
Abstract
Anthropogenic activity, especially modern apiculture, has considerable impact on the natural distribution of the Western honeybee, Apis mellifera, leading to the spread, replacement and fragmentation of many subspecies. This creates demand for the conservation of some subspecies, in particular, Apis mellifera mellifera, which once was widely distributed in Western Europe and nowadays is endangered through habitat loss and fragmentation. Moreover, A. m. mellifera may be further endangered by hybridisation in populations that now occur in artificial sympatry with other subspecies. Here, we quantify and compare individual hybridisation between sympatric and allopatric honeybee populations of A. m. mellifera and A. m. carnica using microsatellite markers and a Bayesian model-based approach. We had a special focus on pure breeding populations, which are a major tool in honeybee conservation. Our results demonstrate that subspecies are still highly differentiated, but gene flow is not prevented by the current management strategies, creating urgent demand for an improved conservation management of A. m. mellifera. However, the occurrence of a high number of pure individuals might suggest that some sort of hybrid barrier acts against the complete admixture of the two subspecies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Human activity has a major impact on the distribution of the Western honeybee, Apis mellifera. While the species’ original range in Africa, western Asia and Europe (Ruttner 1988) has enormously extended to new continents over the last centuries, this process has not affected all subspecies to the same extent. Some of the 24 allopatrically distributed subspecies (Ruttner 1988) have experienced massive range expansions, while others faced contractions and fragmentation of original populations after the introduction of non-native honeybees by humans (Ruttner 1988; Garnery et al. 1998; Franck et al. 2000; De la Rua et al. 2002; Schneider et al. 2004; Jensen et al. 2005). These honeybee movements certainly involved admixture between different subspecies belonging to different evolutionary lineages, like lineage M from Western Europe (A. m. mellifera), lineage C from south-eastern Europe (A. m. carnica and A. m. ligustica) and lineage A from Africa (Cornuet et al. 1991; Garnery et al. 1992; Franck et al. 2001).
Introductions in Europe
The black honeybee, A. m. mellifera, used to occupy an area expanding from the Pyrenees to the Ural, which is the largest territory occupied by any European subspecies (Ruttner 1988). This large distribution suggests a high potential of adaptation to changing environments and the occurrence of various different ecotypes (Garnery et al. 1992; Jensen et al. 2005). Therefore, its protection is of considerable conservational interest. However, major introductions of other honeybee subspecies have occurred over 150 years in the original range of A. m. mellifera (Fig. 1). In large parts of Western Europe, the two sister subspecies A. m. carnica and A. m. ligustica of evolutionary lineage C have continuously been introduced. In Germany, A. m. mellifera was almost completely replaced by A. m. carnica (Moritz 1991) and in Denmark by A. m. ligustica (Jensen et al. 2005). In France, distinct mtDNA haplotypes and nuclear alleles of evolutionary lineage C (A. m. carnica and A. m. ligustica) were detected, indicating admixture of south-eastern subspecies with the original population of lineage M (A. m. mellifera, Garnery et al. 1998). In Northern Europe, admixture was detected in a British population analysing nuclear and mitochondrial markers (Jensen et al. 2005). In Spain, eastern mtDNA haplotypes were detected in populations of A. m. iberica, a sibling subspecies of A. m. mellifera (Radloff et al. 2001; De la Rua et al. 2002, 2003). In conclusion, A. m. mellifera is potentially endangered mainly due to the strong reduction and fragmentation of its original distribution range (Büchler and Pechhacker 2005; Jensen et al. 2005).

