
Abstract
Three segmented poly(urethane–urea) block copolymers were synthesized using polydimethylsiloxane diol (PDMS), polytetramethylene ether glycol diol (PTMG), and a combination of PDMS/PTMG with a molar ratio of 0.2/0.8. The polymers were prepared using 4,4′-methylenediphenyl diisocyanate followed by chain extension with 4,4′-methylene-bis(3-chloro-2,6-diethylaniline). The polyurethane-ureas were characterized by ATR-FTIR spectroscopy and their thermal degradation behavior was investigated using thermogravimetric analysis at four different heating rates of 5, 10, 15, and 20 °C/min. The degradation of the polymers was found to consist of three steps. The first step could be related to the depolycondensation of urethane linkages. The second degradation step was explained by the adsorption of volatile fragments, and the third step was accounted for the polyol segment chain scission which happened at a significantly high temperature. The apparent activation energy was calculated by the thermogravimetric data. The dependence of activation energy on conversion (E a) for all synthesized copolymers was derived by five isoconversional methods including Kissinger–Akahira–Sunose, Starink, Flynn–Wall–Ozawa, Friedman, and Vyazovkin. The obtained mathematical results of all methods showed similarities except for PUUS. The results obtained by Friedman and Vyazovkin methods were in good agreement with three poly(urethane–urea) block copolymers.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polyurethanes (PUs) are among the most useful commercial classes of polymers which are widely used in both engineering and consumer products like coatings, adhesives, reaction molding plastics, fibers, foams, rubbers, thermoplastic elastomers, and composites [1, 2]. Polyurethane elastomers (PS) show excellent mechanical and elastic properties because of their microphase separated structure [3], but they have low thermal stability. From an application point of view, thermal stability is one of the most important properties, which has attracted considerable attention in the literature over the past decades [4, 5]. PUs are commonly known to be thermally stable up to 250 °C, and their decomposition is initiated at hard segments of urethane linkages [6].
Degradation may cause serious problems to these polymeric materials during service life at elevated temperatures. Preparing polyurethane block copolymers modified with polyamides, polycarbonate-polyurethanes, hard segments based on aromatic diamine groups, and soft segments based on polybutadiene, polyisobutylene, and polydimethylsiloxane is one of the most promising strategies to overcome these disadvantages [7].
Polysiloxanes are versatile materials used in many applications due to the diversity of properties and processing technologies. They have unique combination of properties, which is related to their chemical structure and macromolecular architecture [8–10].
Polydimethylsiloxane (PDMS) is an important example of this class of polymers. Such materials may find uses as materials with desired thermal, optical, electrical, and mechanical properties.
According to the literature, polyurethane–siloxane polymers could be obtained in form of linear copolymers, interpenetrating polymer networks as well as classical polymer blends. These materials contain the main advantages of both urethane and siloxane moieties, i.e., good elasticity and tensile strength which are characteristic of PUs, and high elasticity (especially at low temperatures) combined with good thermal and chemical stability which are attributed to the structure of polysiloxanes [11]. The influence of different polydimethylsiloxanes on lowering the value of free surface energy of PUs is also significant [6].
The thermal degradation and stability of polydimethylsiloxane-based PUs have been extensively investigated because of the great importance of the PU which lies in its ease of processing and diversity of applications [12, 13].
Saunders has shown that a urethane segment may degrade in three general degradation steps: (1) dissociation to isocyanate and alcohol, (2) formation of primary amine, carbon dioxide, and olefin, and (3) formation of secondary amine [14]. However, the mechanism of degradation of PU, that forms products such as amines, olefins, and carbon dioxide, remains somewhat unclear [15].
Lal and co-workers calculated the degradation activation energy of PUs containing various feed loadings of non-linear optical chromophore [16]. They concluded that values of the activation energies are directly proportional to feed concentration. Gupta has studied the thermal degradation profile of hydroxyl-terminated polybutadiene-based polyurethane–urea as a function of chain extender concentration [17]. The accelerated aging of polyurethane in air has shown to be a source of reduction in tensile strength with time.
Gopalakrishnan and co-worker calculated the activation energy of polyurethane by two kinetic models and compared the results [18].
Yeh et al. investigated the thermal degradation of poly(ether-urethane) and poly(siloxane-urethane) copolymer by TG-FTIR analysis [7].
Król and co-workers focused on the degradation steps of a polyurethane siloxane. They revealed that the activation energy increased as a result of siloxane introduction into the PU chains [6].
