
Abstract
In this study, we reexamined the patterns of divergence and phylogeographic structure among Apis cerana populations on Hainan island and southern mainland China. Analysis of 10 microsatellite loci from 12 geographically distinct A. cerana populations identified a total of 151 alleles, with a range of 7 to 24 at each locus. The level of genetic variation (observed heterozygosity) within each population ranged from 0.603 to 0.661, while variation within populations contributed most (93.78–98.84 %) of the molecular variance. Microsatellite data revealed three differentiated groups, one including all the mainland populations while the other two containing one of and the other three island populations, respectively. Severe genetic introgressions from mainland China into Hainan island were apparent, and these were characterized at both population and individual levels using combined analysis of microsatellites and mitochondrial DNA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The Eastern honeybee (Apis cerana Fabricius) is native throughout Asia and nests in distinct habitats of complex topography, divergent climate, and varied flora (Abrol 2013b). This species is a vital component of natural ecosystems that pollinates numerous flowering plants and agricultural crops (Sasaki et al. 1991; Verma and Partap 1993; Partap and Verma 1994; Klein et al. 2007; Kremen et al. 2007). Moreover, management of A. cerana is widely practiced in Asian countries and provides financial support to local people (Songram et al. 2006). However, despite the ecological and economic importance, Eastern honeybee colonies have undergone considerable decline in recent times (Abrol 2013a), and knowledge on the intra-specific structure of A. cerana is urgently required to guide conservation of this crucial species (Zayed 2009).
The within-species variability of A. cerana was explored originally using morphological methods, and more recently using molecular markers. Morphometric measurements were combined with multivariate analysis in a pioneering approach for determining honeybee diversity (DuPraw 1964; DuPraw 1965), and this methodology was used in a comprehensive examination of this species (Ruttner 1988). Extensive morphological investigations have now been performed on populations from most of the native habitats in which this species is found (Hepburn et al. 2001; Tan et al. 2003; Radloff et al. 2005a; Radloff et al. 2005b; Tan et al. 2008). Radloff et al. (2010) summarized these studies and inferred six highly differentiated major morphoclusters, building up a robust within-species systematics. However, inconsistencies between regional populations reported in different studies remain unexplained. For example, in mainland China, the work of Yang et al. (1986) identified five subspecies including Eastern (A. c. cerana), Tibetan (A. c. skorikovi), Aba (A. c. abansis), Indian (A. c. indica), and Hainan subspecies (A. c. hainana) (Yang et al. 1986), whereas morphological analysis by other groups identified four and three morphoclusters, respectively (Tan et al. 2008; Radloff et al. 2010). In particular, reports on bee populations from Hainan island differed markedly between studies.
More recently, molecular markers mostly based on mitochondrial DNA sequences have been used to investigate A. cerana diversity, and a framework structure consisting of four deeply divergent groups was suggested (Smith 2011b). Although mtDNA and morphological analysis do not always agree entirely, data on A. cerana differentiation from these approaches are in broad agreement (Smith 2011a). The mainland Asian group is comprised of three major morphoclusters: Northern, Himalayan, and Indo-Chinese. The Yellow Indian group corresponds to the Indian Plains morphocluster, the Sundaland group corresponds to the Indo-Malayan morphocluster, and the Oceanic Philippines group corresponds to the Philippine morphocluster. Sampling and molecular characterization of many regional populations of mainland China and its neighboring islands remains inadequate (Tan et al. 2007), as is the case for islands of the Sunda shelf (Smith 2011a). Most mtDNA studies on honeybee species to date have focused on mitochondrial genes of the intergenic region between the 3′ end of cytochrome oxidase subunit I and the 5′ end of cytochrome oxidase subunit II (COI-COII), and this region has proven to be highly informative for elucidating the Apis mellifera divergence (Arias and Sheppard 1996; Franck et al. 2000; Palmer et al. 2000; Clarke et al. 2001; de la Rua et al. 2001; Iiyasov et al. 2011). However, this fragment is not optimal for examining genetic variation of the Eastern honeybee as it is relatively small in size and is completely absent in samples from some A. cerana populations (Smith and Hagen 1996). Therefore, further studies using more suitable molecular markers (either mitochondrial or nuclear) are required.
Microsatellites, also referred to as simple sequence repeats (SSRs), are stretches of DNA sequence consisting of 1–6 tandem nucleotide repeats (Rasmus and Per 1999; Li et al. 2002). This type of molecular marker is abundant in eukaryotic genomes (Hiroshi et al. 1982; Tautz and Renz 1984), genetically codominant, and highly polymorphic (Tautz 1989; Hans 2004). In virtue of these genomic and genetic characteristics, it is widely used to interpret within-species variability of a variety of organisms (Paetkau et al. 1995; Van Hooft et al. 2000; Juan et al. 2008; Serrano et al. 2009; Boykin et al. 2010; Tashima et al. 2010). However, to the best of our knowledge, relatively few population genetic analyses have been performed on A. cerana using SSR (Sittipraneed et al. 2001; Ji et al. 2011; Rueppell et al. 2011).
