
Abstract
In the -omics era, bioinformatics technologies allowed the development of several approaches useful to analyze biological systems. To provide a deep insight into a biological system, the interactions between molecules are usually modeled as a dynamic network (also known as “temporal network” or “time-varying network”). The latter allows investigating how the interactions evolve over time, contrary to a static network. This survey presents an assessment of the software tools for network alignment and motif discovery in dynamic networks. We considered a set of criteria belonging to the following macro areas: (i) methodology, (ii) functionality, and (iii) availability. For instance, we investigated the objective functions and the scores used for the processing, alignment methods, use of a method for the alignment of static networks adapted to the dynamic context, network discrimination performance, and other additional information. We reported how several issues may be transferred from static to dynamic networks by taking into account the temporal information. Furthermore, we encountered a systematic convergence toward iterative strategies both for network alignment and motif discovery, justified by the fact that a dynamic network is usually analyzed through the sub-analysis of its time points.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Over the past few decades, bioinformatics has made possible the development of novel strategies in molecular biology. A cell is a system composed of highly integrated subsystems interacting among them; thus, the analysis of these interactions is crucial to understand the molecular-level dynamics. For instance, the analysis of interactions between genes and proteins is useful to describe the cellular functions or to predict protein functions (Barabási et al. 2011), as well as to study the disease–gene associations (Cinaglia et al. 2018). The biological networks are inherently dynamic structures that are represented through a graph model where the set of nodes (V) are the molecules, and the set of edges (E) are the known interactions between these: \(G=(V,E)\).
In biology, depending on what the nodes and edges represent, we may classify a network as a Protein Interaction Network (PIN) (Athanasios et al. 2017), a gene regulatory network (Huynh-Thu and Sanguinetti 2019), or a signaling network (Ju and Wei 2017). A PIN concerns a mathematical representation of the physical contacts between the proteins; specifically, the nodes represent the proteins, and each edge is a protein–protein interaction (PPI) (Rao et al. 2014).
In the real-world systems, the interactions between molecules evolve over time. The static representation of a network does not report if the different interactions take place contemporarily, or at different times. A static biological network reconstruction should be interpreted as a potential network where all edges and nodes will be hardly present all together in vivoFootnote 1 (Klein et al. 2012).
Therefore, the static approach loses important temporal information, including both changes in the network topology and flow on the network. A dynamic network is a time-varying network where each instance is a network of interacting molecules at time t. A relevant task for biological dynamic network analysis concerns the development of network alignment (NA) methodologies. NA allows finding important genes and temporal functional changes in the biological networks, e.g., to study PPIs.
As all networks, a dynamic network is affected by the following operations that may perturb its topology: (i) node addition, (ii) node deletion, (iii) edge addition, and (iv) edge deletion. Furthermore, it is called “probabilistic” when it contains at least one uncertain interaction, otherwise it is “deterministic”. An important observation is that a probabilistic network is a summary of all possible deterministic networks that are determined by the subset of interactions that take place (Ghoshal et al. 2013). The evaluation of the functional distance between two nodes (e.g., molecules) may be performed by calculating the average of the shortest paths between these. The average of the shortest paths over all node pairs (i.e., mean path length) has been used as a measure for the network navigability. The search of the shortest paths is used to find functional clusters in biological systems, as well as to identify core pathways. The number of the shortest paths that go through an edge in a network is called “Edge Betweenness Centrality”. The latter is crucial to understand how the biological networks operate, as well as how these could be fixed or manipulated. Note that the counting of the shortest paths is a polynomial-complex time problem (Ren et al. 2018a).
Even though the genome-scale computational analysis of interactions on a dynamic network allows extracting a greater set of information related to PPIs, the static network analysis is still the most applied and studied, in that the most widely applied large-scale technologies for PPI detection do not provide temporal data. In several domains, the static networks are used as model for complex real-world systems both static and dynamic, independently. This approach is useful when the temporal aspect is not relevant. Otherwise, it is not optimal to identify the causes and the consequences of external stimuli.
Temporal protein complexes are typically constructed by the dynamic assembly or disassembly of proteins. Gene expression data provides potential insights into the dynamics of PINs (Zahiri et al. 2020). For instance, data acquired by DNA microarray technologies under different conditions may be used simultaneously to construct time series. The latter is applied in several fields to report data changes over time; to give an example, a trend (e.g., in epidemiology, or in economy) is usually analyzed by using a representation based on time series (Cinaglia and Cannataro 2022).
This survey presents an overview of software tools available in the literature for NA and motif discovery applied to dynamic networks. We performed an extensive non-systematic review methodology. First and foremost, we introduced an analysis of static networks. Usually, the analysis of dynamic networks is based on existing methodologies for static networks. Subsequently, the articles presenting the software tools for network alignment and motif discovery in dynamic networks have been investigated to extract the relevant information, according to a set of criteria belonging to the following macro areas: (i) technology, (ii) functionality, and (iii) availability.
Our investigation used several well-known databases, such as: PubMed, Google Scholar, Medline, Crossref. The main keywords used for searching are reported as follows: network alignment, motif discovery, dynamic network, temporal network, time-varying network, network analysis, software, tool, method. The keywords were combined to improve the matching, according to the topic of interest. In addition, we also included the software tools used for comparison and/or testing, within the articles found. Only articles presenting original research published in English (American or British, indifferently) have been considered.
Software tools for NA and motif discovery in dynamic networks have been included or excluded based on the criteria reported as follows.
Inclusion criteria:
-
full-text articles;
-
availability;
-
software tools often used for performance comparison (e.g., “DynaMAGNA++”), or as reference implementation by other methodologies.
As an alternative to a software tool, we included articles reporting an exhaustive set of information for the following features:
-
network alignment in dynamic networks: objective, score, strategy, and method;
-
motif discovery: typology of network, strategy, and method.
In addition, articles referring to objects of particular interest for the topic (e.g., topological measures, metrics, patterns) were included in any case.
Exclusion criteria: the few articles that do not report a resource for downloading of the software tool, or the information described above, were excluded.
Our investigation identified the following candidate software tools and methodologies for dynamic networks:
-
network alignment: DynaMAGNA++, DynaWAVE, Got-WAVE, Twadn, Tempo++, and Tempo;
-
motif discovery: COMMIT, DynaMIT, SNAP, Mukherjee, Kovanen.
An extensive assessment of these is reported in Sects. 4.1 and 4.2 for network alignment and motif discovery, respectively.
Our contribution may be summarized as follows:
-
presenting the state of the art for NA and motif discovery in dynamic networks;
-
investigating the tools and algorithms of interest for NA and motif discovery in dynamic networks, by focusing on the strategies and methods used to implement these;
-
performing a comparison between the NA tools for dynamic networks based on network discrimination performance and strategies;
-
performing a comparison between the motif discovery tools for dynamic networks based on strategies and methodologies.
The rest of the paper is organized as follows: Sect. 2 introduces dynamic networks; Sect. 3 reports the principles related to the static network analysis with a focus on NA and motif discovery in static network; Sect. 4 surveys the software tools for both NA and motif discovery in dynamic networks, by discussing a comparison between these within Sect. 5.
1.1 Related works
The software tools for static networks are widely discussed in literature, but the same is not true for dynamic networks. However, we reported a series of studies related to NA and motif discovery based on topics discussed in this paper. Ma and Liao (2020) presented a review focused on the alignment of PPI networks. The authors reported the methodologies applied for each software tool, by focusing on the following main features: local/global, pairwise/multiple, one-to-one/many-to-many. This review is limited to static networks and introduced the dynamic networks only as example of a possible extension of the presented tools. For instance, DynaMAGNA++ is mentioned as an extension of MAGNA++. The former is better explained in this paper, being a software tool developed for dynamic networks, contrary to the latter that is developed for static ones. Similarly, Milano et al. (2017) presented an extensive assessment of NA algorithms by focusing on the biological aspects. The same issue is also investigated at a lower level. For instance, Kim et al. (2018) investigated the dynamic network models by focusing on the ones with latent variables. This study focused on the mathematical representation of the models applied in the field of dynamic networks, excluding the algorithmic development. Hulovatyy et al. (2015) discussed how the methodologies for static analysis may be applied on each snapshot (i.e., a single time point at time t) to preserve partially time-varying network). Otherwise, Patra and Mohapatra (2020) presented a list of reviews on network motif discovery based on the mentioned criteria, by focusing attention on the contribution and the limitations of each one. However, the authors limited the investigation to the static networks.
