
Abstract
Chlorophyll (Chl) is present in many plant organs and plays vital roles during growth and development. Degradation of Chl causes the loss of green colour that typically occurs during senescence, and fruit ripening. The present study addresses genome-wide identification and bioinformatics analyses of tomato (Solanum lycopersicum), SlSGR1 (Solyc08g080090.2.1) SlSGR2 (Solyc12g056480.1.1) and SlSGR-like (Solyc04g063240.2.1) genes. Multiple sequence alignment indicated that the three tomato SGR proteins have conserved domains. Motif, sequence, and protein structure analysis showed that SlSGR-like differentially evolved from SlSGR proteins. Co-expression analyses were performed for each SlSGR using transcriptomic data of two fruit ripening stages [mature green (MG) and ripe fruit (R)] of Ailsa Craig (AC) tomato cultivar. According to the co-expression network analyses, SlSGRs participate in sulphur homeostasis, fatty acid biosynthesis, and biological processes of plant development during ripening stages of tomato. Six common genes were identified in the merged co-expression network. Also, 38 transcription factor families (TFFs) were searched in the co-expression network. Only 13 transcription factors, belonging to seven TFFs, were found to be involved in regulation of these two genes. Of these TFFs, GRAS and GeBP had five and three members for SlSGR-like and SlSGR1 in the network. The expression profiles of SlSGR1/2 and SlSGR-like in different tissues and different fruit ripening stages showed that SlSGR1/2 are highly expressed in ripening fruits, whereas SlSGR-like is more involved in leaf maturation. Moreover, expression of SlSGR1/2 and SlSGR-like in the leaves of ‘Ciko’ and ‘Black’ tomato varieties under 200mM salt stress indicated that significant expression changes occurred in SlSGR2 suggesting that SlSGR genes may be involved in signalling responses to abiotic stress. The findings provide new insight into the functions of these genes in growth and salinity stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Many immature fleshy fruits and young leaves undergo dramatic changes during maturation and ripening stages in terms of chemical composition and ultrastructure (Abdelrahman et al. 2017). Chlorophyll (Chl), including Chla and Chlb molecules, plays vital roles in absorption, transmission, and transformation of light energy in the process of photosynthesis in green plants (Hörtensteiner 2009). Environmental stress-induced leaf senescence is also characterized by degradation of Chls, proteins, nucleic acids and lipids, as well as lower metabolic activities (Abdelrahman et al. 2017). Fruit ripening and leaf senescence involve the degradation of chlorophylls, and enhancement of carotenoids and anthocyanins (Matile et al. 1999). The breakdown of chl-protein complexes, leading to chlorophyl degradation, is a prerequisite of the de-greening process. It also helps meet the nutritional needs of fast-growing vegetative tissues and reproductive organs (Lim et al. 2007). The ‘Stay Green’ trait is one of the most physiologically and agronomically important traits allowing plants maintain photosynthetic activity and improve stress tolerance.
The biochemical pathway of Chl breakdown, called the PAO pathway, has been characterized by the functions of Chl catabolic enzymes (CCEs) (Kuai et al. 2018). Before the Chl degradation pathway begins, chl b is converted to chl a through a two-step reaction (Kusaba et al. 2007; Meguro et al. 2011). Chl a is then converted into a primary fluorescent Chl catabolite (pFCC) by several CCEs in four successive steps (Jiao et al. 2020). The genes (SGR, RCCR, PAO, PPH, NYC1 and NOL), involved in these processes, which encode the enzymes involved in leaf senescence and fruit ripening, have been identified (Shimoda et al. 2016; Schelbert et al. 2009; Pružinská et al. 2007). In the first four successive steps of the conversion, magnesium (Mg) in Chl a is removed by a Mg-dechelatase named STAY-GREEN (SGR) in Arabidopsis, and Chl a is converted into pheophytin a (Phein a) (Shimoda et al. 2016). The functional mechanism and regulatory networks of SGRs and SGR homologs showed that SGR removes Mg not only from free chlorophyll but also from chlorophyll-protein complexes (Shimoda et al. 2016).
The stay-green phenotype, induced by the sgr mutation was initially identified in pea by Mendel (1901). After that, the SGRs and SGR homologs were identified in multiple species, including rice (Rong et al. 2013), Arabidopsis (Sakuraba et al. 2012, 2014b), tomato (Barry et al. 2008; Luo et al. 2013), pepper (Barry et al. 2008) and alfalfa (Zhou et al. 2011). In recent years, Chl breakdown in response to several abiotic/biotic stresses has become a hot topic in the scientific community due to the relationship between “STAY GREEN” and beneficial agricultural characteristics such as tolerance to abiotic/biotic stresses as well as improved yield production. In this regard, while gr1/nye1-1 and sgrl mutants displayed a stay-green phenotype under abiotic stress conditions, including high salinity, drought and heat, the overexpression of SGR1 and SGR-like results in early leaf yellowing (Sakuraba et al. 2014a). On the other hand, Arabidopsis plants overexpressing SGR2, exhibited a ‘Stay Green’ phenotype (Sakuraba et al. 2014b), suggesting that whilst SGR1 and SGR-like positively regulate Chl degradation, SGR2 negatively regulates the process during senescence. Tomato fruit ripening leads to fruit colouration and is dependent on the accumulation of carotenoids accompanied by the degradation of Chl (Liu et al. 2015). Silencing of SlSGR1 inhibited chlorophyll degradation, exhibited a stay-green phenotype and extended the shelf-life of tomato (Hu et al. 2011; Luo et al. 2013a). In a recent study, lycopene levels were increased in CRISPR/Cas9- multiplex genome edited tomato, with edited SISGR1 and four other genes (Li et al. 2018). The overexpression of the SGR-like gene results in earlier leaf yellowing than wild-type (WT) lines under ABA treatment (Yang et al. 2020). Therefore, these results suggest that SGR genes play a pivotal regulatory role in Chl degradation and colour formation during fruit ripening. Although the functions of SGR/SGR-like genes during Chl degradation and the interactions between SGR/SGR-like genes and other CCEs have been demonstrated in various organisms, little is known about how SGR/SGR-like genes interact and which transcription factors are involved in their regulation of fruit ripening, and how SGR/SGR-like genes are expressed under both fruit ripening and salt stress conditions in tomato. For this reason, we performed comprehensive bioinformatics analyses of SlSGR genes in tomato. By conducting co-expression network analyses, we investigated how SlSGRs are interacted with each other and on which transcription factor families (TFFs) are more involved in their regulations under fruit ripening conditions. We also tried to identify gene expression profiles of SlSGR genes in different tomato organs and ripening stages. Lastly, we investigated the expression of SlSGR genes under salt stress using two different tomato varieties, ‘Ciko’ and ‘Black’.
