
Abstract
Plant tissue culture is an essential tool in biotechnology. However, tissue-cultured plants often exhibit variations that are either genetic or epigenetic in origin, termed somaclonal variations. Among these variations, DNA methylation is an important heritable epigenetic modification that plays a role in a wide variety of biological processes, including gene expression. In this study, we performed bisulfite sequencing of regenerated Chinese cabbage (Brassica rapa ssp. pekinensis) lines to identify DNA alterations induced by tissue culture. Sequencing data from each regenerated line were compared with reference genome sequences, and common differentially methylated regions (DMRs) were detected in the regenerants. To determine changes in expression levels of DMR-containing genes, we performed quantitative reverse transcription-polymerase chain reaction (qRT-PCR) of the target genes and PCR amplification with bisulfite-converted DNA. We identified DMRs between a non-regenerant line and regenerant lines and selected 10 DMR-associated genes that presented annotation information in Arabidopsis or Brassica rapa. Their expression levels were verified by qRT-PCR to determine the relation between methylation state and gene expression. We observed that genes positioned in DMRs significantly correlated with differential gene expression. We also observed similar methylation patterns in the selected DMRs by PCR-based methylation analysis. The results of this study are a valuable resource for the epigenetic analysis of regenerated lines, especially for Chinese cabbage.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Plant tissue culture is a fundamental tool of plant biotechnology that allows the production of large numbers of genetically identical plantlets. This in vitro technology is widely used in various research fields such as the mass production of secondary metabolites and genetic transformation for crop improvement. Plant cells are cultured under sterile conditions and some of them undergo dedifferentiation based on the plant’s power of regeneration, called totipotency. The stability of regenerated plant production and gene expression are the most important issues regarding this technique. However, in vitro culture and regeneration often lead to genetic and epigenetic changes, which are referred to as somaclonal variations. Variations derived from tissue cultures have been reported in various crops (Larkin and Scowcroft 1981; Bajaj 1990) and are manifested as changes in chromosome numbers or structures, nucleotide sequence, gene expression, transposon activation, and phenotype (Kaeppler and Phillips 1993; Miguel and Marum 2011). These undesired variations of in vitro-grown plants may compromise the objectives of tissue culture. Therefore, it is necessary to determine the epigenetic variations that can potentially occur in in vitro-cultured plants to study the potential long-term consequences of this phenomenon.
DNA methylation is an essential epigenetic modification that involves a variety of biological processes (Niederhuth and Schmitz 2014; Seymour and Becker 2017). It has been described as being involved in gene regulatory mechanisms (Zilberman and Henikoff 2007; Teixeira and Colot 2009; Maunakea et al. 2010; Bucher et al. 2012; Xing et al. 2015; Seymour and Becker 2017), morphological development (Jacobsen and Meyerowitz 1997; Cubas et al. 1999; Soppe et al. 2000; Manning et al. 2006; Hsieh et al. 2009), and agronomic trait formation (Manning et al. 2006; Miura et al. 2009; Quadrana et al. 2014). Changes in DNA methylation patterns are frequently observed in regenerated plants and have been suggested to cause phenotypic variation through the modulation of gene expression (Kubis et al. 2003). Although epigenetic changes are often temporary and may be easily reverted to their normal status, epigenetic inheritance has also been reported in plants (Iglesias and Cerdán 2016). For these reasons, increasingly more studies are now focusing on the epigenetic aspects of somaclonal variation (Kaeppler et al. 2000; Miguel and Marum 2011).
There are three types of DNA methylation according to the sequence context. A cytosine can be methylated when it is located in the following sequences: CG, CHG, and CHH (H can be A, C, or T) (Feng et al. 2010; Law and Jacobsen 2010). Various studies have been carried out to locate CpG loci considered essential for gene regulation, and changes in DNA methylation in promoter and gene body regions affect gene expression and phenotype (Miguel and Marum 2011; Yang et al. 2014; Taiko et al. 2015). Therefore, profiling DNA methylation across the genome is vital to understand the impact of epigenetics (Laird 2010). The hypomethylation within a promoter region allows transcription factors to bind to the DNA strand and transcription to occur, while hypermethylation results in an opposite effect. Epigenetic variations have been assessed by various methods, including restriction fragment length polymorphism (RFLP), methylation-sensitive amplification polymorphism (MSAP), and methylation-specific polymerase chain reaction (MSP) (Müller et al. 1990; Smulders and De Klerk 2011; Coronel et al. 2018). Diverse computational tools and resources for the analysis of DNA methylation have been developed, including next-generation sequencing (NGS) (Bock and Lengauer 2008). These tools and resources enable characterization of genome-wide DNA methylation and methylation statuses at a single-base resolution.