Original (after Ruttner 1988) (a) and assumed (www.sicamm.org) modern (b) distribution areas of A. m. mellifera
Hybridisation
The ability of the subspecies to hybridise represents another potential threat to A. m. mellifera. Although hybridisation occurs (Franck et al. 2000; Jensen et al. 2005; Schneider et al. 2004), it is not yet known to what extent it actually leads to genetic introgression by the ability of the hybrids to reproduce. In order to propose measures for the conservation of A. m. mellifera, it appears necessary to understand the mechanisms underlying hybridisation and to quantify hybridisation among the subspecies, which now occur in artificial sympatry. Besides conservation issues, artificial secondary contact zones are of considerable interest in order to study evolutionary processes and speciation mechanisms among evolutionary lineages and subspecies. For example, African subspecies displayed a high reproductive advantage in neotropic climates that resulted in the replacement of European colonies (Hall and Muralidharan 1989; Clarke et al. 2001, 2002). The aim of our study is therefore to quantify the amount of introgression and the hybridisation potential between the two main European evolutionary lineages of the honeybee (M and C). We chose two areas in Switzerland. In the first one, the indigenous A. m. mellifera now occurs in artificial sympatry with A. m. carnica. In the second area, A. m. mellifera has been replaced area-wide by A. m. carnica over the last 40 years. In both areas, so-called pure breeding populations have been established by apiculturists for A. m. mellifera and/or A. m. carnica, respectively. In these pure breeding populations, virgin queens are mated with selected males (drones) at isolated mating stations in order to prevent hybridisation between subspecies (Böttcher 1947). In case of A. m. carnica, they serve as stock improvement tools. For A. m. mellifera, pure breeding populations are also a major conservation instrument to avoid gene flow from A. m. carnica. In this study, we quantify genetic differentiation and hybridisation among pure Swiss breeding populations using nuclear markers. We further quantify the amount of introgression at the individual and population level that may occur despite pure breeding management. Given that the current management strategies are sufficient, we expect no hybridisation in the pure breeding populations. If however, hybridisation will be detected, conservation efforts for the black honeybee, A. m. mellifera, must be improved.
Methods
Honeybee samples
In 2003, one worker each was sampled from a total of 274 colonies in 15 Swiss pure breeding populations (Fig. 2; Table 1). Six A. m. mellifera and five A. m. carnica populations were located in eastern Switzerland, where both subspecies now occur in sympatry. Four A. m. carnica populations were located in western Switzerland, where A. m. mellifera has been replaced area-wide by A. m. carnica. Additionally, one worker each was sampled from 208 colonies in 11 reference populations throughout Europe (Fig. 2; Table 1). Reference populations for A. m. mellifera originated from Lozère, France (F); Dalsland, Sweden (S) and Flekkefjord, Norway (N). Samples from Norway were obtained from a legally protected conservation area for A. m. mellifera. The sample from France originated from a very secluded area with no known introduction of foreign subspecies. Reference samples for A. m. carnica originated from Slovenia (SL) with its country-wide legal protection of A. m. carnica and from some Austrian counties (AC) that also have legal protection for this subspecies. One pure breeding population from Austria (AZ) was included, because many imports into Switzerland originated from this specific population. We included three population samples from A. m. ligustica from Italy (Emilia Romagna (ER), Lazio (LA) and Lombardi (LO), Fig. 2; Table 1). We obtained a mean sample size of 18.5 workers per population. To ensure that all sampled workers originated from different colonies; they were randomly sampled from brood cells and stored in pure ethanol until further DNA processing.

European reference samples (a) and Swiss sample sites (b). Sample sites are indicated with their code from Table 1. Black labels indicate samples of A. m. mellifera, grey of A. m. carnica, white of A. m. ligustica. Within Switzerland, the line indicates the geographic separation of western and eastern Switzerland
DNA extraction and microsatellite analyses
Whole genomic DNA was extracted with MagneSil® BLUE beads (Promega) using the following modified protocol of the producer: About 20 μl of MagneSil® BLUE beads were added to 200 μl digestion solution and shaken for 5 min. The beads were then bound to the side wall of the tube with a MagnaBot® Magnetic Separation Device (Promega) and the digestion solution was removed. The beads were washed twice with 200 μl of 80% EtOH for 5 min. After a drying step of the beads for 20 min at 55°C, the elution was performed by adding 50 μl of ddH2O for 5 min and subsequent removal as described above. DNA concentrations ranged between 63.5 and 165.5 ng/μl with a mean of 97.9 ng/μl (n = 16). Nine microsatellite loci (A007, A28, A43, Ac306, Ap33, Ap273, Ap226, Ap289, B24; Solignac et al. 2003) were amplified in one multiplex reaction. Reactions were performed in a 10 μl reaction volume using a Multiplex PCR Kit (QIAGEN) following the protocol of the producer with continuously modified primer concentrations and running 33 amplification cycles. Fragments were run on a 3100 Prism Genetic Analyser (Applied Biosystems) with an internal LIZ size standard (ABI) and scored with the software Genemapper 3.0 (ABI).