2 Materials and methods
2.1 Identification and annotation of SGR genes in S. lycopersicum
To identify SlSGR genes in tomato, we obtained all the SGR protein sequences as query sequences from the TAIR (https://www.arabidopsis.org/), SGN (https://solgenomics.net/) and PMRD (http://bioinformatics.cau.edu.cn/PMRD/) databases. The proteins, encoded by the putative SlSGR genes, were analysed for domain analysis with the Pfam database (http://pfam.xfam.org/).
2.2 Sequence analysis of SlSGR genes
The physico-chemical characteristics of SlSGR genes, including protein length, isoelectric point (pI), and molecular weight (MW) were predicted by ExPASy ProtParam (https://web.expasy.org/protparam/) (Gasteiger et al. 2003). Generic phosphorylation sites in SlSGRs were searched online using NetPhos − 3.1 (https://services.healthtech.dtu.dk/ service.php?NetPhos-3.1) (Blom et al. 1999). Amino acid sequences of SlSGR proteins from different plant species were aligned using BioEdit (Hall et al. 2011). CELLO (http://cello.life. nctu.edu.tw/) was used to predict the subcellular localizations (Yu et al. 2006).
2.3 Phylogenetic evolution and gene structure analyses
The phylogenetic tree was constructed using MEGA-X software with the maximum likelihood estimation (MLE) method and was analysed with 1000 bootstrap replications (Kumar et al. 2018). Predicted functional annotation of SlSGR proteins was performed on the PANNZER server (http://ekhidna2.biocenter.helsinki.fi/sanspanz/) (Törönen et al. 2018). The exon-intron organizations were constructed according to the Gene Structure Display Server (http://gsds.gao-lab.org/) (Guo 2007). To search conserved motifs in SlSGR protein sequences, Multiple Expectation Maximization for Motif Elicitation (https://meme-suite.org/meme/tools/meme) was used (Bailey et al. 2009).
2.4 Promoter and miRNA analyses
To search for cis-acting elements in the promoter sequences of SlSGR genes, genomic DNA sequences in the promoter region (1500 bp) were downloaded from Phytozome and the promoter regions were scanned using the PlantPAN 3.0 server (http://plantpan.itps.ncku.edu.tw/) (Chow et al. 2019). The potential target miRNAs of SlSGRs were predicted by psRNA target server (http://plantgrn.noble.org/psRNATarget/) based on default parameters (Dai and Zhao 2011).
2.5 Co-expression gene network analyses
The co-expression networks were created using a sub-dataset of the study conducted by Silva et al. (2021). The full raw data and its count matrix are available with the GSE148217 number on Gene Expression Omnibus (GEO), NCBI (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE148217). Three SlSGR genes SlSGR1 (Solyc08g080090), SlSGR2 (Solyc12g056480.1.1) and SlSGR-like (Solyc04g063240) were searched in the count matrix file and only two SlSGR genes (SlSGR1 and SlSGR-like) were found. The correlation analyses were conducted with about 19, 000 genes after filtration of the sub-dataset and using the read counts of mature green (MG) and ripe fruit (R) stages of tomato cultivar Ailsa Craig (AC), selected from the count matrix of the study. The details of correlation analysis were described by (Contreras-López et al. 2018). Then the most highly correlated genes with each SlSGR were filtered from the correlation result file at a > 0.9 correlation coefficient (r) level using Pandas package of Python programming language (Mckinney 2010). Then, each SlSGR gene file was merged into a single “result file” and the members of 38 transcription factor families, obtained from iTAK TF database (http://itak.feilab.net/cgi-bin/itak/index.cgi) (Zheng et al. 2016), were searched and labeled within the result file. The data on the file was visualized on Cytoscape 3.8.2 (Shannon et al. 2003).
2.6 Plant materials and salt stress treatments
Laboratory experiments were conducted at Burdur Mehmet Akif Ersoy University, Turkey; between June-September in 2020. Flower, young/mature leaf, and pericarp at four developmental stages including IMG (immature green), MG (mature green), BR (Breaker) and Ripe (R) were collected from ‘red cherry’ tomato cultivar to determine SlSGR gene expression profiles at fruit ripening stages.
Two tomato (Solanum lycopersicum L.) hybrids ‘Ciko F1’ and ‘Black’ were used for salt treatment. Plants were grown with Hoagland solution for two months and salt treatment was applied gradually (50 mM, 100 mM, 150 mM and finally 200 mM NaCl) over 3-day intervals to avoid osmotic shock (Akbudak and Filiz 2019). After 24 h of the final treatment of 200 mM NaCl, plant leaves were picked for RNA isolation. For each stage, three biological replicates were prepared. All the samples were immediately frozen in liquid nitrogen and were transferred to -80 °C until RNA isolation.