According to our literature investigation, the software tools for dynamic networks are rarely investigated in a survey or a review, especially about the NA. The mentioned studies begin with an overview of the static models, and these only introduce the dynamic extensions. Briefly, it is also clear how many of these studies have included more than 5 years, or have not specifically related to dynamic networks. We extended the assessment of software tools for static networks to dynamic networks. The state of the art is reported at the beginning of each section. The methodologies are almost always classified into various categories based on the mapping and the pattern growth. We explored the software tools by focusing our attention on (i) methodology, (ii) functionality, and (iii) availability (Sect. 4). For instance, we evaluated the objective function and the metrics/scores used for the preprocessing, the alignment method, and the eventual re-use of a method for static alignment in the dynamic context, as well as the network discrimination performance, and other additional information. Furthermore, we explored how the methodologies for static networks have been adapted to a time-varying context (Sect. 3).
2 Dynamic biological networks
The dynamics of a network over time may be described through the temporal networks (also known as “dynamic network”, or “time-varying networks”) (Mertzios et al. 2013). Temporal networks are based on a mathematical structure modeled to represent a link between nodes over time (i.e., “contact”). A contact is an extension of a static link that allows to represent more than one relationship between two nodes, thus being connected is not a transitive mathematical relation (Fisher and Pinter-Wollman 2021). Any system represented by pairwise interactions that evolve over time and of which the information about time is known could be modeled as a dynamic network.
Usually, “dynamic networks” and “temporal networks” are used mutually with reference to dynamic network, especially in bioinformatics and biology. However, network science differentiates the original meaning of the term “dynamic network” from “temporal network”: a dynamic network is used to investigate how a dynamic system affects the related network evolving over time; otherwise, a temporal network is typically more data oriented and used to investigate a data set (e.g., it is particularly appropriate to study epidemic outbreaks) (Holme 2015). Note that if the dynamic systems on the network are faster than the contact dynamics, and the network at any given moment of time is non-trivial, then we may define a static network evolving in time through the terms “dynamic network” and “temporal network”, indifferently.
More generally, the dynamic networks may be considered as a way of representing temporal networks; other lossless representations are based on graph sequence (or multilayer networks), time-node graph, time series of contacts on a static graph, timelines of contacts, and adjacency tensors. Therefore, the term “temporal networks” concerns all formalisms proposed to represent evolving networks without loss of information (e.g., temporal networks, time-varying networks, interaction networks) (Rossetti and Cazabet 2018). According to Joakim et al. (2020), dynamic network representations may be grouped into the following representations ordered by temporal granularity:
-
edge-weighted: the temporal information is included as labels on the edges and/or nodes of a static network;
-
discrete: it is represented by multiple snapshots at successive time points (discrete time intervals);
-
continuous: it has no temporal aggregation and is the most complex for representation.
The edge-weighted model is able to represent only the actual state of a network, and its significant loss of temporal information makes it not suitable to represent the evolution over time. Discrete and continuous models are able to represent the changes over time. However, continuous model is the most complex and rarely applied. Biological dynamic networks are almost always based on the discrete model, as well as the solutions presented in this paper.
In biology, dynamic networks are used to study the interaction between molecules (Doria-Belenguer et al. 2020), as well as to investigate the genetic control of metabolism or to evaluate the temporal aspects during cell differentiation.
2.1 Construction of dynamic networks
Cellular systems are highly dynamic in as much as a protein is not always active over a whole cell cycle, and time factor is crucial to understand its evolution. A static network does not consider time variable, being an overall representation of the evolution of the entire system under examination. Otherwise, the construction of a dynamic network must take into account time factor, thus allowing to identify time point at which a protein exhibits its activity (Zhang et al. 2019).
A dynamic PIN (DPIN) is constructed by involving proteomics, genomics, and transcriptome analysis. Therefore, the information related to protein expression may be acquired from gene expression data. To give an example, Li et al. (2020) proposed a method for the construction of DPINs by integrating time-course gene expression data and subcellular location data. A DPIN may be constructed by applying several approaches, mainly based on the following methodologies: (i) methods based on protein presence dynamics and (ii) methods based on co-expression alterations.
The first one constructs a DPIN by generating multiple static subnetworks. Each subnetwork is a static PIN constructed by mapping the interactions identified at different time points or conditions. The second one constructs a DPIN analyzing highly co-expressed PPIs with a correlation coefficient over a defined threshold. Usually, this method is applied by using the Pearson correlation coefficient (PCC) as correlation method to evaluate the co-expression of coding genes between each pair of interacting proteins in expression profiles.
Briefly, a method based on protein presence dynamics reflects the changes in protein presence over time, while a method based on co-expression alteration reflects the differences of co-expression under different conditions. Therefore, as previously mentioned, a dynamic network is one whose links are active only at certain points in time (i.e., time point) (Wang et al. 2014).
According to Holme and Saramäki (2012), a dynamic network may be considered as a series of “static” networks, where each one is a snapshot observed at t consecutive time points. Previously, a generic network has been reported as \(G=(V,E),\) where V and E denote the set of nodes and set of edges, respectively. In a snapshot-based representation, we may denote a dynamic network as \(G=[{G}_1,{G}_2,\ldots ,{G}_t]\), where \({G}_i=(V_{i},E_{i})\) represents the i-th snapshot and \(0 \le i \le t\) with t the total number of time points. Usually, nodes are considered invariant over time (i.e., \({G}_i=(V,E_{i})\)). A snapshot-based representation may be converted to an event duration-based representation, to give an example: if an edge connects two nodes in \(t_{1}\), \(t_{2}\), and \(t_{3}\), then the event between these two nodes is active from \(t_{1}\) to \(t_{3}\). The node set of the snapshots is combined in an event duration-based representation. Formally, it may be explained as follows: given a dynamic network H(V, T), with V a set of nodes and T a set of events, we may define an event as a temporal edge consisting of 4-tuple: u, v, \(t_{s}\), \(t_{e}\). Where two nodes (i.e., u and v) interact from a time \(t_{s}\) to \(t_{e}\), the event is active at time t (\(t_{s} \le t \le t_{e}\)). Therefore, the duration of the event is \(t_{e} - t_{s}\). Two nodes may have more than one event, while two events cannot be active on the two same nodes within the same period. If two events between the same two nodes are active at the same time, then they are combined into a single event. For illustrative purposes only, a synthetic dynamic network consisting of ten time points is reported in Fig. 1 by representing its evolution over time, as described.

The figure shows for illustrative purpose only an example related to a synthetic dynamic network consisting of ten time points. Assuming invariant the number of nodes, the network evolves over time by varying its interactions (i.e., edges)
In addition, Fig. 2 shows a static network (i.e., PIN) and a dynamic network (i.e., DPIN) having a set of time points \(t_{i}\), with \(0 \le i \le 2\). In this figure, time points represented in the figure concern the states of the network over time, where each time point \(t_{i}\) evolves at \(t_{i+1}\) by changing its topology. Consequently, the set of the shortest paths evolve over time, by changing the functional distance between molecules at each time point. The analysis is performed on a dynamic network given time-dependent representation, contrary to static networks that provide an output temporally decontextualized. In a static network, an interaction that occurred for a very short period of time is represented in the same way as an interaction that occurred for a more relevant period of time. This issue does not afflict the dynamic networks that take into account the time variable.