In the present study, a set of 10 SSR markers from A. mellifera were genotyped against Eastern honeybee DNA samples from a previous study (Zhao et al. 2014). Genetic variation within and between these geographic populations was investigated at the molecular level, and combined analysis of mitochondrial and microsatellite data was used to probe phylogeographic structure.
2 Materials and methods
2.1 Sampling and DNA extraction
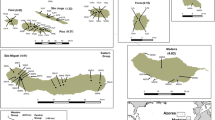
Bee samples were identical to those previously described in Zhao et al. (2014). A total of 360 A. cerana colonies representing 12 geographic populations (30 for each population) were collected from natural nests or semi managed log-hives in Guangdong province (GD), Guangxi autonomous region (GX), and Hainan island (HN), China. Geographic coordinate information is displayed in Figure 1. For each bee colony, one worker bee was randomly selected and total genomic DNA was extracted from its thorax using a standard phenol-chloroform extraction protocol (Smith and Hagen 1996). DNA was treated with ribonuclease A (Roche, Basel, Switzerland) and frozen at −75 °C until needed.

a Location of main sampling regions: GD Guangdong province, GX Guangxi autonomous region, HN Hainan island. b Location of sampling sites within the main sampling regions. Circles represent the following sampling sites: GDJL (N24.67°, E116.06°), Jiaoling County in GD; GDLM (N23.77°, E114.29°), Longmen County in GD; GDYD (N24.21°, E113.77°), Yingde City in GD; GXBH (N21.88°, E109.44°), Beihai City in GX; GXCZ (N22.02°, E107.52°), Chongzuo City in GX; GXGL (N24.98°, E110.42°), Guilin City in GX; GXHZ (N23.88°, E111.04°), Hezhou City in GX; GXLB (N23.87°, E109.31°), Laibin City in GX; HNBS (N19.34°, E109.46°), Baisha County in HN; HNHK (N19.95°, E110.30°), Haikou City in HN; HNTC (N19.35°, E110.07°), Tunchang County in HN; and HNWN (N18.75°, E110.35°), Wanning City in HN. Patterns within circles represent phylogeographical analysis of 12 bee populations based on individuals defined by mitochondrial mito-type (M) and microsatellite genotype (G). CH denotes the mainland group, HN denotes the island group.
2.2 Screening of microsatellite markers
A set of 48 microsatellite markers (listed in Supplementary file 1) originally developed from A. mellifera (Solignac et al. 2007) were prescreened and 10 (Table I) showed superior data quality and polymorphism when applied to A. cerana samples and were subsequently used in this study. For each of the 10 loci, the 5′ end of the corresponding forward primer was labeled with fluorescent dye (6-FAM) by Sangon Biotech Co., Ltd, Shanghai, China.
2.3 PCR and SSR genotyping
PCR was performed in a final volume of 20 μL, and reactions contained 11–11.4 μL sterile deionized water (depending on SSR primers), 2 μL of 10 × PCR buffer (Mg2+-free), 1.2–1.6 μL 25 mM MgCl2 (depending on SSR primers), 1.6 μL dNTPs (2.5 mM each), 0.8 μL each primer (10 μM), 0.2 μL Taq DNA polymerase (5U/μL, Takara), and 2 μL DNA template (50 ng/μL). All PCR amplifications were carried out on a Bio-Rad T100 thermocycler with the following conditions: initial denaturation at 95 °C for 3 min, followed by 25 cycles of denaturation at 94 °C for 30 s, annealing at 56–61 °C (depending on SSR primers) for 30 s, elongation at 72 °C for 30 s, and a final elongation at 72 °C for 10 min. A mixture of the 0.5 μL fluorescent PCR products, 0.25 μL size standard (GeneScan 500LIZ, Applied Biosystems, USA) and 9.25 μL highly deionized formamide was prepared, denatured at 95 °C for 10 min, cooled at −20 °C for 5 min, and then subjected to capillary electrophoresis using an ABI 3730xl DNA analyzer (Applied Biosystem Inc, the USA).