2.7 RNA isolation and gene expression analysis by RT-qPCR
Total RNA was extracted from harvested plant tissues (flower, young / mature leaf, fruit pericarps and salinity treated plants) using PureLink™ Plant RNA Reagent (Thermo Fisher Scientific) in accordance with the manufacturer’s instructions. Total RNA was dissolved in 50 µL of elution buffer, the concentration and quality of RNA were determined by NanoDrop (Epoch Microplate, Biotek). Total RNA was reverse transcribed into cDNA by the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). The mean of tomato Actin (Diretto et al. 2020) and Elongation Factor 1-α (Pokalsky et al. 1989) genes were used as the internal control for normalization of the expression of the target genes. The RT-qPCR reaction was performed in a 20 µl reaction mixture using the CFX96™ Real-Time System (Bio-Rad). The thermal program for PCR was set using the following conditions: 95 °C for 5 min, 35 cycles of 5 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C. The relative expression level of SlSGR genes from three biological and two technical replicates was calculated by the 2− ΔCt method (Livak and Schmittgen 2001). Primers used in this experiment are listed in (Supplementary Table S1). Changes in relative expression levels of the gene were checked for statistical significance in accordance with the one-way ANOVA and the means and standard deviation of the replications were compared by the least significant difference (LSD) test at the 5% level.
3 Results and discussion
3.1 Identification of the SGR genes
Members of the SlSGR family were downloaded from the Solanum lycopersicum genome (https://solgenomics.net/) and SGR domains (PF12638) were identified using the Pfam database (http://pfam.xfam.org/) profile of the SGR domain. The protein properties of SlSGRs including ID number, exon number, protein length, theoretical isoelectric point (pI), molecular weight, domain family, subcellular localization and phosphorylation sites are presented in Table 1. The protein lengths of SlSGRs ranged from 242 to 334 amino acids (aa) with an average length of 271 residues. Molecular weight varied from 27.4 kDa to 30.8 kDa, and the pI values varied from 6.66 to 9.32, with an average of 8.5, suggesting that most SlSGR proteins were weakly basic. The subcellular localization predictions for SlSGRs indicated their localizations to be in the chloroplast, nucleus, extracellular matrix, and mitochondria. The number of predicted phosphorylation sites of SlSGR proteins ranged from 8 to 33 with an average of 25. The highest prediction values for phosphorylation sites are chloroplast, nucleus, EM and mitochondria, suggesting dynamic regulation of SlSGR proteins in cell metabolism.
3.2 Conserved motif and phylogenetic analyses
To better understand the SlSGR proteins, the identity matrix of the SlSGR protein sequences of tomato, Arabidopsis and rice were calculated, and ranged between 0.699 and 0.281 (Supplementary Table S2). The highest identity was about 70% between AtSGR1 and AtSGR2, followed by about 62% between AtSGR1 and SlSGR1, and about 61% between SlSGR1 and SlSGR2. The lowest sequence identity was determined between AtSGR1 and OsSGR1. Consequently, the motif analysis showed that there were sequence variations among SGR proteins.
To identify conserved domains of SGR proteins, amino acid sequences of eight proteins from Arabidopsis, tomato, and rice were aligned with BioEdit. The results showed that SGR domains had three different conserved sites, including chloroplast signal peptides, SGR domains and variable C-terminal regions (Fig. 1). SGR proteins have four Cys-rich motifs (C-X3-C-X-C2-F-P-X5-P) at the C-terminus, but SGR-like proteins, whose unstable region is 12 to 38 amino acids long, lack this motif (Pilkington et al. 2012). This result is consistent with that of Bade et al. ( 2016) suggesting that SlSGR-like proteins do not have the CRM domain, which plays unique roles in the conformational change and Mg-dechelating activity (Xie et al. 2019).

Multiple alignments of SGR proteins in tomato, Arabidopsis, and rice using BioEdit program. Black shading indicates the identical regions of SGR proteins. Blue, red, and orange lines indicate the chloroplast signal peptide, the STAY-GREEN protein domain structure (PF12638), and variable C-terminal region, respectively. CRM is shown in the red frame
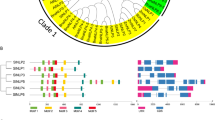
To explore the evolutionary relationship of the SlSGR family, SGR protein sequences of S. lycopersicum, A. thaliana, and O. sativa were used to generate the phylogenetic tree (Fig. 2). A total of eight SGRs were clearly divided into two groups, called SGR and SGR-like with well-supported bootstrap values (1000 replicates). The phylogenetic tree showed that SlSGR-like belonged to the SGR-like subfamily.

Phylogenetic tree of SGR genes in S. lycopersicum, Arabidopsis and O. sativa. The rootless phylogenetic trees of S. lycopersicum, Arabidopsis and O. sativa SGR protein sequences
3.3 Gene structure and protein motif composition analyses of SGR genes
Protein motifs are conserved amino acid sequences, which are functionally important portions of proteins. To identify the structural diversification of SGR proteins, four conserved motifs were identified by MEME server and annotated with SMART and Pfam database (Fig. 3a). The lengths of these conserved motifs varied from 29 to 50 amino acids. All SGR proteins had similar motifs (Fig. 3b). Of these, motif 1 was found in all the SGRs, except for LOC_Os04g59610.2. This motif similarity indicates that SGR genes may share similar functions (Fig. 3c.).