Among the computational analysis techniques, NGS is a powerful tool applied in epigenetic research as well as in genetic analyses due to its high sensitivity, specificity, and scalability. The characterization of genome-wide DNA methylation on a large scale and single-base resolution has been enabled by NGS technology, including whole-genome bisulfite sequencing (WGBS), small RNA sequencing, and chromatin immunoprecipitation sequencing (ChIP-seq) (Laird 2010). Whole-genome DNA methylation analyses have been conducted in plants such as Arabidopsis thaliana, Oryza sativa (rice), Populus trichocarpa (poplar), and Glycine max (soybean) (Feng et al. 2010; Schmitz et al. 2013). These techniques have contributed not only to the obtainment of information about differentially methylated regions (DMRs), but also to the study of gene regulation mechanisms at the epigenetic level. In addition, the time required to obtain and characterize DNA methylomes has been reduced, while their accuracy has been improved in comparison to previous methods.
Chinese cabbage (Brassica rapa ssp. pekinensis) is one of the most important vegetables worldwide. The B. rapa reference genome (variety ‘Chiifu-401-42’) was published in 2011 (Brassica rapa Genome Sequencing Project Consortium 2011) and our group has constructed a pseudomolecule genome of B. rapa ‘CT001’ for precise genome research (Park et al. 2019).
In this work, we performed WGBS in a comparative manner between non-regenerant (control) and regenerant lines. DMRs were selected from R0, R1, and R2 lines of R0C22 and R0C31 and compared to those from a non-regenerant plant; those that were present in the same regions were selected and considered to be regenerant-specific DMR candidates. As DNA methylation in gene body and promoter is knowingly associated with gene expression levels, DMRs located 1-kb upstream of a gene and in its exons were selected for analysis. Our study provides an overview of the DNA methylome patterns in regenerated lines and highlights the importance of an epigenetic perspective on somaclonal variation.
2 Materials and methods
2.1 Plant materials and genomic DNA extraction
Regenerated lines of the Chinese cabbage inbred line ‘CT001’, which is widely used for tissue culture and plant transformation, were produced as follows. The seeds of ‘CT001’ were sterilized and germinated in MS (Murashige and Skoog 1962) basal medium. The upper part of each seedling was used as a non-regenerated control line and the hypocotyls were used to induce calluses and shoots. The regenerated lines R0C22 and R0C31 were self-pollinated to generate progeny lines. The regenerated lines R0C22 and R0C31 and their progenies (R1C22, R2C22, R1C31, and R2C31) had their genomes sequenced.
Total genomic DNA of the non-regenerant and regenerants was extracted from young leaves using sodium dodecyl sulfate lysis buffer following a modified method described by Dellaporta et al. (1983). The integrity and quality of DNA were evaluated using the Trinean DropSense instrument (Trinean, Belgium) and the PicoGreen assay (Molecular Probes, USA).
2.2 Bisulfite sequencing and mapping
Genomic DNAs of a non-regenerant line, two R0 regenerants (R0C22 and R0C31), and their progeny lines (R1C22, R1C31, R2C22, and R2C31) were fragmented to 200–300-bp sizes and bisulfite-converted using the EZ-DNA Methylation-Gold Kit (Zymo Research, Orange, USA) following the manufacturer’s instructions. Bisulfite-converted libraries were constructed using the Nextflex bisulfite-seq kit (Illumina, USA). Fragments were ligated to adaptors with a unique index sequence. The ligated products, with a length of approximately 550 bp, were used as templates for PCR amplification. Quality control (QC) was performed using a Bioanalyzer (Agilent, Santa Clara, USA) instrument and the library was subjected to sequencing using NextSeq 500™ (Illumina, USA).
The raw reads were cleaned by removing adaptor sequences, while reads with more than 10% of unknown bases and low-quality reads were removed using Trimmomatic software (Bolger et al. 2014). High-quality reads with a maximum of 2-bp mismatches were mapped to the ‘CT001’ pseudomolecule reference sequences (Park et al. 2019) using Bismark (Krueger and Andrews 2011), which employs a three-letter mapping algorithm for bisulfite read mapping with Bowtie2 (Langmead and Salzberg 2012) for recovered read mapping. The outputs were converted into sequence alignment map (SAM)/binary alignment map (BAM) formats and imported to genome browsers to be visualized and directly explored. Using the bisulfite sequencing data, we calculated DNA methylation with sequences that were mapped to a reference sequence with 95% mapping coverage levels. In addition, we only analyzed cytosines that were mapped with a depth of more than nine reads to identify the methylated cytosine among the mapped sites.