An example of DPIN, with the related static PIN where all edges and nodes are present all together. In the figure, the original PIN is constructed by the interactions observed at different time points. The DPIN changes its topology, as well as the functional distance between molecules, at each time point. In a static network, an interaction occurred for a very short period of time and could be considered that it occurred for a more relevant period of time. Otherwise, this issue does not affect the dynamic networks that take into account the time variable
3 Static networks
This section reports an overview related to two crucial issues for network analysis in bioinformatics (i.e., network alignment, and motif discovery) by focusing attention on static networks, to evaluate how several methodologies have been extended into a dynamic context. The latter is detailed in Sects. 4 and 5. Network alignment aims to transfer biological knowledge between organisms by using a well-studied model organism to investigate a less well-studied organism. It is applied to find regions of topological and functional similarities (or dissimilarities) between molecular networks of different organisms. Otherwise, motif discovery is applied to gene regulation networks to mine the building blocks of a complex network. These are particularly useful to identify the fundamental patterns in networks.
3.1 Network alignment
Network alignment (NA) is a crucial challenge for computational analysis of biological systems. It aims to find a node mapping that conserves similar regions between compared networks. NA approaches may be categorized as local or global: local NA (LNA), and global NA (GNA), respectively. LNA looks for small local regions of high similarity (i.e., small subnetworks), admitting a many-to-many mapping. GNA looks for the best overlapping of the entire networks in input by mapping large suboptimal subnetworks (Guzzi and Milenkovic 2018). It performs a one-to-one (or injective) mapping, admitting a suboptimal matching among local network regions. NA may also be categorized as one-to-one, many-to-many, or one-to-many (Faisal et al. 2015). A one-to-one NA maps a node from a given network to at most one unique node from another network. On the other hand, in a many-to-many NA, a node from a given network can be mapped to several nodes from another network. In addition, the latter may categorize as one-to-many NA if a node from the latter network can be mapped to at most one node from the former network. To give an example, Fig. 3 shows a one-to-one node mapping between two static networks, as follows: given two static networks \(G1=(V_{1}, E_{1})\) and \(G2=(V_{2}, E_{2})\), with \(\left| V_{1}\right| \le \left| V_{2}\right|\) (no loss of generality), the NA between G1 and G2 produces a set of aligned node pairs (v, f(v)), with \(v \in V_{1}\), by performing a one-to-one node mapping \(f : V_{1} \rightarrow V_{2}\). Meng et al. (2016) introduced a systematic comparison of LNA and GNA. The authors reported that GNA is superior to LNA in topological alignment quality. However, with respect to biological alignment quality, GNA is superior to LNA for topological information only (T), while LNA is superior to GNA both for sequence information only (S), combined topological and sequence information (T &S), and in the best of T, S, and T &S. Briefly, GNA outperforms LNA both topologically and biologically, if only topological information is used, while LNA is superior to GNA (only in biological quality) if also sequence information is included. In any case, GNA is always the best in terms of topological alignment. In addition to the aforementioned categorizations, an NA may be categorized as pairwise or multiple: the former aligns two networks, while the latter is able to align more than two networks (Vijayan et al. 2020).

The figure shows a one-to-one node mapping between G1 and G2. The latter is shown through dashed lines, while conserved edges with green lines, and no-conserved edges with red lines
Alignment measures are crucial to evaluate the similarity among mapped nodes, and the number of conserved edges, as well as its cost function. Usually, the following measures are applied: edge correctness (EC) (Zaslavskiy et al. 2009), induced conserved structure (ICS) (Patro and Kingsford 2012), and symmetric substructure score (\(S^{3}\)) (Saraph and Milenković 2014).
Let f be the injective mapping (described previously) between the nodes of two given networks G1 and G2, with \(G1=(V_{1}, E_{1})\) and \(G2=(V_{2}, E_{2})\).
EC is defined as the number of edges conserved by f on the total number of edges of G1 (i.e., \(E_{1}\)). Therefore, it is equal to 1 only if G2 contains an isomorphic copy of G1.
Formally, EC is defined as follows:
ICS measures the ratio of the number of edges aligned between G1 and G2 to the number of edges from the induced subnetwork (i.e., \(f(V_{1}\Vert G2)\)). It is defined as follows:
\(S^{3}\) was introduced to overcome the asymmetries of EC and ICS by considering the edges of both networks (Vijayan et al. 2015). It is defined as follows:
\(S^{3}\) maximizes the accuracy of the alignment (i.e., GNA) directly during its construction, with respect to the amount of conserved edges, and it turns out to be a good compromise between EC and ICS. Briefly, EC penalizes the misaligned edges in the smaller network, while ICS penalizes the misaligned edges in the larger network (Guerra and Guzzi 2020).
NA is usually applied between two networks, even if some software tools are extended to multiple NA (Gligorijević et al. 2016). Methods for multiple NA are generally based on traditional NA strategies. For instance, Singh et al. (2008) carried out a pairwise-alignment method to evaluate the scores between every pair of networks, before performing a spectral partitioning method (i.e., clustering). Other solutions have been developed to find subnetwork by mapping pathways, e.g., applying a one-to-many mapping between each molecule in a pathway and a set of molecules of another (Ay et al. 2011).
NA may also be evaluated indirectly. A common approach consists of measuring the frequency of Gene Ontology (GO) terms (Consortium 2006) to improve the alignment between PPI networks. The scores based on GO terms usually do not take into account the global dependency, evaluating each pair of proteins independently. Therefore, the semantic similarity of a mapping may be combined to GO, in order to investigate if a network topology encodes significant biological information, and to evaluate the biological similarity between all pair of proteins. To give an example, GO could be used to improve the quality of a similarity matrix built on local structural information, by taking into account the biological processes (Zhu et al. 2020).
3.2 Motif discovery
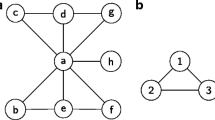
As mentioned previously, the second key issue of this section concerns the motif discovery. A motif is a small subnetwork which occurs frequently in a given network. It may be considered as the basic building block of a biological network useful to uncover both functions and local properties related to this one. Therefore, it is a pattern of interconnections occurring in complex networks at numbers that are significantly higher than those in randomized networks (Ivarsson and Jemth 2019). Figure 4 shows a non-exhaustive example of a motif within a static network.

A non-exhaustive example of a motif consisting of four nodes (vertices) and four edges in a static network. The box on the left highlights the motif
In bioinformatics, the analysis of motifs is used to investigate the large-scale structure of a biological network, as well as to detect its homology. The motif discovery allows studying proteins functions in the PPI complex, e.g., to discover the biological meanings of unknown proteins (Cai et al. 2020). It is crucial to explore the properties of a biological network.
Given a motif topology, counting the number of embedding is a subgraph isomorphism problem (NP-complete). It is afflicted by the explosion in the number of distinct mappings (i.e., combinatorial explosion due to occurrence isomorphism) that occurs when more vertices in the motif are identical. Consequently, identical vertices may be mapped to the input graph in different possible combinations. A possible solution is to recognize the indistinguishable vertices. The conventional approach for motif discovery generates a set of candidate patterns, by applying statistical or structural measures, e.g., based on the frequency of the motifs in the input network on a randomized version of this one. It performs a preliminary computation to identify an initial set of candidates that is used to generate other novel ones as long as a given condition is respected by an iterative strategy (e.g., limiting the computation to a specific subclass of motifs) (Jazayeri and Yang 2020). In this context, the edge/node-disjoint subgraph embeddings may also be performed to evaluate the frequency and significance.
The frequency of a motif is generally used to infer the statistically significant subgraph matches. In a set of networks, the frequency of a motif is considered as the number of items of the set containing the pattern. Similarly, in a single network, it is defined as the number of embeddings of the pattern in the network. Usually, a methodology defines a threshold to discriminate the frequent/significant candidates. The measures applied to motifs may be classified as (i) statistical measures and (ii) structural measures mainly. The statistical significance of a motif in a network is usually measured by using two main parameters: Z-score and P-value (Gupta et al. 2007). Given a motif m and a target network tn, the Z-score is the difference between the frequency of m in tn and its mean frequency in a sufficiently large set of randomized networks, divided by the standard deviation (SD) of the frequency values for the randomized networks. The P-value is the probability (P) that the frequency of m in a randomized network is equal to or larger than the frequency of m in tn. Otherwise, the structural measures may be categorized into two main groups: (i) motif-based node degree and (ii) motif-based edge degree (Xia et al. 2019). The first denotes the number of motifs that include the same node. The second is based on the ratio between the number of triangles including a specific edge in a motif, and the number of all existing triangles for the same edge in the network.