Motif composition and gene structure of the SGR genes of tomato. (a) Motif patterns of the tomato SGR proteins generated using MEME. (b) Motif locations of SGR proteins of A. thaliana, S. lycopersicum and O. sativa. (c) Consensus motif symbol of SGR proteins. (d) Gene structure of SGR genes generated using Gene Structure Display Server of A. thaliana, S. lycopersicum and O. sativa. Upstream/downstream regions are represented by a blue box, coding regions (CDS) are shown as yellow round-corner rectangles, and introns are shown as red lines
In order to compare the diversity of gene structure, the exon-intron organization of SGR genes was analysed (Fig. 3d). The number of introns in SGRs varied from two to four. Although AtSGRs contain two introns, SlSGR1 contains three introns, SlSGR2 consists of three introns with no UTRs and SlSGR-like is comprised of four introns. Overall, SGR genes show a complex gene structure with varying intron positions and lengths.
3.4 Gene ontology enrichment analysis
In terms of GO categories, we predicted the functions of all SlSGR proteins, including biological process, molecular function, and cellular component using PANNZER server (Törönen et al. 2018). A total of three biological processes, three molecular functions, and four cellular components were identified (Fig. 4). Chloroplast organization (GO:0009658), chlorophyll metabolic processes (GO:0090056) and tetrapyrrole catabolic processes (GO:1,901,404) were identified in the biological process category indicating that SlSGR genes mostly function in chlorophyll metabolism. Protein binding (GO:0005515), tRNA 2’-phosphotransferase activity (GO:0000215), and ribosyltransferase activity (GO:0003950) were predicted for the molecular function category. Therefore, SlSGR genes may be involved in tRNA metabolism. Plastid thylakoid membrane (GO:0055035), integral component of membrane (GO:0016021), plastid membrane (GO: 0042170) and extracellular region (GO: 0005576) were identified under the cellular component category. In conclusion, these SGR proteins may have a wide range of roles in chlorophyll metabolism.

Functional annotation of SlSGR proteins in terms of biological process, molecular function, and cellular components
3.5 Prediction of miRNA targets
MicroRNAs (miRNAs) are small noncoding single-stranded RNA molecules that play important regulatory roles by targeting mRNAs for cleavage or translational repression (Li et al. 2017). To identify potential miRNA target sites within the putative SlSGR genes, the mRNA sequences were analysed with the psRNATarget server. By querying the miRNA database, we identified two miRNAs, sly-miR9469-3p and sly-miR156e-5p, targeting SlSGR-like and SlSGR2 genes, respectively, for translation and cleavage (Supplementary Table S3). Consequently, transcriptional regulation of SlSGRs may be controlled by miRNAs.
3.6 Cis-regulatory elements analysis
cis-regulatory elements are special motifs involved in gene transcriptional regulation during plant development and in the response to stress (Le et al. 2012). To explore the transcriptional regulation and potential functions of SlSGRs, the putative cis-regulatory elements in the 200 bp upstream region of the initiation codon were analysed using the PlantCARE database and, eight cis-regulatory elements were identified (Fig. 5). AT-hook transcription factors, are found specifically in land plants, and are involved in stress responses and regulation of growth and development (Favero et al. 2020). MYB TFs are characterized by a highly conserved DNA-binding domain and regulate plant-specific processes like hormone responses, differentiation, biotic and abiotic stresses, and phenylpropanoid metabolism (Segarra et al. 2009). WRKY TFs regulate many different plant processes such as biotic/abiotic stresses, seed development, and senescence (Zou et al. 2004). Additionally, different functions related to cis-elements were found in the promoter region of SlSGRs, such as GATA (phytohormone), DOF (light, seed maturation and germination), bHLH (regulation of fruit dehiscence, stress responses), bZIP (photomorphogenesis and energy homeostasis), and homeodomain (responses to environmental stresses) (Perotti et al. 2017). Overall, the cis-regulatory elements found in the SlSGRs indicate that SlSGR genes may participate in responses to hormones and stress signals.

Predicted transcription factor binding sites (TFBSs) in promotor sequences of SlSGR genes
3.7 Co-expression network analyses of SlSGR genes
Co-expression network analysis can reveal modules of co-expressed genes, which share similar biological functions in large transcriptomic datasets (Rao and Dixon 2019). In the present study two co-expression networks were set up for SlSGR1 and SlSGR-like genes. Later, these two networks were amalgamed to identify co-expressed common genes. Each co-expression network was constructed using transcriptomic data of two fruit ripening stages, mature green (MG) and ripe fruit (R) stages, of the AC tomato cultivar. After correlation analyses, 3875 and 2287 genes clustered in the networks of SlSGR1 and SlSGR-like, respectively, at r > 0.9. The top ten identified neighbours of SlSGR1 in the co-expression network functioned in autophagocytosis, calcium signalling, carbon fixation and sucrose metabolism, cell wall matrix polysaccharide biosynthesis, arginine metabolism, cysteine oxidation and proteins phosphorylation (Table 2). The top ten neighbour genes in the network of SlSGR-like were found to take part in magnesium (Mg) metabolism, DNA methylation, oxidoreductase activity, anthocyanin biosynthesis, acyl lipid metabolism, extracellular signaling and ubiquitin-mediated proteolysis (Table 3). The association of SlSGR genes with very broad metabolic pathways supports the importance of the gene family in cell metabolism.
In addition, six common genes were the identified when two co-expression networks were merged. The functional annotations of these genes are shown in Table 4. Of these genes, thioredoxin proteins play vital roles in catalysing the thiol-disulfide interchange and regulation of the redox environment of the cell (Gelhaye et al. 2005). Enoyl-acyl carrier protein (ACP) reductases (ENRs) are involved in fatty acid biosynthesis, catalyzing the fatty acid elongation cycle (Massengo-Tiassé and Cronan 2009). Cupin family protein family like germin and germin-like proteins (GLPs) are involved in plant development and defence (Wang et al. 2014; Hu et al. 2014) stated that Pollen Ole e 1 domain-containing proteins act as developmental regulators as well as major allergens in many plant tissues. The co-expression network of the SlSGR genes showed that they may be involved in sulphur homeostasis and fatty acid biosynthesis, during fruit ripening of tomato.