2.3 Data analysis
We determined the mC density and average methylation level of each line. The mC density refers to the number of cytosine methylation in each sequence context of the aligned reads. We calculated each type of cytosine methylation in the regenerated lines. Average cytosine methylation level was calculated based on the ratio between the number of methylated cytosines and total cytosines within a mapped read. Methylation density was determined using the percentage of methylated cytosines in relation to the total analyzed cytosines. The average methylation level of all cytosines was calculated based on the methylated cytosines in relation to the total cytosines present in the ‘CT001’ pseudomolecule.
In addition, the percentage of methylation was measured. For instance, the percentage of methylation per CpG site was calculated by dividing the number of methylated CpG sites by the total number of CpG sites in the CT001 genome.
2.4 Identification of differentially methylated regions (DMRs)
The DSS package (http://bioconductor.org/packages/release/bioc/html/DSS.html) (Park and Wu 2016) was used to detect DMRs between the non-regenerant and the regenerants, and we selected a raw p value threshold of 0.05. Cytosines within methylation loci that presented an average fivefold coverage were used to calculate methylation levels. The regions where methylation levels differed by more than 10% between the non-regenerant and regenerants were defined as DMRs. DMR calling was performed with each regenerant line. We analyzed the progeny set of R0C22 (R0C22, R1C22, and R2C22) and R0C31 (R0C31, R1C31, and R2C31) with the callDMR function using a P-value threshold of 0.05, a delta of 0.1, and otherwise default parameters. Overlapping DMRs from the two sets were selected and analyzed. In addition, DMRs overlapping among R0 regenerated lines were investigated using an in-house script.
Genes associated with DMRs of R0C22 and R0C31 were sorted individually. Regarding the functional annotation of the genes, enriched terms of their corresponding Arabidopsis genes, such as GO terms and UniProtKB keywords, were assigned using the DAVID functional gene clustering tool (Huang et al. 2009) and enrichment p values were corrected using the Benjamini–Hochberg method. In addition, expression analyses were performed with The Arabidopsis Information Resource (TAIR) ID of the methylation-related genes using the eFP browser (http://bar.utoronto.ca/efp/cgi-bin/efpWeb.cgi). A homology search was performed using the basic local alignment search tool (BLAST) software.
2.5 Gene expression analysis
Gene expression analyses were performed to analyze a possible correlation between DNA methylation status and expression of genes within the candidate DMRs. The relationship between methylation and gene expression levels in DMRs was investigated in the regenerated lines (R0C22, R0C31, R1C22, R1C31, R2C22, and R2C31). We selected 10 DMRs located in the exonic or 1-kb upstream regions of annotated genes with different methylation levels in both regenerant lines: R0C22 and R0C31 (Table 5).
Total RNA was isolated from the non-regenerant and the regenerants using the plant total RNA extraction kit (TaKaRa, Otsu, Japan) according to the manufacturer’s instructions. cDNA was synthesized in 20-μL reaction mixtures with cDNA synthesis premix (iNtRON Biotechnology, Seongnam, Korea) and under the following conditions: 50 °C for 60 min and 95 °C for 5 min for reverse transcriptase (RTase) inactivation. Flanking sequences of the selected genes within DMRs were identified in the ‘CT001’ pseudomolecule reference genome sequences, and primers were designed using Vector NTI software (Invitrogen Carlsbad, CA, USA). The primers used for quantitative reverse transcription-PCR (qRT-PCR) are listed in Table 1.
RT-PCR analysis was performed using Maxime™ i-Star-Taq PCR Pre-Mix (iNtRON Biotechnology, Seongnam, Korea) to evaluate the expression level of genes within DMRs. The qRT-PCR assay was performed using TransStart® Top Green qPCR SuperMix (TransGen Biotech, Beijing, China) with a Rotor-Gene 6000 (Corbett Robotics, Brisbane, Australia) according to the manufacturer’s recommended protocol. PCR conditions were as follows: pre-incubation for 10 min at 95 °C, followed by 40 cycles of 10 s at 95 °C and 30 s at 60 °C. The melting curve analysis of PCR products was performed by increasing the temperature from 60 to 95 °C. Fluorescence intensity data were collected at the end of each cycle and analyzed using the instrument software. The cycle threshold (Ct) value of each sample was used for calculating relative gene expression levels by the ΔΔCt method (Livak and Schmittgen 2001). Actin was used as the endogenous housekeeping gene for normalization.