In the present work, first, polyurethane–ureas based on PDMS diol (PUUS), PTMG diol (PUUR), and a combination of PDMS/PTMG diols with a molar ratio of 20/80 (PUUSR) were synthesized by reaction with a diamine chain extender, 4,4′-methylene-bis(3-chloro-2,6-diethylaniline) (M-CDEA), and the chemical characterization of the polymers obtained was carried out using ATR-FTIR. Then, the thermal stability of the polymers was examined by thermogravimetric analysis (TGA), and the activation energy for each step of degradation was established by five isoconversional approaches, namely Flynn–Wall–Ozawa (FWO), Kissinger–Akahira–Sunose (KAS), Starink, Friedman, and Vyazovkin methods.
Although many works have been reported on the thermal stability of PUs in the literature [19, 20], to the best of our knowledge, this is the first report on the study of PDMS and PTMG-based polyurethane synthesized with M-CDEA chain extender.
Experimental
Materials
Hydroxyl-terminated PDMS (with number average molecular weight of 2500 g/mol) was obtained from Evonik (Germany) and methylenediphenyl diisocyanate (MDI) of Merck (Germany) was used as received. Polytetramethylene ether glycol (with number average molecular weight of 2000 g/mol) was purchased from Merck. All the diols were dried thoroughly under vacuum for at least 12 h prior to synthesis. M-CDEA was purchased from Lonza group (Switzerland) and used as received. Tetrahydrofuran (THF) and toluene were dried over sodium, and dimethylformamide (DMF) was dried over CaH2.
Synthesis of polyurethane–urea thermoplastic elastomers
The polyurethane–urea thermoplastic elastomers (PUUs) were synthesized by a two-step solution polymerization method. Molar ratios of polyols, isocynate, and chain extenders were kept constant at 1:2:1. A typical two-step solution polymerization procedure was performed as was reported in our previous work [11]. Briefly, molten MDI was poured in a three-necked round flask equipped with a stirrer, nitrogen inlet, and an additional funnel, and the flask was placed in an oil bath at 68–70 °C. The macrodiol, PDMS (1 mol), in the mixture (50:50 v/v) of dried THF and toluene was added to the flask. After mixing for 2 h, the solution of M-CDEA in dried DMF was added into the reactor and the obtained viscous polymer was immediately cast in a Teflon mold and cured for 24 h in an oven at 100 °C PUUS. The same method was applied for the preparation of PUUR, a polyurethane–urea based on PTMG; and PUUSR, a mixture of PTMG/PDMS diols (0.8/0.2 mol). The formulations of the prepared polyurethane samples are tabulated in Table 1 and their chemical structures and the reaction of PUU are given in Scheme 1.

Chemical structures of PUUs
Characterization
Fourier transform infrared spectroscopy (FTIR)
FTIR analysis was performed on a Bruker Equinox spectrometer (Germany) equipped with a Golden Gate single reflection ATR-FTIR attachment (attenuated total reflection) accessory. The resolution for all the infrared spectra was 4 cm−1, and there were 16 scans for each spectral run. The test specimens were in the form of polymeric sheets.
Thermogarvimetric analysis (TGA)
The thermal stability of PUUs was investigated using a Mettler Toledo/DSC/TGA1 (UK). The heating rates of 5, 10, 15, and 20 °C/min were applied up to 700 °C under nitrogen gas. The weight of the sample was around 10 mg for each heating rate.
Results and discussion
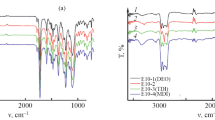
Figure 1 shows ATR-FTIR spectra for polyurethane–urea samples containing different soft segments. As it can be seen in this figure, the absorption peaks of NCO groups at 2270 cm−1 and the wide peaks related to diol hydroxyl groups at 3300–3500 cm−1 are disappeared after completion of synthesis. Sharp absorption bands appeared at 3300, 1729, 1645, and 1530 cm−1 are, respectively, related to NH, urethane, urea, and –CNH groups [21]. These results confirm the reaction between polyol and isocyanate. The absorption bands in a typical ATR-FTIR spectrum of siloxane-containing copolymer consist of 1260 cm−1 (sym. CH3 bending), 1020 and 1100 cm−1 (Si–O–Si stretching), 803 cm−1 (CH3 rocking), 3320 cm−1 (urea N–H stretch), 1645 cm−1 (H-bonded urea C=O), which are assigned to the urea linkage, 3331 cm−1 (urethane N–H stretch), 1703 cm−1 (H-bonded urethane C=O), 1080 cm−1 (C–O–C aliphatic ether stretching), and 1595 cm−1 and 1412 cm−1 (C–C aromatic ring stretching).