3.8 TF genes in the co-expression network of SlSGR genes
Metabolic networks and their regulations are vital for fitness of an organism. In addition, transcriptional control of enzyme-encoding genes plays essential roles in response to environmental and developmental stimuli (Gaudinier et al. 2015). In this study, transcriptional networks of SlSGR genes were analysed using gene co-expression networks. A total of 13 TF genes, directly connected to SlSGR genes, were identified. Of these TFs, three TFs were found in the SlSGR1 network, and 10 TFs were identified for the SlSGR-like network (Table 5; Fig. 6B). Two of the three identified TFs in the SlSGR1 network belonged to the GRAS TF family. In addition, three GRAS TF members were identified as first neighbours of SlSGR-like. The GRAS family is a plant‑specific TF family involved in regulation of plant growth and development. The GRAS family is divided into eight subfamilies, SHR, SCR, SCL3, DELLA, HAM, LS, LISCL and PAT1 (Hirsch and Oldroyd 2009). DELLA proteins, which negatively regulate senescence, increase the expression of downstream senescence-related SGR genes (Zhang et al. 2018). In addition, members of the GeBP, ERF, MYB and bHLH TFs were also found in the co-expression networks of two SlSGR genes. It was reported that an ethylene response factor, CitERF13, trans-activated the CitSGR promoter during citrus fruit de-greening (Yin et al. 2016). In conclusion, SGR genes may be either in the same pathway or have molecular interactions with GRAS TF family in cellular metabolism. In addition, SGR genes are co-expressed with different TFs involved in cellular metabolism.

The merged co-expression networks of SlSGR1 and SlSGR-like (A) and the identified members of transcription factor families in each network (B). Six genes (shown in magenta) in each network are identified as common genes between the networks. The networks were constructed based on correlation analyses and genes in the networks were selected at the r > 0.9 level
3.9 Expression patterns of SlSGR genes in different tissues and under salt stress
The expression profiles of SlSGR genes in seven different tissues/organs and under 200 mM salinity treatment (two tomato cultivars) were analysed using RT-qPCR (Fig. 7). Each gene showed a range of expression patterns in different tissues/organs. SlSGR1/2 and SlSGR-like were moderately expressed in flower tissues. However, while SlSGR1 expression was almost undetectable in leaves, the expression of SlSGR-like was the highest in photosynthetically active leaves. The expression SlSGR1/2 increased steadily during fruit ripening and peaked in ripe fruits (R stage). In contrast, SlSGR-like expression was almost undetectable in ripening fruit.

The expression profiles of SlSGR1/2 and SlSGR-like in different tomato organs and salinity treated tomato cultivars. Light grey bars: Flower (F), young leaf (YL), mature leaf (ML), immature green (IMG), mature green (MG), breaker fruit (BR), ripe fruit (R). Dark grey bars: ‘Ciko’ variety control (CC), ‘Ciko’ variety 200 mM salinity treatment (CT), ‘Black’ variety control (BC), ‘Black’ variety 200 mM salinity treatment. Mean values and SE ± were obtained from three biological and two technical replicates. The bars with different letters indicate significant differences, p < 0.05
In contrast to its expression in vegetative and reproductive tissues/organs SlSGR2 seemed to be more active than the others under salinity stress in tomato. While the expression patterns of SlSGR1 and SlSGR-like did not change under salinity in the two different tomato cultivars, SlSGR2 exhibited a drastic increase under salinity stress compared to the controls in both cultivars. These results indicated that expression of SlSGR1 and SlSGR2 increases during fruit ripening and CHL degradation, and SlSGR2 is involved in salinity stress.
It was previously suggested that the SGR subfamily proteins play a critical role in the initiation of Chl degradation and senescence in various plant species (Barry et al. 2008; Zhou et al. 2011; Jiao et al. 2020). Upregulation of Chl degradation and downregulation of Chl biosynthesis result in de-greening. The upregulation of SlSGR1/2 during fruit ripening and of SlSGR-like during leaf senescence coincides with Chl breakdown (Park et al. 2007). Previous comparative investigations of the expression patterns of SlSGR1 in tomato have shown that unlike SlSGR-like, SlSGR1 was strongly expressed in ripening fruits (Hu et al. 2011). To the best of our knowledge, there are no studies about on the role of SlSGR2 in ripening tomato fruit. In this study, the expression of SlSGR1 and SlSGR2 began to increase in MG fruits and reached a very high level in ripe (R) fruits. This expression pattern indicated that SlSGR1/2 play a vital role in Chl degradation in ripening fruits. Similar to this finding, the repression of SlSGR1 in transgenic tomato fruits elevated SlPSY1 mRNA accumulation, which encodes a key carotenoid synthesis enzyme, at the early stages of fruit ripening, resulting in increased lycopene and β-carotene levels in red ripe fruits (Luo et al. 2013). Moreover, RNAi silenced SlSGR1 fruits had a longer shelf-life with the decreased expression of ethylene response (ETR) 1, 3, 4 and 5 genes and delayed activity of PG2A and pectinesterase (PE) enzymes up to seven days post‐breaker stage. However, more research is needed to clarify the exact role of SlSGR2 in tomato ripening. It was also previously shown that SlSGR1 mRNA was present mainly in senescent leaves, but almost undetectable in young and mature leaves (Hu et al. 2011). This result was well-supported in this study as SlSGR1/2 expression levels were very low in young and mature leaves. In contrast to SlSGR1/2, SlSGR-like was hardly detected in tissues with little or no Chl, such as the mature fruits at BR and R stages. Overall, these results imply that SlSGR1/2 and SlSGR-like may function at different stages, and they have opposite expression patterns in tomato leaves and ripening fruit.