2.6 PCR-based methylation analysis
To investigate the methylation state of the selected genes, genomic DNA (1 μg) of each line was treated with bisulfite using the EZ DNA Methylation-Gold™ Kit (Zymo research, CA, USA) in accordance with the manufacturer’s instructions. PCR was then performed in 20-μL reaction mixtures using hot-start Taq (Maxime™ i-Star-Taq PCR Pre-Mix; iNtRON Biotechnology, Seongnam, Korea). The bisulfite-converted DNAs of the control plant ‘CT001’, non-regenerated line R0C0, R0 regenerated lines (R0C21, R0C22, R0C24, and R0C31), R1, and R2 progeny lines of R0C22 and R0C31 were used as template. Primers were designed using MethPrimer 2.0 (http://www.urogene.org/methprimer2/) (Li and Dahiya 2002). The primer sequences and amplicon lengths are shown in Table 2. An overview of the methylation-specific PCR (MSP) approach is presented in Fig. S2. PCR conditions were as follows: 5 min at 95 °C, 40 cycles at 95 °C for 30 s, 60 °C for 30 s, 72 °C for 30 s, and final extension at 72 °C for 10 min. PCR amplicons were loaded onto 1% agarose gel and then visualized under ultraviolet light.
3 Results and discussion
3.1 DNA methylation profiling in the regenerated lines
We conducted WGBS of the genomic DNA of non-regenerant and regenerant lines to analyze the patterns of epigenetic regulation associated with the unintended variation produced by regeneration. On average, 6 Gb of raw WGBS data was generated for each genome (Table S1). The high-quality reads with two allowed mismatches were mapped to the ‘CT001’ pseudomolecule reference genome with Bismark, using Bowtie 2 as the aligner for methylation analysis. Approximately 90% of clean reads could be independently mapped to the reference genome (Table S2). The ambiguously mapped or duplicate reads were removed, and only uniquely mapped reads were retained for further analyses. As a result, an average of 8 million properly mapped paired reads were retained, covering more than 90% of the ‘CT001’ reference genome with an average depth of 10x. Regarding genome coverage, approximately 96% of cytosines was covered by at least one uniquely mapped read.
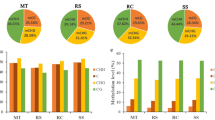
DMRs of each regenerated plant were categorized in the CpG, CHG, and CHH contexts (where H corresponds to A, T, or C). The levels of DNA methylation in these three contexts were determined for each region of each regenerated line and compared to the control line R0C0. DMR calling in regenerated lines resulted in the identification of an average of 36,682,978 methylated CGs (mCG) (53.9% of all CGs), 13,590,933 mCHGs (19.4% of all CHGs), and 11,678,796 mCHHs (26.7% of all CHHs) (Fig. 1). The proportion of methylated cytosines was similar to those in studies on soybeans (Shen et al. 2018). For example, CG methylation was the most predominant followed by CHG and CHH methylation (Lister et al. 2008; Schmitz et al. 2013).

In silico analysis of the methylation patterns of the regenerant-conserved DMRs (RCDs) in the regenerated lines of the R0, R1, and R2 generations. Black arrows indicate lengths and directions of the genes. The green boxes indicate hypomethylated DMRs, and the orange boxes indicate hypermethylated DMRs in the regenerated lines
DNA methylome profiles of the non-regenerated and regenerated lines are shown in Fig. S1. The methylation percentage of CpG sites in every line was higher than those of CHG and CHH sites. Each percentage was similar among the analyzed lines. In addition, as observed in prior studies in rice and maize, hypomethylation was more common than hypermethylation in regenerants (Stroud et al. 2013b; Stelpflug et al. 2014).
3.2 DNA methylation patterns in the regenerated lines
The chromosomal distributions of DMRs of the regenerants were analyzed (Table S3). The number of DMRs present on the chromosome was generally similar for every R0 regenerated line. Subsequent analysis was performed mainly on the DMRs present in the exon and 1-kb upstream of the gene, which was expected to be directly related to the phenotype. DMRs overlapped among R0 regenerated lines were identified using an in-house script, and their distributions are shown in Table S3. The number of overlapping DMRs in four subjects was significantly lower than the number of DMRs identified in individual subjects.