2.4 Data analysis
GeneMapper v.3.7 (Applied Biosystems, USA) was used to score allelic sizes according to the 500LIZ size standard, and allelic data were introduced into Microsoft Office Excel 2007 which was used to estimate diversity indicators including the mean number of alleles per locus (S) and geographic population (Na). The observed (Ho) and expected (He) heterozygosity were calculated using GenAlEx 6.5 (Peakall and Smouse 2006), while the polymorphic information content (PIC) and null allele frequency F(Null) were computed with Cervus 3.0 (Kalinowski et al. 2007). Using Genepop ver.4.0 (Rousset 2008), we performed linkage disequilibrium tests (with Fisher’s method and gametic phase unknown) for each pair of loci considering linkage equilibrium the null hypothesis. We also determined the inbreeding coefficient (Fis) of each population and performed the Hardy-Weinberg equilibrium (HWE) tests with heterozygote deficit considered as the alternative hypothesis using Genepop ver.4.0 (Rousset 2008). Four analysis of molecular variance (AMOVA) tests were carried out and their fixation indices (FCT, FSC, FST) of the corresponding levels (among groups, among populations within group, within populations) were determined by Arlequin 3.11 (Excoffier et al. 2005), with grouping patterns as follows: (1) mainland populations vs. island populations; (2) GD populations (including GDJL, GDLM and GDYD) vs. GX populations (including GXBH, GXCZ, GXGL, GXHZ, and GXLB); (3) HNHK vs. island populations HNBS, HNTC, and HNWN; and (4) mainland populations vs. HNHK vs. island populations HNBS, HNTC, and HNWN. To further explore patterns of divergence, we performed Bayesian inference using STRUCTURE 2.3.1 (Evanno et al. 2005) with the following parameters: length of burn-in period = 2,000,000; number of MCMC repeats after burn-in = 3,000,000; “Admixture model” and “Allele frequency correlated” selected; K values predefined from 1 to 12; and number of iterations = 20. The results were used by Structure Harvester v0.6.94 to generate log probability diagrams http://taylor0.biology.ucla.edu/structureHarvester/(Earl 2012), also by CLUMPP v1.1.2 (Jakobsson and Rosenberg 2007) with “Greedy algorithm” employed and repeats = 10,000 and DISTRUCT v1.1 (Rosenberg 2004) to draw a multicolored figure. MEGA v.6.06 (Tamura et al. 2013) was used to reconstruct neighbor-joining (NJ) relationships based on genetic distance matrices of the present microsatellites (Nei 1972) and previous mitochondrial (Kimura-2-parameter model) DNA data. Further individual assignment tests were performed using GENCLASS 2.0 (Piry et al. 2004). The previously determined mtDNA clustering pattern (all mainland populations represented as the CH group, HNBS + HNWN represented by the HN group) was treated as the reference, and Bayesian (Rannala and Mountain 1997), frequency-based (Paetkau et al. 1995) and genetic distance-based (Nei et al. 1983) algorithms were employed. Final assignment of an individual was dependent on the mean probability of the three methods. Detailed phylogeographic characterization could then be performed when both the microsatellite genotype and mitochondrial mito-type of each individual was determined.
3 Results
3.1 Polymorphism at microsatellite markers
Individuals GDJL-4 and GXGL-5 could not be genotyped at more than eight loci and were excluded from subsequent analysis. A total of 151 alleles were identified at all 10 SSR loci from 358 bee samples (the frequency and distribution of each allele is listed in Supplementary file 2). The number of alleles per locus varied from 7 (AP226) to 24 (AC139), with an overall mean of 15.1 (Table I). All markers proved highly informative (PIC > 0.5) with the exception of AP226 (PIC = 0.231) and are therefore useful for estimating genetic diversity. The results of linkage disequilibrium tests in Supplementary file 3 demonstrated large p values (>0.05) for all loci pairs but AC139 to AT170 (p = 0.003762), suggesting random genetic association among microsatellite markers except AC139 to AT170. Polymorphism at these markers was also determined by estimating heterozygosity, and observed (Ho) and expected (He) heterozygosity ranged from 0.230 to 0.879 and 0.227 to 0.870, respectively. Null allele frequency estimation F(Null) peaked at 9.95 % for AP187 and averaged 6.55 % across all loci, while the HWE test with heterozygote deficit considered as the alternative hypothesis reached a significant level at eight loci, but this threshold was not reached for AP226 (PHWE = 0.4831) or AT141 (PHWE = 0.2808).
3.2 Genetic variation within populations
The overall level of variability across the 10 loci in the 12 A. cerana populations and the mainland and island population was investigated (Table II). The mean number of alleles (Na) ranged from 6.8 to 13.8, and observed and expected heterozygosity was 0.603–0.661 and 0.646–0.712, respectively. These narrow ranges indicated homogeneously distributed variation among different populations. All populations except GXLB were significantly deviated according to the Hardy-Weinberg equilibrium test results (P HWE <0.05) when heterozygote deficiency was chosen as the alternative hypothesis. This result implied a higher degree of inbreeding for most populations. Though the inbreeding coefficients (Fis) displayed the same implication, the Wahlund effect could also be an explanation to present results.