To gain more insight about SlSGR genes in response to salinity stress, gene expression levels were analysed in two tomato varieties using RT-qPCR. Interestingly, while the expression levels of SlSGR1 and SlSGR-like did not significantly change under salinity stress, SlSGR2 expression was upregulated 5-fold in both varieties. Similar to the finding of the present study, previous studies reported that the SlSGR genes are regulated differently under other stress conditions. For example, the expression level of SlSGR1 was enhanced under water-deficit and flooding (Hu et al. 2011). Five days of heat treatment in tomato upregulated the transcript abundance of SlSGR1 and SlSGR2 up to 11.79- and 18.82-fold for the genes, respectively, relative to no heat treatment (0 h) (Jahan et al. 2021). Moreover, the expression of SlSGR-like was induced by darkness, PEG, and ABA, and downregulated by ethylene and H2O2 (Yang et al. 2020). While the expression of SlSGR genes needs to be further studied under different stress treatments, SlSGR2 might have a role in the breakdown of chlorophyll under various abiotic stresses.
Genome-wide identification, gene structure and protein sequences analyses, and expression profile analyses of SlSGR genes in different tissues and under salt stress were performed in the present study. The findings showed that SlSGR2 is not only involved in fruit ripening and Chl degradation but participates in salt response mechanisms in tomato. The upregulation of SlSGR2 under salt stress and of SlSGR1 in fruit ripening may be related to their roles in stress-signalling and senescence pathways, respectively. Additionally, the roles of SlSGR genes in fruit ripening may be regulated by several TFs, among which GRAS family members may have more specific roles.
References
Abdelrahman M, El-Sayed M, Jogaiah S, Burritt DJ, Phan Tan LS (2017) The “STAY-GREEN” trait and phytohormone signaling networks in plants under heat stress. Plant Cell Rep 36:1009–1025
Akbudak MA, Filiz E (2019) Whirly (Why) transcription factors in tomato (Solanum lycopersicum L.): genome-wide identification and transcriptional profiling under drought and salt stresses. Mol Biol Rep 46:4139–4150. https://doi.org/10.1007/s11033-019-04863-y
Bade RG, Bao ML, Jin WY, Ma Y, Niu YD, Hasi A (2016) Genome-wide identification and analysis of the SGR gene family in Cucumis melo L. Genet Mol Res 15. https://doi.org/10.4238/gmr15048485
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res 37. https://doi.org/10.1093/nar/gkp335
Barry CS, McQuinn RP, Chung MY, Besuden A, Giovannoni JJ (2008) Amino acid substitutions in homologs of the Stay-Green protein are responsible for the green-flesh and chlorophyll retainer mutations of tomato and pepper. Plant Physiol 147:179–187. https://doi.org/10.1104/pp.108.118430
Chow CN, Lee TY, Hung YC, Li GZ, Tseng KC, Liu YH, Kuo PL, Zheng HQ, Chang WC (2019) Plantpan3.0: A new and updated resource for reconstructing transcriptional regulatory networks from chip-seq experiments in plants. Nucleic Acids Res 47:D1155–D1163. https://doi.org/10.1093/nar/gky1081
Contreras-López O, Moyano TC, Soto DC, Gutiérrez RA (2018) Step-by-step construction of gene co-expression networks from high-throughput Arabidopsis RNA sequencing data. Methods in Molecular Biology. Humana Press Inc., pp 275–301
Dai X, Zhao PX (2011) PsRNATarget: A plant small RNA target analysis server. Nucleic Acids Res 39. https://doi.org/10.1093/nar/gkr319
Diretto G, Frusciante S, Fabbri C, Schauer N, Busta L, Wang Z, Matas AJ, Fiore A, Rose JKC (2020) Manipulation of β-carotene levels in tomato fruits results in increased ABA content and extended shelf life. Plant Biotechnol J 18:1185–1199. https://doi.org/10.1111 /pbi.13283
Favero DS, Kawamura A, Shibata M, Takebayashi A, Jung JH, Suzuki T, Jaeger KE, Ishida T, Iwase A, Wigge PA, Neff MM, Sugimoto K (2020) AT-Hook Transcription Factors Restrict Petiole Growth by Antagonizing PIFs. Curr Biol 30:1454–1466e6. https://doi.org/10.1016/j.cub.2020.02.017
Gaudinier A, Tang M, Kliebenstein DJ (2015) Transcriptional networks governing plant metabolism. Curr Plant Biol 3–4:56–64. https://doi.org/10.1016/j.cpb.2015.07.002
Gelhaye E, Rouhier N, Navrot N, Jacquot JP (2005) The plant thioredoxin system. Cell Mol Life Sci 62:24–35
Guo AY (2007) GSDS: a gene structure display server. Hereditas 29:1023. https://doi.org/10.1360/yc-007-1023
Hall T, Biosciences I, Carlsbad C (2011) BioEdit: An important software for molecular biology. GERF Bull Biosci 2:60–61
Hirsch S, Oldroyd GED (2009) GRAS-domain transcription factors that regulate plant development. Plant Signal Behav 4:698–700
Hörtensteiner S (2009) Stay-green regulates chlorophyll and chlorophyll-binding protein degradation during senescence. Trends Plant Sci 14:155–162
Hu B, Liu B, Liu L, Liu C, Xu L, Ruan Y (2014) Epigenetic control of Pollen Ole e 1 allergen and extensin family gene expression in Arabidopsis thaliana. https://doi.org/10.1007 /s11738-014-1597-6