ATR-FTIR spectra of polyurethane–ureas
TGA was used to examine the thermal degradation steps of the materials. TGA decomposition information can be used to predict the useful life times of polymeric materials. Samples are heated at three or more different heating rates, because the use of the different heating rates changes the time scale of the decomposition event. The faster the applied heating rate, the higher the given decomposition temperature becomes. This approach establishes a link between time and temperature for the polymer decomposition and provides beneficial information which can be used to model the decomposition kinetics [22].
The TGA measurements of the polymers were performed at four different heating rates of 5, 10, 15, and 20 °C/min. In Fig. 2, a typical TGA thermogram of the synthesized PUUs at 10 °C/min is illustrated. As it can be seen in this figure, all samples show a three-step degradation process. The first and second steps of decomposition for all polymers occurred at more or less similar temperatures. The first degradation step occurred between about 270 and 330 °C which can be attributed to the thermal cleavage of the urethane bonds [22, 23]. The second step between 330 and 390 °C can be assigned to the thermal cleavage of the chain extender in the PUUs [24]. The corresponding overall weight loss for these two stages is about 30 % with a slightly smaller value registered for PUUS. The results are in good agreement with the theoretical calculated data of hard segments for each individual polymer (Table 1). In contrast, the third degradation step of PUUS occurred at a much higher temperature compared to that of the PUUR and PUUSR. Since the degradation of the third step is accounted for the polyol segment chain scission, a significantly higher degradation temperature is predictable for the PDMS-containing formulations because of the relatively high thermal stability of PDMS.

A typical TGA/DTG thermogram of the synthesized polyurethane–ureas at a heating rate of 10 °C/min
Onset temperatures of the first, second and third steps of degradation as well as ash content are deduced from the TGA curves and presented in Table 2 for all samples. As it is expected, the initial degradation temperatures of all the three degradation steps increase with heating rate. As it can be seen, the thermal stability of the polymer based on just siloxane diol is significantly higher than the others.
The thermal degradation kinetics of the synthesized polyurethane–ureas were investigated, and the activation energy of each polymer was estimated using TGA data. The activation energy of degradation at each single step of weight loss was established through five methods, namely FWO, KAS, Starink, Friedman, and Vyazovkin, and the last was used to provide valid data for activation energy.
Flynn–Wall–Ozawa method (FWO)
FWO is an isoconversional method based on the measurement of the temperature (T) in a given value of conversion (α). In this method, a set of experiments conditioned by different rates of heating (β) is used to calculate the activation energy at any particular value of α (E α). The E α is calculated using the following equation:
where R is universal gas constant. Here, it is noticeable that a change in E α value as a function of α can be explained by a multi-stage reaction mechanism [25, 26].
Kissinger–Akahira–Sunose (KAS)
The next isoconversional method used in this study is the KAS method developed based on Eq. (2):
Similar to FWO, the E α can be calculated without the need for the conversion-dependence function (f (α) or g (α)). The activation energy is calculated using the slope of the curve of ln (β/T 2) versus 1/T [27].
Starink method
Starink showed that the KAS equation (Eq. 2) can be improved to a somewhat more accurate expression [27]. Starink assumed that the transformation rate is the product of the absolute temperature and the fraction transformed and derived the following equation:
Friedman method
The Friedman method, a differential isoconversional method, is used to calculate the activation energy at any given conversion using the following equation [28]:
In this method, the value of E α is determined from the slope of the curve of ln (β dα/dt) against 1/T.
Vyazovkin method
The Vyazovkin method is an advanced integral isoconversional method for analysis of E α-dependent mechanisms and process kinetics prediction [29]. For avoiding problems and inaccuracies associated with most isoconversional computational methods, such as KAS, FWO and Starink methods, this advanced approach is adopted in our work. According to this method for a set of n experiments, carried out at different arbitrary heating programs T i (t), the activation energy is determined at any particular value of α by fitting the value of E α that minimizes the Ф(E α ) given in the following equation [30, 31]:
Equation (6) is evaluated numerically, using the trapezoidal rule, for a set of experimental heating programs. A computer code was developed based on genetic algorithm to minimize Φ(E α ), as defined in Eq. (5), for α values between 0.01 and 0.99 with a step of ∆α = 0.01.