3.3 Phylogeographic structure
The results of AMOVA tests (Table III) revealed that variation within populations always contributed the biggest proportion (93.78–98.84 %), while less but significant variation (FCT = 0.0494**) was detected between mainland and island population groups, indicating genetic partitioning between populations on either side of the strait. A moderate level of differentiation (FCT = 0.0241) was apparent between the Hainan island population (HNHK) and the other three populations (HNBS, HNTC, and HNWN), indicating three main groups (mainland, HNHK, and HNBS + HNTC + HNWN). Bayesian analysis (Figure 2) gave the highest Ln probability L(K) of −11633.3 (averaged over 20 replicates) when three groups were assumed, in agreement with the AMOVA results. Unrooted NJ trees based on either microsatellite or mitochondrial DNA data displayed a similar topology (Figure 3). Specifically, mainland clustered together into one branch, while the HNHK population forms an independent branch. The only significant difference between the two trees is the positioning of the HNTC population, which is clustered with HNBS + HNWN in the microsatellite tree, but is more closely associated with mainland populations in the mitochondrial DNA tree. The results in Supplementary file 4 are from individual assignment tests and previous mtDNA data (Zhao et al. 2014) that we used to list the mitochondrial mito-type and microsatellite genotype of each individual bee based on four bee types (Figure 1b): CH mito-type and genotype M(CH) + G(CH); CH mito-type and HN genotype M(CH) + G(HN); HN mito-type and genotype M(HN) + G(HN); and HN mito-type and CH genotype M(HN) + G(CH). Almost all mainland individuals belonged to M(CH) + G(CH), whereas most island bees (mostly from HNBS and HNWN) were M(HN) + G(HN), indicating a clear phylogeographic structure. However, 13 individuals from HNHK and four from HNTC belonged to M(CH) + G(CH), consistent with genetic introgression in the two island populations. Interestingly, 10 individuals from HNTC and three from HNHK were M(CH) + G(HN), a hybrid between CH and HN, which may explain why HNTC clustered together with HNBS + HNWN in the microsatellite tree but was closer to mainland populations in the mitochondrial tree (Figure 3). The other hybrid type M(HN) + G(CH) was seldom detected.

a Log probability (L(K)) of K values ranging from 1 to 12 for the admixture and correlated frequencies model based on 20 replicates from Apis cerana populations (K = number of predefined groups). Length of burn-in = 2,000,000. MCMC repeats after burn-in = 3,000,000. Vertical lines indicate the standard deviation. b Graphic result of structure analysis with ‘Greedy algorithm’ employed and repeats = 10,000 in CLUMPP, K = number of predefined groups, and colors indicate the predefined groups. Populations are separated by thin black lines and each of the 358 bee individuals is represented by a thin vertical line.

Unrooted neighbor-joining trees of 12 geographic populations based on different genetic distance matrices a based on 10 microsatellite loci using Nei’s standard genetic distance (1972) as shown in the lower triangle of c; b based on mitochondrial DNA sequences using the Kimura-2-parameters distance shown in the upper triangle of c; and c genetic distance matrices based on both microsatellite DNA (lower triangle) and mitochondrial DNA (upper triangle). Circles and triangles represent mainland and island populations, respectively.
4 Discussion
Trans-species microsatellite markers from A. mellifera to other Apis species have been used successfully in the past, e.g., Oldroyd et al. (Oldroyd et al. 1997; Sittipraneed et al. 2001; Cao et al. 2012). In the present paper, we employed 10 SSRs from A. mellifera to investigate A. cerana, of which all except AP226 (PIC = 0.231) are highly polymorphic (PIC >0.5) according to a previous report (Vanhala et al. 1998) and are therefore suitable for evaluating genetic variation. A much larger PIC value (0.8966) was reported previously for the AP226 locus, and 21 alleles were described (Ji et al. 2011). This apparent difference from the present study may be explained by different sampling strategies, alternative methods of allele discrimination, or a combination of both factors. It should be noted that our results have little direct comparability with previous SSR studies, since different microsatellites have been used for analysis, with only AP226 in common with (Ji et al. 2011). All comparisons are therefore tentative.
The genetic diversity (average expected heterozygosity) of the geographic populations ranged from 0.646 to 0.712 (average = 0.685), indicating homogeneously distributed variation among different populations. Island populations generally exhibit lower genetic variation than their mainland counterparts, presumably due to the initial loss of diversity upon foundation, and a more constrained population size following foundation (Frankham 1997). However, our results do not fit such a model, suggesting gene flow between mainland and island populations has reduced inter-population differences.