Population genetic analyses
Pairwise Fst values between populations were calculated using the program Arlequin 2001. To calculate the average number of alleles per locus and population the software Genepop (Raymond and Rousset 1995) was used. A neighbour-joining tree was constructed with the program Populations 1.2.28 using Nei’s Da distance (Nei et al. 1983). Bootstrap was performed using 2,000 replicates and edited with the program TreeExplorer 2.12. To quantify the degree of genetic divergence among the groups of populations, Analyses of Molecular Variance (AMOVA) (Excoffier et al. 1992) were performed with the program Arlequin 2001. To test for the assignment to different subspecies the number of groups was evaluated following Evanno et al. (2005) using the program Structure 2.0 (Falush et al. 2003) including all of the Swiss and reference data with no prior information about subspecies association or population groupings. The resulting groups where then associated to the subspecies of the respective reference populations therein and the other populations assigned accordingly. To estimate admixture proportions, many methods have been developed, some using Maximum-Likelihood approaches (Chikhi et al. 2001; Wang 2003), others using Bayesian model-based clustering methods and the most recent method following an Approximate Bayesian Clustering approach (ABC) (Excoffier et al. 2005), performing especially well for more ancient admixture events. However, population estimates do not necessarily give information about the actual amount of introgression into a population. The overall admixture coefficient does give information only on the population but not on the individual basis. Admixture could occur without actual introgression, meaning that hybrids are viable but somehow lack the ability to reproduce. For the management of pure breeding populations the average admixture proportions are of secondary interest. The identification of single individuals as pure or hybrids is much more valuable, in order to accept or exclude them from breeding programs.
In our study, we concentrated on hybridisation on an individual basis using a Bayesian framework of model-based clustering developed by Anderson and Thompson (2002) that was specifically developed for the identification of hybrid individuals in a potentially admixed population. The method is implemented in the program NewHybrids 1.1 beta, which assumes that hybridisation could have arisen during two generations and estimates the probability of an individual to belong to one of six different hybrid classes (pure 1, pure 2, F1 = pure 1 × pure 2, F2 = F1 × F1, backcross 1 = F1 × pure 1, backcross 2 = F1 × pure 2). The contribution of one group to the hybrid classes are the following; pure 1 and 2 = 100% and 0%, respectively, F1 and F2 = 50%; backcross 1 and 2 = 75% and 25%, respectively. Computations were run for each population separately including the populations from Norway (N), France (F), Slovenia (SL) and Austria (AC) as references. Besides the reference populations, no prior information about subspecies, population or group associations were used. The threshold for hybrid individuals was set to P < 0.95 within the hybrid class “pure”. We chose to use an even more stringent threshold than the one proposed by Vähä and Primmer (2006) in order to reduce the misclassification of backcrossed individuals as pure ones to a minimum. Q-values (individual hybrid proportions) for Hybrid index scores were calculated with the program Structure 2.0 assuming three groups (K = 3) and summing the P values for the contributions of the two C-groups.
Results
The analyses of population differentiation revealed an Fst value of 36.8% among the evolutionary lineages M and C. Within A. m. carnica we found evidence for a distinct phylogeographic substructure.
Pairwise Fst
Overall, the results showed a highly significant differentiation among most of the population pairs (Table 2). In general, lower Fst values were observed between populations from the same evolutionary lineage. A clear increase in differentiation was observed among populations from different regions (eastern-western Switzerland) irrespective of the evolutionary lineage (Table 2).
Population tree
The construction of a phylogenetic tree using Nei’s genetic distance (Da) showed a distinct separation of the two evolutionary lineages (Fig. 3) supported by bootstrap values of 65% for the branch with the C-Group populations and 82% for the branch with the A. m. mellifera populations. The Tyrolean A. m. mellifera population clusters between the C-Group and the A. m. mellifera populations. The Austrian samples form a sub branch within the C-Group supported by a bootstrap value of 63%. Another sub branch on the tip of the C-group is formed by the A. m. ligustica populations, supported by a bootstrap value of 58%. Within the M branch, the two reference populations from Norway and France formed a sub cluster at the tip of the branch, supported by a bootstrap value of 60%.