The activation energy values of degradation for the PUU samples calculated by OFW, KAS, Starink, Friedman, and Vyazovkin methods are presented in Fig. 3. The plots imply that the thermal degradation kinetics of the samples comprised at least three stages. The obtained results of Friedman method are in agreement with the Vyazovkin results due to introduction of conversion rate in the both equations, while the results of KAS, Starink, and FWO methods are more or less similar. The first step is actually associated with depolycondensation of urethane linkages (–OCONH–) [23]. In the second degradation step, i.e., in the conversion interval 0.2–0.4 which is associated to the adsorption of volatile fragments, the activation energy increases, since the process is diffusion controlled. The degradation of polyol occurs in the third step.

Activation energy versus conversion for the degradation of PUUS, PUUR, and PUUSR
In the second and third steps, the activation energies obtained by Friedman and Vyazovkin methods are larger than those obtained by KAS, Starink, and OWF methods, while in the first step, this is vice versa.
As it is expected, the thermal degradation of PUUS in the third step takes place at much higher temperature compared to the PUUR and PUUSR, but the associated activation energy is lower for PUUS polymer. This is due to the nature of the depolymerization of PDMS which is mainly governed by the molecular structure and kinetic considerations instead of bond energies. As Camino et al. reported, PDMS thermally degrades into volatile cyclic oligomers via chain-folded scission of Si–O bonds by oxygen-catalyzed depolymerization [32]. Silicon d-orbital participation was postulated with siloxane bond rearrangement leading to the elimination of cyclic oligomers and chain shortening. As it is shown in Scheme 2, this mechanism is clarified by formation of the smallest cyclic product, hexamethylcyclotrisiloxane [33].

Depolymerization mechanism of PDMS by random scission [32]
Conclusion
A series of segmented poly(urethane–urea) block copolymers were synthesized via a two-step solution polymerization which PDMS, PTMG and mixture of PDMS/PTMG were used as diol and MDI and M-CDEA were, respectively, used as diisocyanate and chain extender. FTIR spectra confirmed that the reaction had taken place between the polyol and isocyanate. Degradation of the synthesized copolymers were investigated using TGA experiments at 4 different heating rates, and the degradation kinetics were studied by five isoconversional methods including KAS, Starink, FWO, Friedman, and Vyazovkin. The results showed that the thermal degradation process of the copolymers comprised at least three stages. It was found that there were relatively similar results had obtained by the five methods. The differential isoconversional Friedman method was in a good agreement with the integral isoconversional Vyazovkin method.
References
Belva F, Bourbigot S, Duquesne S, Jama C, Le Bras M, Pelegris C, Rivenet M (2006) Heat and fire resistance of polyurethane-polydimethylsiloxane hybrid material. Polym Adv Technol 17:304–314
Prisacariu C (2011) Polyurethane elastomers: from morphology to mechanical Aspects. Springer, New York
Shokrolahi F, Yeganeh H (2014) Soft segment composition and its influence on phase-separated morphology of PCL/PEG-based poly(urethane urea)s. Iran Polym J 23:505–512
Levchik SV, Weil ED (2004) Thermal decomposition, combustion and fire-retardancy of polyurethanes—a review of the recent literature. Polym Int 53:1585–1610
Chattopadhyay D, Raju K (2007) Structural engineering of polyurethane coatings for high performance applications. Prog Polym Sci 32:352–418
Król P, Pielichowska K, Byczynski L (2010) Thermal degradation kinetics of polyurethane–siloxane anionomers. Thermochim Acta 507–508:91–98
Yeh JT, Shu YC (2010) Characteristics of the degradation and improvement of the thermal stability of poly (siloxane urethane) copolymers. J Appl Polym Sci 115:2616–2628
Tyagi D, Yilgor I, McGrathm JE, Wilkes GL (1984) Segmented organosiloxane copolymers: 2 thermal and mechanical properties of siloxane-urea copolymers. Polymer 25:1807–1816
Adhikari R, Gunatillake PA, McCarthy SJ, Meijs GF (1999) Effect of chain extender structure on the properties and morphology of polyurethanes based on H12MDI and mixed macrodiols (PDMS–PHMO). J Appl Polym Sci 74:2979–2989