We observed notable differences in the distribution of alleles between mainland and island populations (Supplementary file 2), implying genetic partition between the mainland (CH) and island (HN) groups. The results of AMOVA (Table III) together with unrooted NJ trees (Figure 3) also exhibited this pattern of genetic partitioning, illustrating the peculiarity of Hainan bee populations that was first noted by a morphometric study (Yang et al. 1986) and subsequently confirmed using mtDNA analysis (Zhao et al. 2014). The present study therefore reconfirmed this at the nuclear molecular level. Hainan island is separated from mainland China by the Qiongzhou Strait, and it has been suggested that various island species have been separated from and reunited with their mainland counterparts several times due to rising and falling sea levels during the Pleistocene (Voris 2000). Differentiation between species on either side of the strait may therefore reflect independent evolution since the last rise in sea level.
Within the Hainan island population, a moderate level of variation was detected between HNHK and HNBS + HNTC + HNWN (Table III; Figure 3). Given that A. cerana colonies have been transported to the island from mainland China on numerous occasions, this intra-island divergence is likely a consequence of gene flow that has resulted in genetic introgression in the HNHK population. We noticed that over half of all HNHK individuals were of the CH (13/30) and hybrid (5/30) types (Supplementary file 4; Figure 1b). Similar introgression has been reported in A. mellifera populations (Garnery et al. 1998; Franck et al. 2000; De la Rúa et al. 2001; Dall’Olio et al. 2007) and is considered a potential threat to indigenous genetic diversity (De la Rua et al. 2003; Cánovas et al. 2011). The unusual Hainan race is particularly sensitive to such introgression owing to its narrow distribution and limited population size. Therefore, it is suggested with some urgency that importation of exotic bee colonies is ceased, and conservation zones for existing Hainan populations are created and protected.
Previous mtDNA data identified a large introgression (14/30) of the CH mito-type in HNTC individuals (Supplementary file 4; Figure 1b), but the present SSR data detected this genotype in only 4/30 HNTC individuals. Using the same samples but different molecular markers can reveal distinct introgression patterns, as has been shown previously (Franck et al. 1998; De la Rúa et al. 2001). It was suggested that repeated importation may result in introgression that is faster in mtDNA than in nuclear genes (Garnery et al. 1998), presumably because all daughters of a newly introduced queen share her mtDNA, while nuclear genes inherited from exotic species are diluted due to polyandric mating. Our HNTC results are consistent with this interpretation; however, it should be noted that the discrepancy in the rate of introgression may impact the results to some extent. Additionally, sample size (number of individuals per colony and population) may also be an important factor. Further investigation is needed in this regard.
Our microsatellite data indicated no significant variation among mainland populations and therefore no inter-population subdivisions. The eight populations sampled essentially share the same gene pool, and this was the conclusion from both mtDNA and SSR data.
In summary, we reexamined microsatellite polymorphism data from A. cerana populations on Hainan island and southern mainland China and detected a distinct phylogeographic structure between mainland and island populations. These results further confirm the peculiarity of the Hainan island race. Our results revealed genetic introgression resulting from gene flow at both population and individual levels. Combined analysis of microsatellites and mitochondrial DNA identified two distinct introgression patterns in HNTC individuals. Together, these results allow some tentative conclusions on A. cerana phylogeographic structure to be made and will inspire further studies.