Unrooted Neighbour Joining tree of populations based on Nei’s Da distance (Nei et al. 1983). Grey indicates populations of A. m. carnica, black of A. m. mellifera, white of A. m. ligustica. Squares indicate populations from western Switzerland, circles of eastern and southern Switzerland and triangles of reference populations. Nodes with Bootstrap values larger than 50% (2,000 replicates) are indicated in percentages
Structure
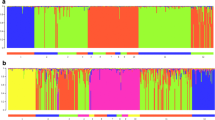
The procedure by Evanno et al. (2005) to assess the correct number of groups for a given dataset with the program Structure 2.0, reveals one distinct partition into two groups of populations separating the A. m. mellifera populations from the populations of lineage C (Fig. 4). The two groups were further analysed separately following Falush et al. (2003) to detect underlying structures that cannot be identified due to more distinct separations. We could find a clear second structure within the C lineage, which was already suggested by the high Fst value within this group (see AMOVA) following the separation into a northern and southern alpine group (Fig. 4). Swiss A. m. carnica populations were assigned to the group with the highest average contribution within the respective population. The populations from Austria (AC, AZ) as well as Gibidum (GI), Greina (GN) and Muotathal (MU) formed one group. The second group includes all the other Swiss A. m. carnica populations, the Slovenian population as well as the A. m. ligustica populations. The vertical bars in the individuals plot in Fig. 4 shows the respective contribution of each group for each individual.

Individuals plot of admixture constructed with the software Distruct displaying the results of the analysis performed with Structure 2.0, assuming K = 3. Vertical bars represent individuals. Colours denominate the relative contribution of one of the three respective parental groups within each single individual
AMOVA
The result of the Analyses of Molecular Variance (AMOVA) revealed a distinct substructure with a significant overall Fst value of 29.3% (P < 0.001) among all populations. Within groups of subspecies, the highest Fst value was detected in the A. m. carnica group with 13.6%, (P < 0.001) followed by the A. m. mellifera group with 8.4% (P < 0.001). A very small, none significant Fst value was detected among the populations of A. m. ligustica (Fst = 0.3%; P = 0.39). No significant differentiation between A. m. carnica and A. m. ligustica could be detected (Table 3). Analyses of the differentiation among groups of populations revealed no significant differentiation of the Swiss A. m. carnica and A. m. mellifera to their respective European reference populations. But within A. m. carnica a significant amount of differentiation could be detected among the northern and southern alpine populations discovered in the Structure analysis (Table 3). The differentiation of Swiss A. m. mellifera populations to each of the Swiss A. m. carnica groups revealed a larger differentiation to the allopatric western Swiss A. m. carnica (Fst = 42.1%; P = 0.005) than to the sympatric eastern Swiss A. m. carnica (Fst = 29.6%; P = 0.037). Within the A. m. mellifera a small but significant differentiation from the northern (N, S) to the southern (Swiss, F) populations was detected (Table 3).
Hybridisation
For A. m. carnica, hybrids were detected in 75% of the pure breeding populations in western and in 80% of the breeding populations in eastern Switzerland. Similarly, in 83.3% of the A. m. mellifera pure breeding populations, hybrids were also detected.
Different admixture patterns
The distribution of hybrid classes within individuals revealed a high relative amount of F1 hybrids in the population Gibidum (GI) and a minor amount in the populations of Greina (GN) and Tyrol (AM). F2 hybrids were detected in the populations Rotbach (RO), Tyrol (AM), Gletsch (GL), Petit-Mont (PM) and Justistal (JU). Backcrosses with A. m. carnica or A. m. mellifera were only detected in A. m. carnica or A. m. mellifera populations, respectively except Tyrol (AM). Backcrosses could be detected in all the populations with hybrids except in Petit-Mont (PM). Overall, the highest amount of backcrossed individuals could be detected in the populations of Swiss A. m. mellifera.