The methylation changes in tissue culture were frequent, and these changes were often inherited by self-pollinated progenies (Stelpflug et al. 2014). Based on DMR data of the regenerants, the genome-wide DNA methylation of each progeny line was investigated. We identified DMRs in the regenerants of two progeny lines of R0C22 and R0C31. In the R0C22 lines (R0C22, R1C22, and R2C22), we identified 440 DMRs, including 142 DMRs in the CG context, 97 DMRs in CHG, and 201 DMRs in CHH. In the R0C31 progeny lines (R0C31, R1C31, and R2C31), 453 DMRs were identified, with 140 DMRs in the CG, 102 DMRs in the CHG, and 211 DMRs in the CHH context.
The changes in the methylation patterns of both groups were analyzed to verify if they were related to the regeneration process. The proportion of methylated cytosine residues in the CG context was bigger than 40% across all lines. The average DMR length in the R0C22 progeny line was 285.52 bp for CG, 223.75 bp for CHG, and 300.99 bp for CHH. The average DMR length in the R0C31 progeny lines was 286.36 bp for CG, 224.63 bp for CHG, and 331.55 bp for CHH. In both progeny lines, DMR length was the longest in the CHH sequence context.
3.3 Analysis of differentially methylated genes
To investigate the relationship between genes with differences in methylation patterns in the regenerated lines, we clustered genes according to DMRs using their corresponding TAIR IDs from the DAVID Bioinformatics resource (https://david.ncifcrf.gov/), adopting the Benjamini–Hochberg method for correction. Table 3 shows results of each gene clustering in both progeny groups of the regenerated Chinese cabbage lines, R0C22 and R0C31, including lists of enriched terms and keywords. As a result, we observed that the DMR-associated genes of the R0C22 lines were mainly related to nucleus, transcription regulation, and sequence-specific DNA binding activity. Functional annotation enriched for DMR-associated genes of the R0C31 lines showed that they were mainly associated with the endoplasmic reticulum and RNA-directed DNA polymerase activity. However, as the results were not statistically significant, we inferred that genes whose methylation patterns changed during the regeneration process did not have any correlation with each other.
Among the DMRs identified in both R0C22 and R0C31 lines, 10 were located at the 1-kb upstream region of known genes, within a promoter region, and 11 were located at exonic regions. The genes that overlapped with the promoter and exonic regions of methylated DMRs were related to the secretory protein, zinc finger CCCH domain-containing protein, serine/threonine-protein phosphatase, receptor-like protein kinase, nudix hydrolase, and the trihelix transcription factor.
We focused our analyses on regions with coverage of at least three reads and with consistent methylation levels among the regenerated lines. A total of 36 DMRs were observed in both the C22 and C31 lines; these were designated as “regenerant-conserved” DMRs (RCD) (Table 4). Among them, CT001_A10358000 has DMRs both in CG and CHH contexts in its intron sequence, while CT001_A06227680 and CT001_A07241390 have DMRs in the CHG and CHH contexts in the intron and 1-kb upstream regions, respectively. Briefly, we identified 33 DMR-associated genes in both groups of R0C22 and R0C31 progeny lines and expected that the methylation levels of these DMRs might be related to the regeneration process, affecting subsequent gene expression. The DNA sequence of the selected DMRs and their adjacent regions were obtained from the ‘CT001’ pseudomolecule genome.
3.4 Validation of the selected DMRs
We selected 10 DMRs between the non-regenerant line and the regenerant lines that were located at the exonic or 1-kb upstream regions of fully annotated genes. In silico analysis of the selected DMRs was performed using the ‘CT001’ pseudomolecule genome browser, which included the bisulfite sequencing data of regenerated lines. We loaded the BAM file of the R0 (R0C0, R0C22, and R0C31), R1 (R1C22 and R1C31), and R2 (R2C22 and R2C31) generations of regenerated lines on the genome browser and compared the methylation status of the selected RCD genes. The CG, CHG, and CHH methylation patterns of each line were compared (Fig. 1). In some cases, the differences in methylation patterns were also identified in contexts other than those expected. For example, RCD1 was expected to bear CG DMRs, but the analysis also showed DMRs in CHG and CHH contexts (Fig. 2). The methylation statuses of 9 out of 10 DMR-associated genes were lower in regenerated lines than in the control lines.

