer.2008.08.061
Li L, Yang X, Liu F, Shang J, Yan G, Li W (2009) Thermosensitive poly (N-isopropylacrylamide)-b-polycaprolactone-b-poly (N-isopropylacrylamide) triblock copolymers prepared via atom transfer radical polymerization for control of cell adhesion and detachment. J Chilean Chem Soc 54:397–400. https://doi.org/10.4067/S0717-97072009000400016
Mishra AK, Patel VK, Vishwakarma NK, Biswas CS, Raula M, Misra A, Mandal TK, Ray B (2011) Synthesis of well-defined amphiphilic poly(ε-caprolactone)-b-poly(N-vinylpyrrolidone) block copolymers via the combination of ROP and xanthate-mediated RAFT polymerization. Macromolecules 44:2465–2473. https://doi.org/10.1021/ma102453h
Yu YC, Kang HU, Youk JH (2012) Synthesis and micellar characterization of thermosensitive amphiphilic poly(ε-caprolactone)-b-poly(N-vinylcaprolactam) block copolymers. Colloid Polym Sci 290:1107–1113. https://doi.org/10.1007/s00396-012-2630-1
Moraes RM, Carvalho LT, Alves GM, Medeiros SF, Bourgeat-Lami E, Santos AM (2020) Synthesis and self-assembly of poly(N-vinylcaprolactam)-b-Poly(ε-caprolactone) block copolymers via the combination of RAFT/MADIX and ring-opening polymerizations. Polymers 12:1252. https://doi.org/10.3390/polym12061252
Kesel C, Lefèvre C, Nagy JB, David C (1999) Blends of polycaprolactone with polyvinylalcohol: a DSC, optical microscopy and solid state NMR study. Polymer 40:1969–1978. https://doi.org/10.1016/S0032-3861(98)00253-5
Dhumure AB, Patil AB, Kulkarni AS, Voevodina I, Scandola M, Shinde VS (2015) Thermoresponsive copolymers with pendant D-galactosyl 1,2,3-triazole groups: synthesis, characterization and thermal behavior. New J Chem 39:8179–8187. https://doi.org/10.1039/C5NJ01334H
Shieh YT, Lin PY, Chen T, Kuo SW (2016) Temperature-, pH- and CO2-sensitive poly(N-isopropylacryl amide-co-acrylic acid) copolymers with high glass transition temperatures. Polymers 8:434. https://doi.org/10.3390/polym8120434
Carvalho LT, Moraes RM, Alves GM, Lacerda TM, Santos JC, Santos AM, Medeiros SF (2020) Synthesis of amphiphilic pullulan-graft-poly(ε-caprolactone) via click chemistry. Int J Biol Macromol 145:701–711. https://doi.org/10.1016/j.ijbiomac.2019.12.207
Wang Q, Tao L, Yang Z, Raj W, Zhang Y, Wang T, Pietrasik J (2020) Macroscopic and microscopic shape memory effects of block copolymers prepared via ATRP. J PolymSci 58:20–24. https://doi.org/10.1002/pola.29435
Wu Q, Yi J, Wang S, Liu D, Song X, Zhang G (2015) Synthesis and self-assembly of new amphiphilic thermosensitive poly(N-vinylcaprolactam)/poly(d, l-lactide) block copolymers via the combination of ring-opening polymerization and click chemistry. Polym Bull 72:1449–1466. https://doi.org/10.1007/s00289-015-1348-x
Yu Y, Sun X, Zhang R, Yuan S, Lu Q (2017) Synthesis and properties of amphiphilic thermo-sensitive block copolymers. J Polym Res 24:55. https://doi.org/10.1007/s10965-017-1219-2
Vihola H, Laukkanen A, Valtola L, Tenhu H, Hirvonen J (2005) Cytotoxicity of thermosensitive polymers poly(N-isopropylacrylamide), poly(N-vinylcaprolactam) and amphiphilically modified poly(N-vinylcaprolactam). Biomaterials 26:3055–3064. https://doi.org/10.1016/j.biomaterials.2004.09.008
Acknowledgements
This research was financially supported by the São Paulo State Research Support Foundation (FAPESP, n° 2013/03355-4).
Author information
Authors and Affiliations
Contributions
RMdM: conceptualization, methodology, formal analysis, investigation, validation, data curation, writing—original draft preparation, and writing—review and editing. LTdC: conceptualization, methodology, formal analysis, investigation, data curation, and writing—original draft preparation. AJRMT: formal analysis and data curation. SdFM: conceptualization, investigation, validation, and writing—review and editing. AMdS: conceptualization, investigation, validation, writing—review and editing, supervision, and project administration.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
de Moraes, R.M., de Carvalho, L.T., Teixeira, A.J.R.M. et al. Well-defined amphiphilic poly(ε-caprolactone)-b-poly(N-isopropylacrylamide) and thermosensitive micelles formulation. Iran Polym J 32, 1627–1641 (2023). https://doi.org/10.1007/s13726-023-01230-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13726-023-01230-4