Number of pure and hybrid individuals
The number of pure and hybrid individuals varied greatly among populations (Fig. 5; Table 4). In nine populations, no hybrids were detected: one A. m. carnica population from western A. m. carnica (MO) and one from eastern Switzerland (SC), one eastern Swiss A. m. mellifera (SI) and the A. m. mellifera reference populations from Norway (N), Sweden (S) and France (F) as well as the A. m. carnica reference populations from Slovenia (SL) and Austria (AC, AZ). While two populations from western Switzerland (BO, TL) did show a small amount of hybrid individuals (4.8%), the Tyrolean supposed to be A. m. mellifera population revealed 80% hybrids and 20% pure A. m. carnica instead. The highest relative number of hybrid individuals beside Tyrol was detected in the Swiss A. m. mellifera populations Rotbach (RO, 70.8%) followed by Gletsch (GL, 68.8%). A comparison of the proportions of pure and hybrids individuals between the three different Swiss groups of populations is shown in Table 4. Within A. m. carnica, the eastern assumed allopatric population group had hybrids but significantly fewer compared to the western one. The Swiss group of A. m. mellifera populations had significantly more hybrids compared to both Swiss A. m. carnica groups.

Number of individuals in each hybrid class for each population. M = pure A. m. mellifera, C = pure A. m. carnica, F1 = pure A. m. mellifera × A. m. carnica, F2 = F1 × F1, BM = Backcross of F1 × A. m. mellifera, BC = Backcross of F1 with A. m. carnica. Population codes are listed in Table 1
Hybrid index score
The histogram of the individual admixture values for each group of pure breeding populations in Switzerland (Fig. 6) revealed a strong skew towards pure individuals. A considerable amount of backcrossed individuals could be detected, with Q-values ranging between approximately 0.05 and 0.3. A small hybrid swarm indicating F1 hybrids (individuals with Q-values around 0.5), was detected in eastern Switzerland, where both subspecies occur in artificial sympatry. Overall, the sympatric population (Fig. 6a) revealed a higher amount of hybrid individuals than the assumed allopatric population (Fig. 6b).

Hybrid index scores for the eastern (a) and western (b) Swiss honeybee populations. The Q-values range from 0 (pure A. m. mellifera) to 1 (pure A. m. carnica). Individuals originating from A. m. mellifera populations are marked in black, individuals from A. m. carnica populations are marked in grey
Discussion
Our data show that despite extensive efforts by beekeepers, artificial sympatry of indigenous A. m. mellifera with introduced A. m. carnica results in considerable hybridisation and subsequent introgression among honeybee populations. Besides habitat loss and fragmentation, this imposes another major threat for the black honeybee A. m. mellifera in admixed environments, thereby creating demand for an improved conservation management. Indeed, our data reveal hybrids in most pure breeding populations, strongly suggesting that management strategies should be adjusted.
Genetic differentiation
The bootstrap values in the population tree support a distinct differentiation between the two main evolutionary lineages (Fig. 3). In fact, the extent of differentiation between A. m. mellifera of the lineage M and the two subspecies of lineage C (A. m. carnica and A. m. ligustica) accounts for 35% of the total variation (AMOVA). This is well within the range observed in other studies (16–55%, Garnery et al. 1998; Franck et al. 2000; Jensen et al. 2005), suggesting that the European evolutionary lineages are still differentiated to a large extent despite human induced artificial sympatry. This strong differentiation might be assigned to the artificial selection pressure on the pure breeding populations thereby reducing genetic differentiation. However, the occurrence of hybrids in most pure breeding populations demonstrates that the conventional methods are not able to identify hybrids and are thus not suitable to prevent gene flow among subspecies.