To illustrate this statement, we describe an example for counting independent motif instances. Just for the sake of simplicity, the method is based on only two main steps: (i) preprocessing and (ii) processing. Let us denote by \(G=(V,E,P)\) the probabilistic network, by M the motif pattern, and by \(G'=(V,E)\) the resulting deterministic network.
The preprocessing builds an overlap graph based on all embeddings discovered in \(G'\). In addition, it counts the non-overlapping motif embeddings, by calculating a score (i.e., priority value) for each node. The latter may be performed by applying a heuristic strategy. Otherwise, the processing extracts the nodes with the highest score from the overlap graph, by populating the result set with the corresponding embeddings. Each of these needs to be evaluated also according to the probability that it exists to the motif count. Note that every time a node is extracted, this is removed along with all of its neighbors from the overlap graph. The processing is performed by using the proposed method, iteratively. Specifically, it operates until all nodes are computed, that is, the overlap graph is empty.
A similar approach is applied by Ren et al. (2018b). Furthermore, Sarkar et al. (2019) presented a mathematical model that can also capture the dependencies between the overlapping embeddings, in order to count the independent instances of a given motif topology in a probabilistic biological network.
4 Dynamic networks
The analysis of dynamic networks is a crucial topic which growing in diverse fields. In biology, the system consisting of interconnected units (e.g., genes, or proteins) may be modeled through a dynamic network based on nodes and edges that evolve over time. Therefore, network theory allows studying both topological and biological hypotheses in order to investigate evolutionary system. It is important to clarify that a “static-temporal approach” allows performing the methods for the analysis of static networks, on dynamic networks (Chen et al. 2014). It considers a dynamic network as a series of snapshots, where each one may be represented through a static network. Starting from this assumption, it analyzes the snapshots independently, joining all results on the original time-series. However, this strategy is limiting, because it discards the relationships between the snapshots. Biological dynamic networks are studied in several fields, e.g., including the temporal dynamics of protein complexes in PPI networks (Ou-Yang et al. 2014), the prediction of protein complex (Dai et al. 2020), or the identification of motifs (Elhesha and Kahveci 2016).
This section presents the software tools and the related approaches for NA and motif discovery. According to Chen et al. (2014), no accepted criteria to perform an overall comparison between algorithms for NA or motif discovery (both for static and dynamic networks) is generally accepted in literature, thus it is hard to say which one is better. Therefore, we based our considerations on the criteria described as follows.
The features used to compare the tools for alignment of dynamic networks have been focused on:
-
the objective function and score calculation;
-
the strategy used to perform the alignment;
-
the tool used as core for alignment where this strategy is implemented (i.e., the alignment of dynamic networks could be performed by aligning each time point within the dynamic network, iteratively);
-
the method used to design the algorithm (e.g., GNA, or LNA);
-
network discrimination performance;
-
other minor additional information (e.g., tool availability, documentation availability).
As in motif discovery in dynamic networks, the features for comparison have been focused on:
-
the type of motif, e.g., DNA sequence motif, RNA sequence motif, or generic motif (not typed);
-
the strategy used to perform the discovering;
-
other minor additional information (e.g., tool availability).
4.1 Alignment of dynamic networks
In the literature, nearly all methods for the alignment of dynamic networks assume that the evolution rate is the same both for the target network and the source network (Zhang et al. 2016). NA (both statics and dynamics) considers an objective function and an optimization strategy to maximize it. As it is easy to see, the alignment of dynamic networks is based on the same notions applied for alignment of static networks. We report the following tools for the alignment of dynamic networks: DynaMAGNA++, DynaWAVE, GoT-WAVE, Tempo, Tempo++, Twadn.
DynaMAGNA++ (Vijayan et al. 2017) is an extension of MAGNA++ (Vijayan et al. 2015). The latter is a software tool for NA of static networks (see Sect. 3). DynaMAGNA++ is able to align dynamic networks in event duration-based representation (i.e., H(V, T), see Sect. 2). To give an example, Fig. 5 shows the NA performed by using DynaMAGNA++, which works as follows: given two dynamic networks \(H1(V_{1}, T_{1})\) and \(H2(V_{2}, T_{2})\), with \(\left| V_{1}\right| \le \left| V_{2}\right|\) (no loss of generality), an NA between H1 and H2 produces a set of aligned node pairs (v, f(v)), with \(v \in V_{1}\), by performing a one-to-one node mapping \(f : V_{1} \rightarrow V_{2}\). This approach makes comparable the NA for static and dynamic networks. According to the authors, DynaMAGNA++ is based on GNA, but this principle is also applicable to LNA. It generalizes an aligned edge to an aligned event, computing both the measure of dynamic edge conservation and the conserved event time (CET). Therefore, DynaMAGNA++ is able to evaluate events with similar end-nodes by calculating also the time during which two dynamic edge pairs are active. The event-based measure is a version adapted to dynamic networks of \(S^{3}\): \(DS^{3}\). Furthermore, the Graphlet Degree Vector (GDV) (Milenković et al. 2010) has been extended to implementing Dynamic GDV (DGDV) to measures NC in dynamic networks. Briefly, DynaMAGNA++ aligns two dynamic networks by evaluating the incident edges and the graphlet changes during the dynamic events, to calculate a value based on DGDV. A pair of nodes belonging to two different networks are similar, if their DGDVs are similar. However, DynaMAGNA++ does not scale well to large networks, in terms of alignment quality and runtime. This problem has been overcome by DynaWAVE.

The figure (Vijayan et al. 2017) shows the alignment performed by using DynaMAGNA++ on two dynamic networks (i.e., H1 and H2), in event duration-based representation. Solid lines represent the events between nodes. For instance, the event between \(u_{3}\) and \(u_{4}\) is active from start time 8 to end time 10. The mapping between two nodes is reported by using a dotted line
DynaWAVE (Vijayan and Milenkovic 2018) works by aligning dynamic networks in event duration-based representation, similarly to DynaMAGNA++. It has been developed extending an existing tool [i.e., WAVE (Sun et al. 2015)] from NA of static networks. WAVE maximizes its objective function by using a greedy seed-and-extend strategy based on the Weighted Edge Conservation (WEC) measure for edge conservation (EC), and a static GDV for node conservation (NC). WEC has been extended in DynaWAVE as Dynamic WEC (DWEC) to support dynamic edges (i.e., event).
Graphlet-orbit Transitions WAVE (GoT-WAVE) (Aparício et al. 2019) is an improvement of DynaWAVE for temporal Global Pairwise NA (GPNA). Both use DWEC to evaluate EC, but GoT-WAVE applies the Graphlet-orbit Transitions (GoT) as measure for NC. GoT is based on the construction of a matrix consisting of the product between the nodes’ vector and the transitions’ vector. Each matrix is used as a feature for the related dynamic network. During the alignment, GoT-WAVE joins the matrices of the two dynamic networks, performing also the principal component analysis (PCA) to reduce the size of resulting matrix. Furthermore, it computes the cosine similarity between nodes’ features, by using the latter as node similarities in the objective function. Briefly, GoT provides to GoT-WAVE more accuracy (\({+}\) 30%) and speed (\({+}\) 60%), compared to DynaWAVE. Note that WAVE, DynaWAVE, and GoT-WAVE use all the same optimization strategy to maximize the objective function.