References
Abrol, D.P. (2013a) Introduction. In: Abrol, D.P. (ed.) Asiatic Honeybee Apis cerana, pp. 21–22. Springer, Berlin
Abrol, D.P. (2013b) Pollination Biology. In: Abrol, D.P. (ed.) Asiatic Honeybee Apis cerana, p. 521. Springer, Berlin
Arias, M.C., Sheppard, W.S. (1996) Molecular phylogenetics of honey bee subspecies (Apis mellifera L.) inferred from mitochondrial DNA sequence. Mol. Phylogenet Evol 5(3), 557–566
Boykin, L.M., Shatters, R.G., Hall, D.G., Dean, D., Beerli, P. (2010) Genetic variation of Anastrepha suspensa (Diptera: Tephritidae) in Florida and the Caribbean using microsatellite DNA markers. J. Econ. Entomol. 103(6), 2214–2222
Cánovas, F., de la Rúa, P., Serrano, J., Galián, J. (2011) Microsatellite variability reveals beekeeping influences on Iberian honeybee populations. Apidologie 42(3), 235–251
Cao, L.-F., Zheng, H.-Q., Hu, F.-L., Hepburn, H.R. (2012) Genetic structure of Chinese Apis dorsata population based on microsatellites. Apidologie 43(6), 643–651
Clarke, K., Oldroyd, B.P., Javier, J., Quezada, E., Rinderer, T.E. (2001) Origin of honeybees (Apis mellifera L.) from the Yucatan peninsula inferred from mitochondrial DNA analysis. Mol. Ecol. 10, 1347–1355
Dall’Olio, R., Marino, A., Lodesani, M., Moritz, R.A. (2007) Genetic characterization of Italian honeybees, Apis mellifera ligustica, based on microsatellite DNA polymorphisms. Apidologie 38(2), 207–217
De la Rúa, P., Galián, J., Serrano, J., Moritz, R.F.A. (2001) Genetic structure and distinctness of Apis mellifera L. populations from the Canary Islands. Mol. Ecol. 10, 1733–1742
de la Rua, P., Galian, J., Serrano, J., Moritz, R.F. (2001) Genetic structure and distinctness of Apis mellifera L. populations from the Canary Islands. Mol Ecol 10, 1733–1742
De la Rua, P., Galian, J., Serrano, J., Moritz, R.F. (2003) Genetic structure of Balearic honeybee populations based on microsatellite polymorphism. Genet. Sel. Evol. 35(3), 339–350
DuPraw, E.J. (1964) Non-Linnean taxonomy Nature 202, 849–852
DuPraw, E.J. (1965) Non-Linnean taxonomy and the systematics of honeybees. Syst. Zool. 14, 1–24
Earl, D.A. (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Ggenet. Resour. 4(2), 359–361
Evanno, G., Regnaut, S., Goudet, J. (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14(8), 2611–2620
Excoffier, L., Laval, G., Schneider, S. (2005) Arlequin (Version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 1, 47–50
Franck, P., Garnery, L., Solignac, M., Cornuet, J.M. (1998) The origin of west European subspecies of honeybees (Apis mellifera): new insights from microsatellite and mitochondrial data. Evolution 52, 1119–1134
Franck, P., Garnery, L., Celebrano, G., Solignac, M., Cornuet, J.M. (2000) Hybrid origins of honeybees from Italy (Apis mellifera ligustica) and sicily (A. m. sicula). Mol Ecol 9(7), 907–921
Frankham, R. (1997) Do island populations have less genetic variation than mainland populations? Heredity 78(3), 311–327
Garnery, L., Franck, P., Baudry, E., Vautrin, D., Cornuet, J.-M., Solignac, M. (1998) Genetic diversity of the west European honey bee (Apis mellifera mellifera and A. m. iberica) II. Microsatellites. Genet. Sel. Evol. 30, 49–74
Hans, E. (2004) Microsatellites: simple sequences with complex evolution. Nature Rev. Genet. 5, 435–445
Hepburn, H.R., Radloff, S.E., Verma, S., Verma, L.R. (2001) Morphometric analysis of Apis cerana populations in the southern Himalayan region. Apidologie 32(5), 435–447
Hiroshi, H., Marianne, G.P., Takeo, K. (1982) A novel repeated element with Z-DNA-forming potential is widely found in evolutionarily diverse eukaryotic genomes. Pro. Natl. Acad. Sci. 79, 6465–6469
Iiyasov, R.A., Kutuev, I.A., Petukhov, A.V., Poskryakov, A.V., Nikolenko, A.G. (2011) Phylogenetic relationships of dark European honeybees Apis mellifera mellifera L. from the Russian ural and west European populations. J. Apic Sci 55(1), 67–76
Jakobsson, M., Rosenberg, N.A. (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23(14), 1801–1806
Ji, T., Yin, L., Chen, G. (2011) Genetic diversity and population structure of Chinese honeybees (Apis cerana) under microsatellite markers. Afr. J. Biotechnol. 10(9), 1712–1720
Kalinowski, S.T., Taper, M.L., Marshall, T.C. (2007) Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. ecol. 16(5), 1099–1106