Besides the strong differentiation among the evolutionary lineages, substructures were also detected within the subspecies indicating a geographic pattern. For A. m. mellifera, Jensen et al. (2005) described a substructure separating the populations from Scandinavia and the British Isles (5.81%). Our results show an additional separation of the Scandinavian and the more southern European populations in Switzerland and France (4.7%, Table 3). Although significant, the amount of differentiation is small and thus indicative for a fast colonisation speed after the last retreat of the ice (Hewitt 1996). The faster a species colonises an empty area through long distance migration or long range leaps, the fewer diversity should be observed in the newly colonised habitats. This supports the results of an mtDNA study and a survey of morphological traits, suggesting that A. m. mellifera has naturally colonised Northern Europe from is southern refuge in France (Ruttner 1988; Garnery et al. 1992). This colonisation pattern has not yet been observed in other species, where northern Europe has always been colonised at least partially out of refuge areas from the Balkan or areas further east (Hewitt 1999, 2000). In the eastern lineage C, the detected substructure was not in agreement with the original subspecies definitions based on the morphological description of Ruttner (1988). The Slovenian A. m. carnica population (southern alpine) is more differentiated from the population of the same subspecies in Austria (northern alpine) than from the A. m. ligustica populations in northern Italy, suggesting that the Alps constitute a natural barrier for gene flow in honeybees. This also indicates that within an evolutionary lineage of honeybees it appears difficult to assign a population to a specific subspecies based on morphometric traits alone. In the case of the southern-alpine group, our results suggest that some amount of gene flow either ancient or recent has lead to the homogenisation of the two subspecies. This supports the results of a study on the Italian honeybee population that proposed the emergence of A. m. ligustica by introgression of the eastern lineage C into a persisting ancient population of lineage M in northern Italy (Franck et al. 2000). It can well be imagined, that the low sea level during glaciation cycles facilitated gene flow among the Italian peninsula and its initial colonisation area in the east (Garnery et al. 1992). The differentiation within A. m. carnica is also detectable in the Swiss populations (Fig. 4) probably reflecting the origin of the queens that have been used to found these populations and that are still imported to supply the pure breeding populations.
Although only two distinct subgroups could be detected within A. m. carnica, all of the populations are significantly differentiated to each other (Table 2), indicating little gene flow among the populations. This could be a result of the different origins of the populations followed by a strong selection pressure and little queen exchange. In contrast, the A. m. mellifera pure breeding population in Switzerland display a quite homogenous group structure. This suggests that either gene flow occurs among these populations or that artificial selection pressures are too weak to alter the structure of the population group.
Hybridisation and introgression
We found clear evidence for hybridisation and subsequent introgression in form of second generation hybrids like F2 and backcrosses. Our results therefore show that besides habitat loss and fragmentation, artificial sympatry of European subspecies of A. mellifera imposes a major threat for the conservation of the black honeybee A. m. mellifera in the remaining population fragments.
Although hybridisation is not reversible and will inevitably end by complete admixture of the parental populations, unless some kind of hybrid barrier prevents this scenario (Allendorf et al. 2001), high proportions of pure A. m. mellifera and A. m. carnica individuals were still found (Fig. 6a). In case of A. m. carnica, the occurrence of pure individuals could be due to the ongoing import of pure queens from abroad. This might be the reason for the reduced amount of hybrid individuals in pure breeding A. m. carnica populations compared to A. m. mellifera, which solely relies on local breeding stocks. However, high levels of pure individuals were also found in A. m. mellifera populations. The formation of a hybrid swarm (Fig. 6a) can stabilise even in the course of hybrid counter-selection (Allendorf et al. 2001). If fertile hybrids are produced but no complete admixture can be observed, either the hybridisation event is very recent or there exist mechanisms that obstruct the hybridisation process. In case of the Swiss sympatric honeybee populations, where only a small hybrid swarm was detected (Fig. 6a), the first possible hybridisation events dates back at least 150 years as suggested by advertisements in the Swiss Bee Journal offering “Carnolian” bees from Austria. Therefore, it is very unlikely that the formation of a small hybrid swarm is due to the young age of the admixed population. Instead, we here propose that the high proportion of pure A. m. mellifera individuals in a sympatric habitat is maintained by some sort of incomplete hybridisation barrier. Assortative mating between subspecies could be one potential mechanism for such a hybridisation barrier and has been reported for A. m. carnica and A. m. ligustica probably due to differential vertical distribution of male sexuals (drones) at mating areas (Koeniger et al. 1989).