Tempo performs the identification of co-evolving dynamic networks. It uses an own score to evaluate the similarities between the aligned nodes, as well as the topology over time. Furthermore, it assigns a penalty for each disconnected component in the aligned subnetworks (Elhesha et al. 2019a). Briefly, Tempo works by performing a pairwise alignment between pairs of static networks belonging to each of the two input networks, respectively. For each alignment, an initial similarity score is calculated. The dynamic alignment score is the sum of all the scores calculated for each static alignment. Subsequently, it performs a series of k swaps (k is an input parameter) between the aligned nodes to maximize this similarity score. Therefore, it works iteratively, limiting its computation to a maximum of k steps. The tool is not provided publicly; we take into account the data reported within the related paper.
Tempo++ is an improvement of Tempo, it aligns dynamic networks with different evolutionary rates (Elhesha et al. 2019b). It is able to identify co-evolving patterns in dynamic networks with varying rates of evolution. Two subnetworks are co-evolving if their topologies remain similar, even though their topologies evolve over time. Tempo++ adopts a dynamic programming technique similar to the dynamic time warping (DTW) (Linke et al. 2020) algorithm to measure the similarity (or dissimilarity) between the two given dynamic networks. Tempo++ bases its dynamic programming technique on a double indexed iterative procedure to map the time points of a network to the multiple consecutive time points of the other one, as opposed to DWT that maps only one time point of a network to multiple consecutive time points of the other one. The tool is not provided publicly; we take into account the data reported within the related paper.
Time Wrapping algorithm for Aligning Dynamic Networks (Twadn) allows the alignment of dynamic PPI networks (Zhong et al. 2020). It uses NetCoffee2 (Hu et al. 2019) and BLASTP (Altschul et al. 1990). The former extracts the topological feature of each node in a series of static networks based on a time sequence, while the latter aligns the proteins evaluating both e-value and bit-score for each protein pair, as sequence similarity scores. E-value and bit-score are usually applied for local sequence comparison for searching protein sequence, in genomics (Pearson 1995). It performs its computation through four main steps: (i) pairwise sequence alignment, extracting only the similar pairs; (ii) topological feature extraction from each static network by using NetCoffee2; (iii) the evaluation of dynamic time warping similarity of all pairs of proteins; (iv) alignment of the dynamic PPI networks applying its simulated annealing technique.
DynaMAGNA++, DynaWAVE, GoT-WAVE, Twadn, and Tempo, assume that the networks have same and uniform evolutionary rates. Unlike, Tempo++ does not base its computation on this assumption (see Fig. 6).

Two dynamic networks (i.e., G1 and G2) evolve at different time rates: from \(t_{1}\) to \(t_{4}\) for G1, and from \(t_{1}\) to \(t_{3}\) for G2. In this figure, the alignment between G1 and G2 has been performed by assuming different evolutionary rates. The alignment is shown through dashed lines, while subnets aligned in G1 are highlighted in bold
4.1.1 Summary of dynamic network alignment tools
Table 1 summarizes the tools discussed above, reporting the following information for each tool: name, year, objective function and/or similarity scores (i.e., “Objective/Score”), strategy, method applied for the alignment (i.e., “Method”), the algorithm used to align snapshots/networks (i.e., “Core”).
Figure 7 depicts the accuracy of the NA performed by DynaMAGNA++, DynaWAVE, Got-WAVE, and Twadn for biological synthetic networks with respect to the Area Under the Receiver Operating Characteristic (AUROC); range [0, 1], the higher score is the best result. Tempo and Tempo++ are not available for download, consequently these have not been tested. Data shown in the mentioned figure is reported in Table 2, based on the analysis described below.

Network discrimination performance of DynaMAGNA++, DynaWAVE, Got-WAVE, and Twadn for biological synthetic networks with respect to AUROC; range [0, 1], the best score is the higher. Tempo and Tempo++ are not available for test; consequently, the calculation of the score was not possible. Data are reported in Table 2
Let i a random node, and \(i^{\prime }\) a node with a link to all neighbors of i, then p is the probability of establishing a link between i and \(i^{\prime }\). Let j a node linked to i and \(i^{\prime }\), q is the probability of removing one of the two links ((j, i), or \((j, i^{\prime })\)). According to Zhong et al. (2020), the comparison has been based on two models consisting of 10 synthetic dynamic networks generated with \([p=0.3, q=0.7]\), and \([p=0.7, q=0.6]\), respectively. AUROC is a performance measurement used to evaluate classification models with different settings. Therefore, we used it to evaluate the performance of each objective function in terms of accuracy (Aparício et al. 2019). Formerly, it is defined as follows:
where TPR is the true positive rate, and FPR is the false positive rate.
In addition, Table 3 reports other additional information: Graphical User Interface (GUI) and/or Command Line Interface (CLI), programming language used for development (i.e., “Language”), code availability, documentation availability (i.e., “Documentation”).
4.2 Motif discovery in dynamic networks
Dynamic networks evolve over time, changing both their nodes and edges configuration. For instance, insertions and deletions change the network’s topology, impacting motif discovery negatively. Usually, dynamic motif discovery algorithms are optimized to handle two main cases: (i) changes in attributes of nodes/edges, or (ii) changes in topology. Let G a dynamic network consisting of a series of static networks \(G=[{G}_1,{G}_2,\ldots ,{G}_t]\), where \({G}_i=(V,{E}_i)\) (\(0 \le i \le t\) with t the total number of time points) represents the i-th snapshot. We assume that all snapshots within the dynamic network maintain a fix number of nodes (V) during its evolution, while topology may change through insertions and/or deletions. We denote \(g=(v,e)\) as a topological subgraph of \({G}_i\), if \(v \subseteq V_i\) and \(e \subseteq E_i\)). Furthermore, we denote g as a frequent dynamic subgraph if it appears more than a predefined number of times in the sequence G. Dynamic subgraphs may start and end at the same time points in the sequence (synchronous), or start/end at different time points within the series (asynchronous).
Motifs in temporal networks (i.e., temporal motifs) are an ordered sequence of timestamped edges conforming both to a specified pattern and to a specified duration of time in which the events must occur; Fig. 8 shows a non-exhaustive example of motifs in a dynamic network by using time-stamped edges.

A non-exhaustive example of motifs in a dynamic network by using time-stamped edges. The instances of the temporal motifs are shown in blue, while in red is shown a non-example of instance of the motif, as it does not satisfy the time requirements/constraints to be considered as such. The box, on the left, highlights the motif consisting of four nodes (vertex) and four events (temporal edges)
In literature, a large number of algorithms have been implemented simply counting motifs in undirected static graphs using an iterative approach. We report the tools related to motif discovery in dynamic networks by focusing attention on the approach used to perform the analysis, as well as on the type of data and graph supported.
COMmunication Motifs in InTeraction networks (COMMIT) (Gurukar et al. 2015) maps the graph in input (i.e., G) identifying all the temporally connected components in its sequence space representation (i.e., S) for frequent subsequence mining. It delegates its activities to three main modules, each one with a specific function: (i) “SEQGROW”, (ii) “ExtensionMiner”, and (iii) “MotifMine”. SEQGROW employs the sequence growth approach by extending S with a set of edges. Initially, all edges within an interval of time \(\Delta T\) in S are considered. Subsequently, SEQGROW calls ExtensionMiner to obtain only the closed edge labels E over a defined threshold and identifies the regions embedding communication motifs. The last step in COMMIT is performed by MotifMine that maps the frequent subsequences, obtained previously, into the original graph space. This modular approach ensures a high scalability.
Dynamic Motif Integration Toolkit (DynaMIT) (Dassi and Quattrone 2016) is a toolkit available as a package on Python Package Index (PIP, pypi.python.org). Formally, it provides a set of strategies based on existing python packages (e.g., matplotlib, scikit-learn, scipy, numpy) implementing custom components developed from scratch. It is based on three main steps supporting alignment, Jaccard similarity calculation, biclustering, co-occurrence evaluation; its steps are reported as follows: (i) motif discovery (i.e., alignment, Jaccard similarity calculation), (ii) motif clustering (i.e., biclustering, co-occurrence evaluation), and (iii) graphical representation of the result. Motif discovery is performed using several existing tools in accordance with the type of motif of interest, such as: DNA sequence motifs, RNA/miRNA sequence motifs, RNA secondary structure motifs, or generic regions of the input network. The pairwise alignment of each motif pair is used to provide scores for clustering component. Furthermore, DynaMIT offers to users the possibility of implementing custom additional components, integrating these within an existing function. This feature has been made available by introducing a set of abstract Python classes.