Klein, A.M., Vaissiere, B.E., Cane, J.H., Steffan-Dewenter, I., Cunningham, S.A., Kremen, C., Tscharntke, T. (2007) Importance of pollinators in changing landscapes for world crops. Proc. R. Soc. B: Biol. Sci. 274, 303–313
Kremen, C., Williams, N.M., Aizen, M.A., Gemmill-Herren, B., Glebuhn, G., Minckley, R., Packer, L., Potts, S.G., Roulston, T., Steffan-Dewenter, I., Vazquez, D.P., Winfree, R. (2007) Pollination and other ecosystem services produced by mobile organisms: a conceptual framework for the effects of land use change. Ecol. Lett. 10, 299–314
Li, Y., Abraham, B.K., Tzion, F., Avigdor, B., Eviatar, N. (2002) Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol. Ecol. 11, 2453–2465
Motamayor, J.C., Lachenaud, P., da Silva, M., Loor, R., Kuhn, D.N., Brown, J.S., Schnell, J. (2008) Geographic and Genetic Population Differentiation of the Amazonian Chocolate Tree (Theobroma cacao L). PLoS One 3(10), e3311
Nei, M. (1972) Genetic distance between populations. Am. Nat. 106, 283–292
Nei, M., Tajima, F., Tateno, Y. (1983) Accuracy of estimated phylogenetic trees from molecular data. J. Mol. Evol. 19(2), 153–170
Oldroyd, B.P., Clifton, M.J., Wongsiri, S., Rinderer, T.E., Sylvester, H.A., Crozier, R.H. (1997) Polyandry in the genus Apis, particularly Apis andreniformis. Behav. Ecol. Sociobiol. 40(1), 17–26
Paetkau, D., Calvert, W., Stirling, I., Strobeck, C. (1995) Microsatellite analysis of population structure in Canadian polar bears. Mol. Ecol. 4, 347–354
Palmer, M.R., Smith, D.R., Kaftanoglu, O. (2000) Turkish honeybees: genetic variation and evidence for a fourth lineage of Apis mellifera mtDNA. J. Hered. 91, 42–46
Partap, U., Verma, L.R. (1994) Pollination of radish by Apis cerana. J. Apic. Res. 33(4), 237–241
Peakall, R., Smouse, P.E. (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes. 6(1), 288–295
Piry, S., Alapetite, A., Cornuet, J.M., Paetkau, D., Baudouin, L., Estoup, A. (2004) GENECLASS2: a software for genetic assignment and first-generation migrant detection. J. Hered. 95(6), 536–539
Radloff, S.E., Hepburn, H.R., Fuchs, S., Otis, G.W., Hadisoesilo, S., Hepburn, C., Ken, T. (2005a) Multivariate morphometric analysis of the Apis cerana populations of oceanic Asia. Apidologie 36, 475–492
Radloff, S.E., Hepburn, H.R., Hepburn, C., Fuchs, S., Otis, G.W., Sein, M.M., Aung, H.L., Pham, H.T., Tam, D.Q., Nuru, A.M., Ken, T. (2005b) Multivariate morphometric analysis of Apis cerana of southern mainland Asia. Apidologie 36, 127–139
Radloff, S.E., Hepburn, C., Hepburn, H.R., Fuchs, S., Hadisoesilo, S., Ken, T., Engel, M.S., Kuznetsov, V. (2010) Population structure and classification of Apis cerana. Apidologie 41, 589–601
Rannala, B., Mountain, J.L. (1997) Detecting immigration by using multilocus genotypes. Proc. Natl. Acad. Sci. 94(17), 9197–9201
Rasmus, N., Per, J.P. (1999) Single-locus tests of microsatellite evolution: multi-step mutations and constraints on allele size. Mol. Phylogenet. Evol. 11(3), 477–484
Rosenberg, N.A. (2004) Distruct: a program for the graphical display of population structure. Mol. Ecol. Notes 4(1), 137–138
Rousset, F. (2008) Genepop’007: a complete reimplementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour. 8(1), 103–106
Rueppell, O., Hayes, A.M., Warrit, N., Smith, D.R. (2011) Population structure of Apis cerana in Thailand reflects biogeography and current gene flow rather than Varroa mite association. Insectes Soc. 58(4), 445–452
Ruttner, F. (1988) Biogeography and taxonomy of honeybees. Springer, Berlin
Sasaki, M., Ono, M., Asada, S., Yoshida, T. (1991) Oriental orchid (Cymbidium pumilum) attracts drones of the Japanese honeybee (Apis cerana japonica) as pollinators. Experientia 47(11–12), 1229–1231
Serrano, M., Calvo, J.H., Martínez, M., Marcos-Carcavilla, A., Cuevas, J., González, C., Jurado, J., Díez de Tejada, P. (2009) Microsatellite based genetic diversity and population structure of the endangered Spanish Guadarrama goat breed. BMC Genet. 10(61), 1–9
Sittipraneed, S., Laoaroon, S., Klinbunga, S., Wongsiri, S. (2001) Genetic differentiation of the honeybee (Apis cerana) in Thailand: evidence from microsatellite polymorphism. J. Apic. Res. 40(1), 9–16
Smith, D.R. (2011a) Asian Honeybees and Mitochondrial DNA. In: Hepburn, R., Radloff, S.E. (eds.) Honeybees of Asia, p. 87. Springer, Berlin