In order to increase the effectiveness of conservation areas and pure breeding populations, more information about the dispersal distances of drones and queens is inevitable to avoid gene flow into the endangered subspecies A. m. mellifera. Topographic structures like mountains and large water-bodies were proposed to inhibit flight paths of drones (Ruttner 1976; De la Rua et al. 2002). Although there are various reports on mating flight frequencies, mating distances and mating success of drones and queens (Ruttner 1956, 1976; Ruttner and Ruttner 1965, 1966, 1968, 1972; Koeniger 1986; Berg et al. 1997; Schlüns et al. 2003, 2004, 2005a, b; Koeniger et al. 2005), detailed knowledge about mating distances is lacking. Because bee breeders use alpine mating apiaries that are isolated by up to 10 km from the surrounding population, our results suggest that either some proportion of drones or queens regularly extend their mating flights beyond this distance. Alternatively, but not mutually exclusive, the identification of hybrid individuals by the bee breeders using conventional morphometric methods is not sufficient. We here suggest improving the conservation management of A. m. mellifera via an enhanced identification of hybrids. The methods applied in this study have shown to be a very useful tool for such identification in order to exclude hybrids from conservational breeding. Deeper insights about the mechanisms of hybridisation and mating distances will further support conservation efforts of honeybee subspecies.
References
Allendorf FW, Leary RF et al (2001) The problems with hybrids: setting conservation guidelines. Trends Ecol Evol 16:613–622
Anderson D, Thompson EA (2002) A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160:1217–1229
Berg S, Koeniger N et al (1997) Body size and reproductive success of drones (Apis mellifera L.). Apidologie 28:449–460
Böttcher FK (1947) Was taugt die Belegstelle? Der Imkerfreund 2:115–116
Büchler R, Pechhacker H (2005) Europäische Aktivitäten und Kooperation im Bereich der Bienenzucht. SBZ
Chikhi L, Bruford MW et al (2001) Estimation of admixture proportions: a likelihood-based approach using Markov chain Monte Carlo. Genetics 158:1347–1362
Clarke KE, Oldroyd BP et al (2001) Origin of honeybees (Apis mellifera L.) from the Yucatan peninsula inferred from mitochondrial DNA analysis. Mol Ecol 10:1347–1355
Clarke KE, Rinderer TE et al (2002) The Africanization of honeybees (Apis mellifera L.) of the Yucatan: a study of a massive hybridization event across time. Evolution 56:1462–1474
Cornuet J-M, Garnery L et al (1991) Putative origin and function of the intergenic region between COI and COII of Apis mellifera L. mitochondrial DNA. Genetics 128:393–403
De la Rua P, Galian J et al (2002) Biodiversity of Apis mellifera populations from Tenerife (Canary Islands) and hybridisation with East European races. Biodivers Conserv 11:59–67
De la Rua P, Galian J et al (2003) Genetic structure of Balearic honeybee populations based on microsatellite polymorphism. Genet Sel Evol 35:339–350
Evanno G, Regnaut S et al (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Smouse PE et al (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondria1 DNA restriction data. Genetics 131:479–491
Excoffier L, Estoup A et al (2005) Bayesian analysis of an admixture model with mutations and arbitrarily linked markers. Genetics 169:1727–1738
Falush D, Stephens M et al (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Franck P, Garnery L et al (2000) Hybrid origins of honeybees from Italy (Apis mellifera ligustica) and Sicily (Apis mellifera sicula). Mol Ecol 9:907–921
Franck P, Garnery L et al (2001) Genetic diversity of the honeybee in Africa: microsatellite and mitochondrial data. Heredity 86:420–430
Garnery L, Cornuet J-M et al (1992) Evolutionary history of the honey bee Apis mellifera inferred from mitochondrial DNA analysis. Mol Ecol 1:145–154
Garnery L, Franck P et al (1998) Genetic biodiversity of the West European honeybee (Apis mellifera mellifera and Apis mellifera iberica): II. Microsatellite DNA. Genet Sel Evol 30:49–74
Hall HG, Muralidharan K (1989) Evidence from mitochondrial DNA that African honey bees spread as continuous maternal lineages. Nature 339:211–213