Hu ZL, Deng L, Yan B, Pan Y, Luo M, Chen XQ, Hu TZ, Chen GP (2011) Silencing of the LeSGR1 gene in tomato inhibits chlorophyll degradation and exhibits a stay-green phenotype. Biol Plant 55:27–34. https://doi.org/10.1007/s10535-011-0004-z
Jahan MS, Shu S, Wang Y, Hasan M, El Yazied AA, Alabdallah NM, Hajjar D, Altaf MA, Sun J, Guo S (2021) Melatonin Pretreatment Confers Heat Tolerance and Repression of Heat-Induced Senescence in Tomato Through the Modulation of ABA- and GA-Mediated Pathways. Front Plant Sci. https://doi.org/10.3389/fpls.2021.650955
Jiao B, Meng Q, Lv W (2020) Roles of stay-green (SGR) homologs during chlorophyll degradation in green plants. Bot Stud 61:25
Kuai B, Chen J, Hörtensteiner S (2018) The biochemistry and molecular biology of chlorophyll breakdown. J Exp Bot 69:751–767
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol 35(6):1547–1549. doi: https://doi.org/10.1093/molbev/msy096
Kusaba M, Ito H, Morita R, Lida S, Sato Y, Fujimoto M, Kawasaki S, Tanaka R, Hirochika H, Nishimura M, Tanaka A (2007) Rice non-yellow coloring1 is involved in light-harvesting complex II and grana degradation during leaf senescence. Plant Cell 19:1362–1375. https://doi.org/10.1105/tpc.106.042911
Le DT, Nishiyama R, Watanabe Y, Vankova R, Tanaka M, Seki M, Ham LH, Shinozaki KY, Shinozaki K, Tran LSP (2012) Identification and expression analysis of Cytokinin metabolic genes in soybean under normal and drought conditions in relation to Cytokinin levels. PLoS ONE 7:42411. https://doi.org/10.1371/journal.pone.0042411
Li S, Castillo-González C, Yu B, Zhang X (2017) The functions of plant small RNAs in development and in stress responses. Plant J 90:654–670. https://doi.org/10.1111/tpj.13444
Li X, Wang Y, Chen S, Tian H, Fu D, Zhu B, Luo Y, Zhu H (2018) Lycopene Is Enriched in Tomato Fruit by CRISPR/Cas9-Mediated Multiplex Genome Editing. Front Plant Sci 9:559. https://doi.org/10.3389/fpls.2018.00559
Lim PO, Kim HJ, Gil Nam H (2007) Leaf Senescence. Annu Rev Plant Biol 58:115–136. https://doi.org/10.1146/annurev.arplant.57.032905.105316
Liu L, Shao Z, Zhang M, Wang Q (2015) Regulation of carotenoid metabolism in tomato. Mol Plant 8:28–39
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408. doi: https://doi.org/10.1006/meth.2001.1262
Luo Z, Zhang J, Li J, Yang C, Wang T, Ouyang B, Li H, Giovannoni J, Ye Z (2013a) A STAY-GREEN protein SlSGR1 regulates lycopene and β‐carotene accumulation by interacting directly with SlPSY1 during ripening processes in tomato. New Phytol 198:442–452. https://doi.org/10.1111/nph.12175
Massengo-Tiassé RP, Cronan JE (2009) Diversity in enoyl-acyl carrier protein reductases. Cell Mol Life Sci 66:1507–1517
Matile P, Hörtensteiner S, Thomas H (1999) CHLOROPHYLL DEGRADATION. Annu Rev Plant Physiol. Plant Mol Biol 50:67–95. https://doi.org/10.1146/annurev.arplant.50.1.67
Mckinney W (2010) Data Structures for Statistical Computing in Python
Meguro M, Ito H, Takabayashi A, Tanaka R, Tanaka A (2011) Identification of the 7-hydroxymethyl chlorophyll a reductase of the chlorophyll cycle in arabidopsis. Plant Cell 23:3442–3453. https://doi.org/10.1105/tpc.111.089714
Mendel G (1901) E p h (1865). Sch Publ 3–47
Park SY, Yu JW, Park JS, Li J, Yoo SC, Lee NY, Lee SK, Jeong SW, Seo HS, Koh HJ, Jeon JS, Park YI, Peak NC (2007) The senescence-induced staygreen protein regulates chlorophyll degradation. Plant Cell 19:1649–1664. https://doi.org/10.1105/tpc.106.044891
Perotti MF, Ribone PA, Chan RL (2017) Plant transcription factors from the homeodomain-leucine zipper family I. Role in development and stress responses. IUBMB Life 69:280–289
Pilkington SM, Montefiori M, Jameson PE, Allan AC (2012) The control of chlorophyll levels in maturing kiwifruit. Planta 236:1615–1628. https://doi.org/10.1007/s00425-012-1723-x
Pokalsky AR, Hiatt WR, Ridge N, Rasmussen R, Houck CM, Shewmaker CK (1989) Structure and expression of elongation factor 1α in tomato. Nucleic Acids Res 17:4661–4673. https://doi.org/10.1093/nar/17.12.4661
Pružinská A, Anders I, Aubry S, Schenk N, Lüthi ET, Müller T, Krautler B, Hörtensteiner S (2007) In vivo participation of red chlorophyll catabolite reductase in chlorophyll breakdown. Plant Cell 19:369–387. https://doi.org/10.1105/tpc.106.044404
Rao X, Dixon RA (2019) Co-expression networks for plant biology: Why and how. Acta Biochim Biophys Sin (Shanghai) 51:981–988
Rong H, Tang Y, Zhang H, Wu P, Chen Y, Li M, Wu G, Jiang H (2013) The Stay-Green Rice like (SGRL) gene regulates chlorophyll degradation in rice. J Plant Physiol 170:1367–1373. https://doi.org/10.1016/j.jplph.2013.05.016
Sakuraba Y, Lee SH, Kim YS, Park OK, Hörtensteiner S, Paek NC (2014a) Delayed degradation of chlorophylls and photosynthetic proteins in Arabidopsis autophagy mutants during stress-induced leaf yellowing. J Exp Bot 65:3915–3925. https://doi.org/10.1093 /jxb/eru008