Kovanen et al. (2011) presented a framework for identification of temporal motifs (reported as appearing as “Kovanen”). Assuming G(V, E) an undirected graph where the nodes (vertices) corresponds to events, and E the related event set, the process for motif discovery is performed by “Kovanen” as follows: (i) find all maximal connected subgraphs in G (i.e., \(G^{*}_{{\mathrm{max}}}=(V^{*},E^{*}_{{\mathrm{max}}})\)); (ii) find all valid edge subsets \(E^{*}\) (\(E^{*} \subseteq E^{*}_{{\mathrm{max}}}\)); (iii) identify motifs corresponding to the set of valid subgraphs \(G^{*}=(V^{*},E^{*})\). This method finds a maximal subgraph iterating (forward, and backward) over the \(\Delta t\)-adjacent events, thus it applies a recursive approach with a worst-case complexity equal to \(\left| E\right|\).
Mukherjee et al. (2018) developed a method for counting motifs in a dynamic biological network (reported as appearing as “Mukherjee”). It focuses on biological dynamic networks that may be modeled as an undirected graph, even if authors report that this method could be applied also to directed graphs as well. It computes the initial counting by applying a static approach on the network at the time \(t_{0}\). Subsequently, the other time points are iteratively investigated to update the initial counting. This solution considers each time point as a static network.
SNAP (Paranjape et al. 2017) provides a method for counting motifs in temporal networks, providing also a formalism for temporal network motifs through a series of timestamped edges, or temporal edges.
Table 4 reports the key points for the discussed tools by focusing attention on typology, strategy, and availability.
5 Discussion
In biology, several open challenges focused their attention on computational approaches for detecting protein complexes from protein interaction networks (i.e., motif discovery). Extending common issues for static network analysis to dynamic networks, the basic concept remains essentially the same. However, a dynamic network introduces further complexities related to static networks, both for alignment and motif discovery (Kim et al. 2021).
Figure 9 shows discussed tools in accordance with the following criteria: static networks or dynamic networks (i.e., network), and the strategy implemented to solve the related problem (e.g., alignment, motif discovery). It shows also the relationship between the objective function and its counterpart for static networks, in order to highlight if the tool is the extension of a solution initially developed for a static context. For instance, the figure shows for DynaMAGNA++ the following information: (i) it performs the NA of dynamic networks, (ii) by applying an evolutionary strategy (iii) based on the \(DS^3\) as objective function for similarity score calculation. Deepening the relationships, (iv) \(DS^3\) is an extension of \(S^3\) that (v) is an objective function applied by MAGNA++ on static networks. Furthermore, (vi) MAGNA++ shares with MAGNA one or more features, e.g., one is the evolution of the other or a version with different applications. Detailed information about the mentioned software tools is available in the previous sections.
Similarly, COMMIT is able to perform (i) motif discovery in (ii) dynamic networks (iii) based on an iterative strategy. Regarding the software tools for motif discovery in dynamic networks, we have also taken into account the type of motifs supported as input. We identified two main types of motifs: generic, and typed. The former concerns all networks within which the nodes are not related to a specific function or biological category. The latter is related to the networks within which the nodes are representative of a specific object. Note that DynaMIT is the only that adopts a variable strategy based on the type of data, such as: DNA sequences, RNA sequences, miRNA sequences, and RNA secondary structure motifs. The other ones apply a single strategy, indifferently. Figure 9 includes this information by indicating the strategy as “variable”, when it is supported. Table 5 better summarizes the strategies implemented by the proposed software tools for dynamic networks.

The figure relates the type of networks in input (static, or dynamic) with the strategy implemented by each discussed tool, both for NA and motif discovery. To give an example, for DynaMAGNA++ the following information is deductible: (i) an evolutionary strategy (ii) based on the \(DS^3\) as objective function for similarity score calculation, (iii) performed on dynamic networks for (iv) alignment. Similarly, the figure reports for COMMIT: (i) a motif discovery tool for (ii) dynamic networks (iv) based on an iterative strategy. Note that a software tool could adapt its strategy according to the input (i.e., “variable”)
It is clear how the iterative strategy is applied by \({\sim }\,83\%\) (5/6) of the proposed tools for motif discovery. As mentioned, DynaMIT is the only one that applies a variable strategy in that it implements several existing motif searchers. This approach allows to better analyze biological networks. However, it needs of a large number of components that should be implemented de novo, or integrating existing tools. From a purely algorithmic point of view, it is inadvisable in this implementation, as the possible type of motif can be much more extensive than reported. In fact, a general-purpose strategy is the most used.
A more uniform distribution is evident among the tools for network alignment. Therefore, we focused attention on the approach applied to align the pair of dynamic networks (refer to Table 1): \({\sim }\,83\%\) (5/6) of the proposed tools are based on G(P)NA, contrary to Twadn that applies a method for LNA. It evidenced how G(P)NA is preferred on generic networks, while LNA is more indicated for networks in which exists a similarity matrix based on defined properties shared between all nodes (e.g., biological similarity for biological networks). However, G(P)NA supports also this feature, while the opposite is not true. According to a test conducted on biological synthetic networks between DynaMAGNA++, DynaWAVE, GoT-WAVE, Twadn, it seems that Twadn (based on LNA) overperformed \({\sim }\,66\%\) of the G(P)NA tools, except for GoT-WAVE. The latter is the only using the Graphlet-orbit Transitions (GoT) as measure for NC. According to Meng et al. (2016), G(P)NA outperforms LNA both topologically and biologically when only the topological information is available. Otherwise, LNA is superior to G(P)NA in biological quality when sequence information is taken into account. LNA is performed by Twadn, which showed a better network discrimination performance on biological synthetic networks, compared to DynaMAGNA++ and DynaWAVE. However, Twadn is limited to dynamic PPI networks, while the other software tools may be applied to all networks.
Investigating the tools for NA in dynamic networks, it emerged that some categories of problems may be transferred from static to dynamic networks by extending an alignment method for static networks in the dynamic context, or by calling the latter on each time point, iteratively. As reported, DynaMAGNA++ is based on MAGNA++, and DynaWAVE is based on WAVE, as well as GoT-WAVE. MAGNA++ and WAVE are two well-known tools for the alignment of static networks (see Sect. 3).
On the other hand, motif discovery in dynamic networks bases its strategy on techniques designed for static networks only partially. For instance, in COMMIT only the module related to mapping frequent subsequences could support also a static network, other modules would need a high degree of re-implementation related to the interaction between time points that cannot be neglected. Therefore, this discrepancy could be due to the fact that a re-implementation ex novo has a decidedly lower development time cost as any existing tool should be excessively updated to work with dynamic networks given the need to relate the various snapshots. However, the methodologies for motif discovery are afflicted from another issue related to the asynchronous sequences. It occurs when dynamic subgraphs start and/or end at different time points within the series. Tools for motif discovery in dynamic networks, presented previously, focused attention on topological changes through insertions and deletions of nodes and edges. In literature, a large number of algorithms have been implemented simply to counting motifs in undirected static graphs by using an iterative approach. This approach may be used also for large networks, even if it is computationally expensive (it is a typical NP-complete problem).
Whether we are talking about the NA or motif discovery applied to dynamic networks, it is evident how the main challenges may be associated with computational resources required to perform the analysis. As discussed, the analysis of graph/subgraph (network/subnetwork) isomorphism is quite computationally expensive, as well as the similarity score evaluations during NA.