The hypomethylation in gene CT001_A02068540. The pattern of the selected DMRs can be visualized through hierarchical clustering of methylation levels for all lines. For each line track, bar height represents the percentage of methylation. The blue arrow and boxes represent the genes. The yellow, purple, and red bars indicate CG, CHG, and CHH methylations, respectively
3.5 Correlation between DMR and gene expression
We analyzed whether the expression of DMR-associated genes changed according to the methylation patterns of regenerated lines. We selected 10 DMRs conserved in R0C22 and R0C31 and their progeny lines and investigated their DNA methylation levels and gene expression patterns. As mentioned above, gene expression levels were positively correlated with the methylation levels within the transcribed regions (Fig. 3). The expression of genes associated with hypomethylated DMRs in the regenerated lines was generally upregulated. In contrast, the expression of genes associated with hypermethylated DMRs in the regenerated individuals was generally downregulated. The DMRs located at the 1-kb upstream and exonic regions of the selected genes were also analyzed. The annotated genes associated with DMRs included those involved in transcriptional regulation, DNA polymerase activity, and signal transduction functions. In addition, we performed in silico analyses of the differences in gene expression during the regeneration process using TAIR IDs and the Arabidopsis eFP browser (http://bar.utoronto.ca/efp/cgi-bin/efpWeb.cgi). Six out of ten genes could be analyzed with the eFP browser. The expression levels of the genes tended to increase by 2–4 times during the callus induction process, therefore these genes were considered to be related to this process. In particular, the expression level of A. thaliana subtilase family protein SBT3.3 (AT1G32960) was increased by 16 times after callus induction and this was probably associated with the regeneration process. When compared with ‘CT001’, the genes were hypomethylated, suggesting that they were more expressed during regeneration.

Gene expression analysis of 10 selected genes within regenerant-conserved DMRs (RCDs) in the regenerated lines. Expression levels of each DMR gene in R0, R1, and R2 generations of R0C22 and R0C31 were compared with those of inbred line ‘CT001’ and non-regenerant R0C0. Y-axis indicates relative mRNA expression, and bars on the graph indicate the SE of the means (n = 3)
For validation of the identified changes in methylation, RT-PCR of the 10 selected DMR-associated genes was conducted using cDNA synthesized from total RNA extracted from each regenerated line and primer sets for the selected DMRs. The amplicon with the expected product size was identified (Fig. S1), and gene expression was examined in non-regenerant and regenerant lines by qRT-PCR using the same primer sets used for RT-PCR (Fig. 3). This analysis revealed that alterations in DNA methylation, either hyper- or hypo-methylation, regulated gene expression levels. Most results indicated that regenerated lines exhibited increased gene expression along with a lower methylation level, while a decrease in the methylation status resulted in increased gene expression.
Among the selected genes, RCD1, RCD2, and RCD3 were expected to be associated with stress response (Table 5). As shown in Fig. 3, the expression of RCD1 in regenerants was higher than that in the non-regenerant, indicating that CG hypomethylation in the promoter region and extensive CHG and CHH hypomethylation in the exonic and promoter regions effectively upregulated RCD1 gene expression. RCD1 was matched with AT3G07460, encoding a putative transmembrane protein containing the DUF538 domain, and has been reported to respond to various biotic and abiotic stresses in plants (Gholizadeh 2011). AT3G21910 matched with RCD2 and encodes a putative cysteine-rich repeat secretory protein, DUF26, that exists in three forms in plants: cysteine-rich receptor-like secreted proteins (CRRSPs), cysteine-rich receptor-like protein kinases (CRKs), and plasmodesmata-localized proteins (PDLPs). The expression of genes corresponding to CRRSPs and CRKs increased when exposed to biotic and abiotic stress (Vaattovaara et al. 2019). RCD3 is matched with AT2G40140, a zinc finger (CCCH-type) family protein that is known to be associated with tolerance to salinity stress and osmotic stress in plants (Han et al. 2014). In Arabidopsis, the expression of the corresponding gene, AtSZF2, rapidly increases under salinity stress (Sun et al. 2007). Therefore, the increased expression of those genes as a result of hypomethylation was expected in response to various environmental stresses, which occurred during the regeneration process, such as stresses caused by culture media, plant hormones, and reagents used for tissue culture.
In addition, RCD4 matched with the AT4G17250 gene (Table 5), whose expression increased after 37 °C heat treatment in Arabidopsis (Lim et al. 2006). AT2G20440 matched with RCD5, which encodes the gyp1p superfamily protein (Table 5). Its expression changes in relation to meiosis in Arabidopsis, but the exact function of the gene has not been revealed (Libeau et al. 2011). RCD6 matched with AT5G59160 (Table 5), which is a protein phosphatase (TOPP) family gene, also known as protein phosphatase 1 (PP1). Genes of the TOPP family have been reported, to be regulators of plant immunity, but the detailed functions in plants have not been revealed (Liu et al. 2020). The AT2G42070 gene matched RCD8 and is described as Arabidopsis nudix hydrolase (AtNUDX23) (Table 5). It has been reported to be involved in flavin homeostasis of plant cells (Maruta et al., 2012). RCD9 matched with AT1G13450, which encodes transcription factor GT-1 (Table 5). GT-1 has been reported to affect gene expression by binding to BoxII, a cis-acting element present in the upstream promoter region of light-responsive genes (Nagata et al. 2010). As the methylation patterns and the expression of these genes changed in regenerated lines, an association between the genes and the tissue culture process adopted is expected, but further studies are required.