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276
Hewitt GM (1999) Post-glacial re-colonization of European biota. Biol J Linn Soc 68:87–112
Hewitt G (2000) The genetic legacy of the quaternary ice ages. Nature 405:907–913
Jensen AB, Palmer KA et al (2005) Varying degrees of “Apis mellifera ligustica” introgression in protected populations of the black honeybee, “Apis mellifera mellifera”, in northwest Europe. Mol Ecol 14:93–106
Koeniger G (1986) Mating sign and multiple mating in the honeybee. Bee World 67:141–150
Koeniger N, Koeniger G et al (1989) Assortative mating in a mixed population of European honeybees, Apis mellifera ligustica and Apis mellifera carnica. Insect Soc 36:129–138
Koeniger N, Koeniger G et al (2005) The nearer the better? Drones (“Apis mellifera”) prefer nearer drone congregation areas. Insect Soc 52:31–35
Moritz RFA (1991) The limitations of biometric control on pure race breeding in Apis mellifera. J Apic Res 30:54–59
Nei M, Tajima F et al (1983) Accuracy of estimated phylogenetic trees from molecular data. J Mol Evol 19:153–170
Radloff SE, Hepburn HR et al (2001) Morphometric affinities and population structure of honeybees of the Balearic Islands in the Mediterranean Sea. J Apic Res 40:97–103
Raymond M, Rousset F (1995) Genepop (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Ruttner F (1956) The mating of the honeybee. Bee World 37:3–15
Ruttner H (1976) Untersuchungen über die Flugaktivität und das Paarungsverhalten der Drohnen. VI. Flug auf und über Höhenrücken. Apidologie 7:331–341
Ruttner F (1988) Biogeography and taxonomy of honeybees. Springer Vlg, New York
Ruttner F, Ruttner H (1965) Untersuchungen über die Flugaktivität und das Paarungsverhalten der Drohnen. II. Beobachtungen an Drohnensammelplätzen. Z Bienenforsch 8:1–9
Ruttner F, Ruttner H (1966) Untersuchungen über die Flugaktivität und das Paarungsverhalten der Drohnen. III. Flugweite und Flugrichtung der Drohnen. Z Bienenforsch 8:332–354
Ruttner F, Ruttner H (1968) Untersuchungen über die Flugaktivität und das Paarungsverhalten der Drohnen. IV. Zur Fernorientierung und Ortsstetigkeit der Drohnen auf ihren Paarungsflügen. Z Bienenforsch 9:259–265
Ruttner F, Ruttner H (1972) Untersuchungen über die Flugaktivität und das Paarungsverhalten der Drohnen. V. Drohnensammelplätze und Paarungsdistanz. Apidologie 3:203–232
Schlüns H, Schlüns EA et al (2003) Sperm numbers in drone honeybees (Apis mellifera) depend on body size. Apidologie 34:577–584
Schlüns H, Koeniger G et al (2004) Sperm utilization pattern in the honeybee (Apis mellifera). Behav Ecol Sociobiol 56:512
Schlüns H, Moritz RFA et al (2005a) Paternity skew in seven species of honeybees (Hymenoptera: Apidae: Apis). Apidologie 36:201–209
Schlüns H, Moritz RFA et al (2005b) Multiple nuptial flights, sperm transfer and the evolution of extreme polyandry in honeybee queens. Anim Behav 70:125–131
Schneider SS, DeGrandi-Hoffman G et al (2004) The African honey bee: factors contributing to a successful biological invasion. Annu Rev Entomol 49:351–376
Solignac M, Vautrin D et al (2003) Five hundred and fifty microsatellite markers for the study of the honeybee (Apis mellifera L.) genome. Mol Ecol Notes 3:307–311
Vähä J-P, Primmer CR (2006) Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol Ecol 15:63–72
Wang J (2003) Maximum-likelihood estimation of admixture proportions from genetic data. Genetics 164:747–765
Acknowledgements
We thank all the beekeepers from the Verein Schweizerischer Mellifera Bienenfreunde (VSMB), the Schweizerische Carnica Imkervereinigung (SCIV) and the Société romande d’apiculture (SAR) for kindly providing samples. We would also like to thank the Swiss Federal Office for Agriculture (BLW), the Verband Schweizerischer Bienenzüchtervereine (VSBV) and the Verein deutschschweizerischer und rätoromanischer Bienenfreunde (VDRB) for financial support. We further like to thank Michel Solignac and Pierre Franck for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Soland-Reckeweg, G., Heckel, G., Neumann, P. et al. Gene flow in admixed populations and implications for the conservation of the Western honeybee, Apis mellifera . J Insect Conserv 13, 317–328 (2009). https://doi.org/10.1007/s10841-008-9175-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10841-008-9175-0