6 Conclusions and future directions
As was shown by investigating the issues related to NA in dynamic networks, some categories of problems may be transferred from static to dynamic networks by extending an alignment method for static networks in the dynamic context. Most of the software tools reported in this paper are based on existing methodologies for NA between static networks. On the other hand, motif discovery does not follow this approach. From what was presented, motif discovery bases its strategy on techniques designed for static networks only partially. We suppose that this discrepancy is mainly due to the fact that a re-implementation ex novo has a decidedly lower development time cost, as any existing tool should be excessively updated to work with dynamic networks given the need to relate the various snapshots. Otherwise, a tool for NA of static networks may be called iteratively by producing an alignment for each pair of successive snapshots (e.g., the pair: \(t_{i}\) and \(t_{i+1}\)). Software tools for NA and motif discovery implement a computation based on iterative optimizations or greedy algorithms. This approach allows the re-use of code from the static to the dynamic context. However, it produces critical tasks that could lead to an NP-complete problem. As described, the latter may occur in the presence of identically overlapping instances. Finally, our investigation encountered a systematic convergence towards iterative strategies both for NA and motif discovery in dynamic networks, justified by the fact that a dynamic network is usually analyzed through the sub-analysis of its time points.
Future directions for networks analysis are moving toward the development of solutions based on deep learning and network embedding (Barros et al. 2021). Chiu and Zhan (2018) proposed a method to build a feature vector for a deep neural network based on weak estimators in addition to tradition similarity measures. It improves the model accuracy, as well as the related prediction accuracy, in order to estimate the changing probabilities in dynamic networks. Furthermore, authors explore another crucial task, consisting of the issue of link prediction in dynamic networks. In literature, this issue is studied to solve several problems. For instance, it may happen that the topologies of dynamic networks are not available at specific time points. Chow et al. (2021) presented an alignment-based network construction algorithm to predict missing target networks, allowing the analysis of incomplete biological dynamic networks. Networks embedding applies graph embedding techniques to analyze complex biological networks that are quite computationally expensive to solve by using common approach for network-based analysis (Mohan and Pramod 2021; Yu et al. 2020). To give an example, weg2vec (or Weighted Event Graph to Vector) (Torricelli et al. 2020) is a temporal network embedding method that is able to represent an entire temporal network in a reduced dimensional abstract space. It samples a higher-order static representation of a temporal network, coding also the complex patterns characterizing its structure and dynamics. It is based on an unsupervised representation learning technique, that identifies similarity between different events (i.e., nodes), by predicting both their activation and their influence on a similar set of nodes in a subsequent time point. The network embedding offers new opportunities to model higher-order network datasets, to perform efficiently data analysis and prediction (e.g., node classification, link prediction, node clustering), with considerable advantages for future studies in the field of (static, dynamic) biological networks.
Notes
In vivo: “Literally, in life, but used generally for procedures or tests done on intact organisms rather than on isolated cells in culture (in-vitro)”. Dictionary of Biomedicine, Oxford University Press. https://doi.org/10.1093/acref/9780199549351.001.0001.
References
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Aparício D, Ribeiro P, Milenković T et al (2019) Temporal network alignment via got-wave. Bioinformatics 35(18):3527–3529
Athanasios A, Charalampos V, Vasileios T et al (2017) Protein–protein interaction (PPI) network: recent advances in drug discovery. Curr Drug Metab 18(1):5–10
Ay F, Kellis M, Kahveci T (2011) SubMAP: aligning metabolic pathways with subnetwork mappings. J Comput Biol 18(3):219–235
Barabási AL, Gulbahce N, Loscalzo J (2011) Network medicine: a network-based approach to human disease. Nat Rev Genet 12(1):56–68
Barros CDT, Mendonça MRF, Vieira AB et al (2021) A survey on embedding dynamic graphs. arXiv:2101.01229
Cai Y, Wang J, Deng L (2020) SDN2GO: an integrated deep learning model for protein function prediction. Front Bioeng Biotechnol 8:391
Chen B, Fan W, Liu J et al (2014) Identifying protein complexes and functional modules-from static PPI networks to dynamic PPI networks. Brief Bioinform 15(2):177–194
Chiu C, Zhan J (2018) Deep learning for link prediction in dynamic networks using weak estimators. IEEE Access 6:35937–35945. https://doi.org/10.1109/access.2018.2845876
Chow K, Sarkar A, Elhesha R et al (2021) ANCA: alignment-based network construction algorithm. IEEE/ACM Trans Comput Biol Bioinform 18(2):512–524. https://doi.org/10.1109/tcbb.2019.2923620
Cinaglia P, Cannataro M (2022) Forecasting COVID-19 epidemic trends by combining a neural network with RT estimation. Entropy (Basel) 24(7):929. https://doi.org/10.3390/e24070929
Cinaglia P, Guzzi PH, Veltri P (2018) Integro: an algorithm for data-integration and disease-gene association. In: 2018 IEEE international conference on bioinformatics and biomedicine (BIBM), pp 2076–2081. https://doi.org/10.1109/BIBM.2018.8621193
Consortium GO (2006) The gene ontology (GO) project in 2006. Nucl Acids Res 34:D322-d326. https://doi.org/10.1093/nar/gkj021
Dai C, He J, Hu K et al (2020) Identifying essential proteins in dynamic protein networks based on an improved \(h\)-index algorithm. BMC Med Inform Decis Mak 20(1):110
Dassi E, Quattrone A (2016) DynaMIT: the dynamic motif integration toolkit. Nucl Acids Res 44(1):e2
Doria-Belenguer S, Youssef MK, Böttcher R et al (2020) Probabilistic graphlets capture biological function in probabilistic molecular networks. Bioinformatics 36(Suppl-2):i804–i812
Elhesha R, Kahveci T (2016) Identification of large disjoint motifs in biological networks. BMC Bioinform 17(1):408
Elhesha R, Sarkar A, Boucher C et al (2019a) Identification of co-evolving temporal networks. BMC Genom 20(Suppl 6):434
Elhesha R, Sarkar A, Cinaglia P et al (2019b) Co-evolving patterns in temporal networks of varying evolution. In: Proceedings of the 10th ACM international conference on bioinformatics, computational biology and health informatics. ACM, New York, NY, USA, Bcb’19, pp 494–503. https://doi.org/10.1145/3307339.3342152
Faisal FE, Meng L, Crawford J et al (2015) The post-genomic era of biological network alignment. EURASIP J Bioinform Syst Biol 2015:3
Fisher DN, Pinter-Wollman N (2021) Using multilayer network analysis to explore the temporal dynamics of collective behavior. Curr Zool 67(1):71–80
Ghoshal G, Chi L, Barabasi AL (2013) Uncovering the role of elementary processes in network evolution. Sci Rep 3:2920. https://doi.org/10.1038/srep02920
Gligorijević V, Malod-Dognin N, Pržulj N (2016) Fuse: multiple network alignment via data fusion. Bioinformatics 32(8):1195–1203
Guerra C, Guzzi P (2020) Evaluation of the topological agreement of network alignments. In: 2020 IEEE international conference on bioinformatics and biomedicine (BIBM). IEEE Computer Society, New York, pp 1785–1792. https://doi.org/10.1109/bibm49941.2020.9313513
Gupta S, Stamatoyannopoulos JA, Bailey TL et al (2007) Quantifying similarity between motifs. Genome Biol 8(2):R24