Unlike other RCD genes, hypermethylation and decreased expression of RCD7 were shown in regenerated plants (Fig. 3; Table 5). This showed that DNA hypermethylation was associated with repressed gene expression. Typically, the presence of methylated cytosines within a promoter region reduces gene expression (Taiko et al. 2015). AT4G10390, which matched with RCD7, encodes a kinase superfamily protein that is known to be related to plant growth and development. In Arabidopsis, its expression decreased threefold after treatment with melatonin which is related to plant stress defense (Weeda et al., 2014). Therefore, it can be assumed that the AT4G10390 gene is associated with a decrease in expression related to plant stress defense. It was predicted that the decrease in gene expression of RCD7, due to hypermethylation, was intended to defend against stress created during the regeneration process. In conclusion, the results of this analysis indicated that the regeneration process might affect DNA methylation in the regenerated lines and subsequently affect their gene expression.
Furthermore, the methylation status of the selected RCDs was maintained in DMRs in the progeny lines obtained by self-pollination. This study shows that epigenetic signals, such as DNA methylation and histone modifications, caused by environmental stress in the previous generation are also conserved in the progeny line; this is known as “transgenerational epigenetic inheritance (TEI)”. In general, epigenetic signatures are known to be removed from progeny by germline reprogramming in mammals (Heard and Martienssen 2014). However, in plants, epigenetic signatures have been reported to be stably inherited for several generations in various organisms (Hauser et al. 2011).
In Arabidopsis, genes involved in methylation in CG, CHG, and CHH sequence contexts have been reported (Stroud et al. 2013a; Zhang et al. 2018). CG methylation is regulated by methyltransferase 1 (MET1) and the plant homolog of mammalian DNA (cytosine-5)-methyltransferase 1 (DNMT1) (Kankel et al. 2003). Chromomethylase 3 (CMT3) is known to regulate CHG methylation and domains rearranged methyltransferases (DRM1 and 2) and plant homologs of mammalian DNMT3 maintain CHH methylation through the RNA-directed DNA methylation (RdDM) pathway (Xie et al. 2004). In addition, it was reported that DNA methylation is regulated by multiple pathways based on studies of mutants of methylation-related genes in Arabidopsis (Stroud et al. 2013a). Since Chinese cabbage belongs to Brassicaceae, like Arabidopsis, it is expected that DMRs of Chinese cabbage may have been regulated by similar DNA methylation-related genes as in Arabidopsis. Therefore, the results obtained in this study can be utilized to study the interplay of genes involved in regulating DNA methylation in Chinese cabbage.
Although the change in methylation pattern between non-regenerants and regenerant was confirmed, the difference in phenotype was not observed. In previous studies in rice (Stroud et al. 2013b) and maize (Stelpflug et al. 2014), most phenotypic variations were observed only in a subset of plants regenerated from tissue culture. Thus, we considered that variation of methylation patterns in the regenerants in this study might not critically affect the phenotype. In a previous study, a significant portion of hypomethylated DMRs identified in tissue-cultured plants was found to be hypomethylated even in the natural state (Stelpflug et al. 2014). Plants display alteration of DNA methylation in response to diverse environmental stress in nature. Accordingly, it was suggested that these DMRs are sensitive to environmental changes, rather than specifically arising because of the tissue culture process.
3.6 Methylation-specific PCR
Different methylation patterns of RCD1 and RCD10 were detected among regenerated lines using methylation-specific PCR (MSP) in which bisulfite-treated DNA was used as a template. For each selected DMR, we designed two primer sets specific to methylated (M pair) and unmethylated DNA (U pair).