Smith, D.R. (2011b) Asian Honeybees and Mitochondrial DNA. In: Radloff, S.E., Hepburn, R. (eds.) Honeybees of Asia, pp. 69–93. Springer, Berlin
Smith, D.R., Hagen, R.H. (1996) The biogeography of Apis cerana as revealed by mitochondrial DNA sequence data. J Kansas Entomol Soc. 69, 294–310
Solignac, M., Mougel, F., Vautrin, D., Monnerot, M., Cornuet, J.M. (2007) A third-generation microsatellite-based linkage map of the honey bee, Apis mellifera, and its comparison with the sequence-based physical map. Genome Biol. 8(4), R66
Songram, O., Sittipraneed, S., Klinbunga, S. (2006) Mitochondrial DNA diversity and genetic differentiation of the honeybee (Apis cerana) in Thailand. Biochem. Genet. 44(5), 256–269
Tamura, K., Stecher, G., Peterson, D., Filipski, A., Kumar, S. (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol Evol 30(12), 2725–2729
Tan, K., Fuchs, S., Koeniger, N., Ruiguang, Z. (2003) Morphological characterization of Apis cerana in the Yunnan Province of China. Apidologie 34(6), 553–561
Tan, K., Warrit, N., Smith, D.R. (2007) Mitochondrial DNA diversity of Chineses Apis cerana. Apidologie 38, 238–246
Tan, K., Hepburn, H.R., Radloff, S.E., Fuchs, S., Xian, F., Zhang, L.J., Yang, M.X. (2008) Multivariate morphometric analysis of the Apis cerana of China. Apidologie 39, 343–353
Tashima, S., Kaneko, Y., Anezaki, T., Baba, M., Yachimori, S., Masuda, R. (2010) Genetic diversity within the Japanese badgers (Meles anakuma), as revealed by microsatellite analysis. Mammal Study 35, 221–226
Tautz, D. (1989) Hypervariability of simple sequences as a general source for polymorphic DNA markers. Nucleic Acids Res. 17(16), 6463–6471
Tautz, D., Renz, M. (1984) Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucleic Acids Res. 12(10), 4127–4138
Van Hooft, W.F., Groen, A.F., Prins, H.H.T. (2000) Microsatellite analysis of genetic diversity in African buffalo (Syncerus caffer) populations throughout Africa. Mol. Ecol. 9, 2017–2025
Vanhala, T., Tuiskula-Haavisto, M., Elo, K., Vilkki, J., Mäki-Tanila, A. (1998) Evaluation of genetic variability and genetic distances between eight chicken lines using microsatellite markers. Poultry Sci. 77(6), 783–790
Verma, L.R., Partap, U. (1993) The Asian hive bee, Apis cerana, as a pollinator in vegetable seed production. International Centre for Integrated Mountain Development (ICIMOD), Nepal
Voris, H.K. (2000) Maps of Pleistocene sea levels in Southeast Asia: shorelines, river systems and time durations. J. Biogeogr. 27, 1153–1167
Yang, G.H., Xu, S.Y., Kuang, B.Y., Liu, Y.D. (1986) Subspecific differentiation and distribution of Apis cerana Fab in China. J. Yunnan Agric. Univ. 1, 89–92
Zayed, A. (2009) Bee genetics and conservaton. Apidologie 40, 237–262
Zhao, W., Tan, K., Zhou, D., Wang, M., Cheng, C., Yu, Z., Miao, Y., He, S. (2014) Phylogeographic analysis of Apis cerana populations on Hainan island and southern mainland China, based on mitochondrial DNA sequences. Apidologie 45, 21–33
Acknowledgments
We wish to acknowledge our gratitude to Yuexiong Luo, Zheng Xu, Junjun Hu, and Jinglin Gao for their generous help in sample collection. This study was financially supported by the “National System of Bee Industry and Technology” Program (CARS-45-kxj14).
Phylogéographie des populations d’ Apis cerana sur l’île de Hainan et dans le sud-est de la Chine continentale mise en évidence par le polymorphisme des microsatellites et l’ADN mitochondrial
Apidae / microsatellite / structure phylogéographique / introgression génétique
Untersuchungen auf der Basis von Mikrosatelliten und mitochondrialer DNA zur Phylogeographie von Apis cerana im südlichen China und auf der Insel Hainan
Apis cerana / Mikrosatelliten / Phylogeographische Struktur / genetische Introgression
Author information
Authors and Affiliations
Corresponding authors
Additional information
Manuscript editor: Marina Meixner
Wenzheng Zhao and Miao Wang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Zhao, W., Wang, M., Liu, Y. et al. Phylogeography of Apis cerana populations on Hainan island and southern mainland China revealed by microsatellite polymorphism and mitochondrial DNA. Apidologie 48, 63–74 (2017). https://doi.org/10.1007/s13592-016-0450-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-016-0450-x