Wang F(1998) Polydimethylsiloxane modification of segmented thermoplastic polyurethanes and polyureas. Ph.D. Dissertation, Virginia Polytechnic Institute and State University
Askari F, Barikani M, Barmar M (2013) Study on thermal stability of polyurethane-urea based on polysiloxane and polycaprolactone diols. Korean J Chem Eng 30:2093–2099
Hamdani S, Longuet C, Perrin D, Lopez-cuesta JM, Ganachaud F (2009) Flame retardancy of silicone-based materials. Polym Degrad Stab 94:465–495
Chuang F, Tsen W, Shu Y (2004) The effect of different siloxane chain-extenders on the thermal degradation and stability of segmented polyurethanes. Polym Degrad Stab 84:69–77
Saunders JH, Frisch KC (1964) Polyurethanes: chemistry and technology. Interscience Publisher, New York
Barikani M, Askari F, Barikani M, Barmar M (2011) Effect of fire retardants in improvement of combustion restriction and thermal decomposition of polyurethane foams: a review. Iran J Polym Sci Technol 24:3–31
Lal S, Kumar S, Kumar M, Arora S (2011) Kinetic study of thermal degradation of polyurethanes with nonlinear optical chromophore. Arch Appl Sci Res 3:309–318
Gupta T, Adhikari B (2003) Thermal degradation and stability of HTPB-based polyurethane and polyurethaneureas. Thermochim Acta 402:169–181
Gopalakrishnan S, Sujatha R (2011) Comparative thermoanalytical studies of polyurethanes using Coats-Redfern, Broido and Horowitz-Metzger methods. Der Chem Sin 2:103–117
Barikani M, Hepburn C (1987) The relative thermal stability of polyurethane elastomers. II: influence of polyol-diisocyanate molar block ratios with a single and mixed diisocyanate system. Cell Polym 6:29–36
Momtaz M, Barikani M, Razavi-Nouri M (2015) Effect of ionic group content on thermal and structural properties of polycaprolactone-based shape memory polyurethane ionomers. Iran Polym J 24(505):513
Park HB, Kim CK, Lee YM (2002) Gas separation properties of polysiloxane- polyether mixed soft segment urethane urea membranes. J Memb Sci 204:257–269
Régnier N, Fontaine S (2001) Determination of the thermal degradation kinetic parameters of carbon fibre reinforced epoxy using TG. J Therm Anal Calorim 64:789–799
Chattopadhyay DK, Webster DC (2009) Thermal stability and flame retardancy of polyurethanes. Prog Polym Sci 34:1068–1133
Lu H, Sun P, Zheng Z, Yao X, Wang X, Chang FC (2012) Reduction-sensitive rapid degradable poly(urethane-urea)s based on cystine. Polym Degrad Stab 97:661–669
Ozawa T (1992) Estimation of activation energy by isoconversion methods. Thermochim Acta 203:159–165
Vyazovkin S, Burnham AK, Criado JM, Pėrez-Maqueda LA, Popescu C, Sbirrazzuoli N (2011) ICTAC Kinetics committee recommendations for performing kinetic computations on thermal analysis data. Thermochim Acta 520:1–19
Albu P, Bolcu C, Vlase G, Doca N, Vlase T (2011) Kinetics of degradation under non-isothermal conditions of a thermo-oxidative stabilized polyurethane. J Therm Anal Calorim 105:685–689
Friedman HL (1964) Kinetics of thermal degradation of char-forming plastics from thermogravimetry: application to a phenolic plastic. Polym Sci Part C 6:183–195
Vyazovkin S, Dollimore D (1996) Linear and nonlinear procedures in isoconversional computations of the activation energy on nonisothermal reactions in solids. J Chem Inf Comput Sci 36:42–45
Vyazovkin S (1997) Evaluation of activation energy of thermally stimulated solid-state reactions under arbitrary variation of temperature. J Comput Chem 18:393–402
Vyazovkin S (2006) Model-free kinetics: staying free of multiplying entities without necessity. J Therm Anal Calorim 83:45–51
Camino G, Lomakin SM, Lazzaria M (2001) Polydimethylsiloxane thermal degradation Part 1: kinetic aspects. Polymer 42:2395–2402
Camino G, Lomakin SM, Lageard M (2002) Thermal polydimethylsiloxane degradation. Part 2. Degrad Mech Polym 43:2011–2015
Acknowledgments
Financial support of Iran National Science Foundation (INSF), under Grant No: 90006173, is highly appreciated and acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Askari, F., Barikani, M., Barmar, M. et al. Study of thermal stability and degradation kinetics of polyurethane–ureas by thermogravimetry. Iran Polym J 24, 783–789 (2015). https://doi.org/10.1007/s13726-015-0367-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13726-015-0367-7