Sakuraba Y, Park SY, Kim YS, Wang SH, Yoo SC, Hörtensteiner S, Paek NC (2014b) Arabidopsis STAY-GREEN2 is a negative regulator of chlorophyll degradation during leaf senescence. Mol Plant 7:1288–1302. https://doi.org/10.1093/mp/ssu045
Sakuraba Y, Schelbert S, Park SY, Han SH, Lee BD, Andres CB, Kessler F, Hörtensteiner S, Paek NC (2012) STAY-GREEN and chlorophyll catabolic enzymes interact at light-harvesting complex II for chlorophyll detoxification during leaf senescence in Arabidopsis. Plant Cell 24:507–518. https://doi.org/10.1105/tpc.111.089474
Schelbert S, Aubry S, Burla B, Agne B, Kessler F, Krupinska K, Hörtensteiner S (2009) Pheophytin pheophorbide hydrolase (pheophytinase) is involved in chlorophyll breakdown during leaf senescence in Arabidopsis. Plant Cell 21:767–785. https://doi.org/10.1105/tpc.108.064089
Segarra G, Van Der Ent S, Trillas I, Pieterse CMJ (2009) MYB72, a node of convergence in induced systemic resistance triggered by a fungal and a bacterial beneficial microbe. Plant Biol 11:90–96. https://doi.org/10.1111/j.1438-8677.2008.00162.x
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. https://doi.org/10.1101/gr.1239303
Shimoda Y, Ito H, Tanaka A (2016) Arabidopsis STAY-GREEN, mendel’s green cotyledon gene, encodes magnesium-dechelatase. Plant Cell 28:2147–2160. https://doi.org/10.1105/tpc.16.00428
Silva CJ, van den Abeele C, Ortega-Salazar I, Papin V, Adaskaveg JA, Wang D, Casteel CL, Seymour GB, Ulate BB (2021) Host susceptibility factors render ripe tomato fruit vulnerable to fungal disease despite active immune responses. J Exp Bot. https://doi.org/10.1093/jxb/eraa601
Törönen P, Medlar A, Holm L (2018) PANNZER2: A rapid functional annotation web server. Nucleic Acids Res 46:W84–W88. https://doi.org/10.1093/nar/gky350
Wang X, Zhang H, Gao Y, Sun G, Zhang W, Qui L (2014) A comprehensive analysis of the cupin gene family in soybean (Glycine max). PLoS ONE 9:110092. https://doi.org/10.1371/journal.pone.0110092
Xie Z, Wu S, Chen J, Zhu X, Zhou X, Hörtensteiner S, Ren G, Kuai B (2019) The C-terminal cysteine-rich motif of NYE1/SGR1 is indispensable for its function in chlorophyll degradation in Arabidopsis. Plant Mol Biol 101:257–268. https://doi.org/10.1007/s11103-019-00902-1
Yang M, Zhu S, Jiao B, Duan M, Meng Q, Ma N, Lv W (2020) SlSGRL, a tomato SGR-like protein, promotes chlorophyll degradation downstream of the ABA signaling pathway. Plant Physiol Biochem 157:316–327. https://doi.org/10.1016/j.plaphy.2020.10.028
Yin XR, Xie XL, Xia XJ, Yu JQ, Ferguson IB, Giovannoni JJ, Chen KS (2016) Involvement of an ethylene response factor in chlorophyll degradation during citrus fruit degreening. Plant J 89:403–412. https://doi.org/10.1111/tpj.13178
Yu C-S, Chen Y-C, Lu C-H, Hwang J-K (2006) Prediction of protein subcellular localization. Proteins Struct Funct Bioinforma 64:643–651. https://doi.org/10.1002/prot.21018
Zhang Y, Liu Z, Wang X, Wang J, Fan K, Li Z, Lin W (2018) DELLA proteins negatively regulate dark-induced senescence and chlorophyll degradation in Arabidopsis through interaction with the transcription factor WRKY6. Plant Cell Rep 37:981–992. https://doi.org/10.1007/s00299-018-2282-9
Zheng Y, Jiao C, Sun H, Rosli HG, Pombo MA, Zhang P, Banf M, Dai X, Martin GB, Giovannoni JJ, Zhao PX, Rhee SY, Fei Z (2016) iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol Plant 9:1667–1670
Zhou C, Han L, Pislariu C, Nakashima J, Fu C, Jiang Q, Quan L, Blancaflor EB, Tang Y, Bouton JH, Udvardi M, Xia G, Wang ZY (2011) From model to crop: Functional analysis of a stay-green gene in the model legume Medicago truncatula and effective use of the gene for alfalfa improvement. Plant Physiol 157:1483–1496. https://doi.org/10.1104/pp.111.185140
Zou X, Seemann JR, Neuman D, Shen QJ (2004) A WRKY gene from creosote bush encodes an activator of the abscisic acid signaling pathway. J Biol Chem 279:55770–55779. https://doi.org/10.1074/jbc.M408536200
Acknowledgements
We would like to thank Graham B. Seymour and Barbara Blanco-Ulate for providing detailed information about RNA-Seq data used for co-expression analysis.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
SU, AK and EF conceived the study; SU and AK conducted the lab experiments, AK, EF, FK generated bioinformatic analysis; All authors wrote the manuscript, read, edited, and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Communicated by Sung-Chur Sim.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Uluisik, S., Kıyak, A., Kurt, F. et al. STAY-GREEN (SGR) genes in tomato (Solanum lycopersicum): genome-wide identification, and expression analyses reveal their involvements in ripening and salinity stress responses. Hortic. Environ. Biotechnol. 63, 557–569 (2022). https://doi.org/10.1007/s13580-022-00419-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13580-022-00419-5