Gurukar S, Ranu S, Ravindran B (2015) Commit: a scalable approach to mining communication motifs from dynamic networks. In: Proceedings of the 2015 ACM SIGMOD international conference on management of data
Guzzi PH, Milenkovic T (2018) Survey of local and global biological network alignment: the need to reconcile the two sides of the same coin. Brief Bioinform 19(3):472–481
Holme P (2015) Modern temporal network theory: a colloquium. Eur Phys J B. https://doi.org/10.1140/epjb/e2015-60657-4
Holme P, Saramäki J (2012) Temporal networks. Phys Rep 519(3):97–125. https://doi.org/10.1016/j.physrep.2012.03.001
Hu J, He J, Li J et al (2019) A novel algorithm for alignment of multiple PPI networks based on simulated annealing. BMC Genom 20(Suppl 13):932
Hulovatyy Y, Chen H, Milenković T (2015) Exploring the structure and function of temporal networks with dynamic graphlets. Bioinformatics 31(12):i171-180
Huynh-Thu VA, Sanguinetti G (2019) Gene regulatory network inference: an introductory survey. Methods Mol Biol 1883:1–23
Ivarsson Y, Jemth P (2019) Affinity and specificity of motif-based protein–protein interactions. Curr Opin Struct Biol 54:26–33
Jazayeri A, Yang CC (2020) Motif discovery algorithms in static and temporal networks: a survey. J Complex Netw. https://doi.org/10.1093/comnet/cnaa031
Joakim S, Musial K, Gabrys B (2020) Foundations and modelling of dynamic networks using dynamic graph neural networks: a survey. CoRR arXiv:2005.07496
Ju J, Wei P (2017) Signaling network-based functional cell design. Sheng Wu Gong Cheng Xue Bao 33(3):386–392
Kim B, Lee KH, Xue L et al (2018) A review of dynamic network models with latent variables. Stat Surv 12:105–135
Kim H, Jo HH, Jeong H (2021) Impact of environmental changes on the dynamics of temporal networks. PLoS ONE 16(4):1–19. https://doi.org/10.1371/journal.pone.0250612
Klein C, Marino A, Sagot MF et al (2012) Structural and dynamical analysis of biological networks. Brief Funct Genom 11(6):420–433
Kovanen L, Karsai M, Kaski K et al (2011) Temporal motifs in time-dependent networks. J Stat Mech Theory Exp 2011(11):P11005. https://doi.org/10.1088/1742-5468/2011/11/p11005
Li M, Meng X, Zheng R et al (2020) Identification of protein complexes by using a spatial and temporal active protein interaction network. IEEE/ACM Trans Comput Biol Bioinform 17(3):817–827
Linke AC, Mash LE, Fong CH et al (2020) Dynamic time warping outperforms Pearson correlation in detecting atypical functional connectivity in autism spectrum disorders. Neuroimage 223(117):383
Ma CY, Liao CS (2020) A review of protein–protein interaction network alignment: from pathway comparison to global alignment. Comput Struct Biotechnol J 18:2647–2656. https://doi.org/10.1016/j.csbj.2020.09.011
Meng L, Striegel A, Milenković T (2016) Local versus global biological network alignment. Bioinformatics 32(20):3155–3164
Mertzios GB, Michail O, Chatzigiannakis I et al (2013) Temporal network optimization subject to connectivity constraints. In: Fomin FV, Freivalds R, Kwiatkowska M et al (eds) Automata, languages, and programming. Springer, Berlin, pp 657–668
Milano M, Guzzi PH, Tymofieva O et al (2017) An extensive assessment of network alignment algorithms for comparison of brain connectomes. BMC Bioinform 18(Suppl 6):235
Milenković T, Ng WL, Hayes W et al (2010) Optimal network alignment with graphlet degree vectors. Cancer Inform 9:121–137
Mohan A, Pramod K (2021) Temporal network embedding using graph attention network. Complex Intell Syst. https://doi.org/10.1007/s40747-021-00332-x
Mukherjee K, Hasan MM, Boucher C et al (2018) Counting motifs in dynamic networks. BMC Syst Biol 12(Suppl 1):6
Ou-Yang L, Dai DQ, Li XL et al (2014) Detecting temporal protein complexes from dynamic protein–protein interaction networks. BMC Bioinform 15:335
Paranjape A, Benson AR, Leskovec J (2017) Motifs in temporal networks. In: Proceedings of the 10th ACM international conference on web search and data mining. https://doi.org/10.1145/3018661.3018731
Patra S, Mohapatra A (2020) Review of tools and algorithms for network motif discovery in biological networks. IET Syst Biol 14(4):171–189
Patro R, Kingsford C (2012) Global network alignment using multiscale spectral signatures. Bioinformatics 28(23):3105–3114
Pearson WR (1995) Comparison of methods for searching protein sequence databases. Protein Sci 4(6):1145–1160
Rao VS, Srinivas K, Sujini GN et al (2014) Protein–protein interaction detection: methods and analysis. Int J Proteomics 2014:147648
Ren Y, Ay A, Kahveci T (2018a) Shortest path counting in probabilistic biological networks. BMC Bioinform 19(1):465
Ren Y, Sarkar A, Kahveci T (2018b) ProMotE: an efficient algorithm for counting independent motifs in uncertain network topologies. BMC Bioinform 19(1):242
Rossetti G, Cazabet R (2018) Community discovery in dynamic networks. ACM Comput Surv 51(2):1–37. https://doi.org/10.1145/3172867
Saraph V, Milenković T (2014) MAGNA: maximizing accuracy in global network alignment. Bioinformatics 30(20):2931–2940
Sarkar A, Ren Y, Elhesha R et al (2019) A new algorithm for counting independent motifs in probabilistic networks. IEEE/ACM Trans Comput Biol Bioinform 16(4):1049–1062
Singh R, Xu J, Berger B (2008) Global alignment of multiple protein interaction networks. Pac Symp Biocomput 2008:303–314
Sun Y, Crawford J, Tang J et al (2015) Simultaneous optimization of both node and edge conservation in network alignment via wave. In: Pop M, Touzet H (eds) Algorithms in bioinformatics. Springer, Berlin, pp 16–39
Torricelli M, Karsai M, Gauvin L (2020) Weg2vec: event embedding for temporal networks. Sci Rep 10(1):7164
Vijayan V, Milenkovic T (2018) Aligning dynamic networks with DynaWAVE. Bioinformatics 34(10):1795–1798
Vijayan V, Saraph V, Milenković T (2015) Magna++: maximizing accuracy in global network alignment via both node and edge conservation. Bioinformatics 31(14):2409–2411
Vijayan V, Critchlow D, Milenkovic T (2017) Alignment of dynamic networks. Bioinformatics 33(14):i180–i189
Vijayan V, Gu S, Krebs ET et al (2020) Pairwise versus multiple global network alignment. IEEE Access 8:41961–41974. https://doi.org/10.1109/access.2020.2976487
Wang J, Peng X, Peng W et al (2014) Dynamic protein interaction network construction and applications. Proteomics 14(4–5):338–352
Xia F, Wei H, Yu S et al (2019) A survey of measures for network motifs. IEEE Access 7:106576–106587. https://doi.org/10.1109/access.2019.2926752
Yu EY, Fu Y, Chen X et al (2020) Identifying critical nodes in temporal networks by network embedding. Sci Rep. https://doi.org/10.1038/s41598-020-69379-z
Zahiri J, Emamjomeh A, Bagheri S et al (2020) Protein complex prediction: a survey. Genomics 112(1):174–183
Zaslavskiy M, Bach F, Vert JP (2009) Global alignment of protein–protein interaction networks by graph matching methods. Bioinformatics 25(12):i259-267
Zhang Y, Lin H, Yang Z et al (2016) Construction of dynamic probabilistic protein interaction networks for protein complex identification. BMC Bioinform 17(1):186
Zhang J, Zhong C, Lin HX et al (2019) Identifying protein complexes from dynamic temporal interval protein–protein interaction networks. Biomed Res Int 2019:3726721
Zhong Y, Li J, He J et al (2020) Twadn: an efficient alignment algorithm based on time warping for pairwise dynamic networks. BMC Bioinform 21(Suppl 13):385
Zhu L, Zhang J, Zhang Y et al (2020) NAIGO: an improved method to align PPI networks based on gene ontology and graphlets. Front Bioeng Biotechnol 8:547
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cinaglia, P., Cannataro, M. Network alignment and motif discovery in dynamic networks. Netw Model Anal Health Inform Bioinforma 11, 38 (2022). https://doi.org/10.1007/s13721-022-00383-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13721-022-00383-1