MSP data of RCD1 showed that methylated PCR products were observed in ‘CT001’ and non-regenerated plant R0C0, while unmethylated PCR products were not (Fig. 4a). On the contrary, the unmethylation of RCD1 was detected from the R0 regenerated lines and from their progenies. These results showed that RCD1 was methylated in the general condition of B. rapa plants, but it was unmethylated in regenerated lines and the altered methylation status was maintained in R1 and R2 generation. In addition, it was revealed that RCD10 was methylated in ‘CT001’ and R0C0, as well as regenerated lines, while partially demethylated in the R1 progeny line of R0C31 (Fig. 4b). This result also showed that methylation status can be visualized by MSP. The difference in methylation status of RCD10 was not clearly shown, which is expected to be due to the slight change in methylation level in regenerated lines.

Analysis of methylation-specific PCR products amplified by primer sets of RCD1 (a) and RCD10 (b). CT001, inbred line; R0C0, non-regenerated plant; R0, R0 regenerated lines including R0C21, R0C22, R0C24, and R0C31; R1 and R2, progeny lines of R0C22 and R0C31. M, methylated PCR products; U, unmethylated PCR products
MSP is a simple and sensitive PCR-based technique to distinguish unmethylated and methylated DNA (Herman et al. 1996). The methylated primers amplify sodium bisulfite-converted methylated DNA, while the unmethylated primers amplify unmethylated DNA. The MSP method has been performed in diverse organisms to distinguish methylation status in the genome (Khraiwesh et al. 2010; Uthup et al. 2011; Mihara et al. 2017). Identification of epigenetic markers can be further used for screening methylation changes in the genome of regenerated lines. In conclusion, these MSP results indicate that the alteration in methylation status might be induced during regeneration or tissue culture, and it can be specifically evaluated by PCR-based methods.
4 Conclusion
Genetic and epigenetic instabilities were often observed in in vitro-cultured and regenerated lines. These phenomena can affect the practical applications of plant tissue culture in various research areas. The epigenetic instability of DNA has been especially suggested to be involved in gene expression control. In this study, we performed WGBS on regenerated lines of the B. rapa ssp. pekinensis inbred line ‘CT001’ to uncover DNA methylation changes resulting from the regeneration process in Chinese cabbage. We identified epigenetic mutations in regenerated lines that could have been induced by tissue culture and could be inherited by progeny lines, and expression levels of gene were positively correlated with the methylation levels within the transcribed regions. We also developed epigenetic markers to be used for screening methylation changes in genome of regenerated lines. In this study, we identified the DMRs consistently observed in the regenerants and confirmed the variation in methylation patterns of the selected DMRs and the expression of the genes containing the DMRs. Further research is needed on the causes of these epigenetic changes in regenerants and their progenies, as well as the consequences that may affect metabolic processes or phenotypes. In conclusion, our findings will contribute to the understanding of epigenetic aspects of somaclonal variation in regenerated lines.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Bajaj YPS (1990) Somaclonal variation—origin, induction, cryopreservation and implication in plant breeding. In: Bajaj YPS (ed) Biotechnology in agriculture and forestry II, somaclonal variation in crop improvement I. Springer, Berlin, pp 3–48
Bock C, Lengauer T (2008) Computational epigenetics. Bioinformatics 24:1–10
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Brassica rapa Genome Sequencing Project Consortium (2011) The genome of the mesopolyploid crop species Brassica rapa. Nat Genet 43:1035
Bucher E, Reinders J, Mirouze M (2012) Epigenetic control of transposon transcription and mobility in Arabidopsis. Curr Opin Plant Biol 15:503–510
Coronel CJ, González AI, Ruiz ML, Polanco C (2018) Analysis of somaclonal variation in transgenic and regenerated plants of Arabidopsis thaliana using methylation related metAFLP and TMD markers. Plant Cell Rep 37:137–152
Cubas P, Vincent C, Coen E (1999) An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401:157–161
Dellaporta SL, Wood J, Hicks JB (1983) A plant DNA minipreparation: version II. Plant Mol Biol Rep 1:19–21
Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME et al (2010) Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci USA 107:8689–8694
Gholizadeh A (2011) Heterologous expression of stress-responsive DUF538 domain containing protein and its morpho-biochemical consequences. Protein J 30:358
Han G, Wang M, Yuan F, Sui N, Song J, Wang B (2014) The CCCH zinc finger protein gene AtZFP1 improves salt resistance in Arabidopsis thaliana. Plant Mol Biol 86:237–253
Hauser MT, Aufsatz W, Jonak C, Luschnig C (2011) Transgenerational epigenetic inheritance in plants. Biochim Biophys Acta 1809:459–468
Heard E, Martienssen RA (2014) Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157:95–109
Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB (1996) Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 93:9821–9826
Hsieh TF, Ibarra CA, Silva P, Zemach A, Eshed-Williams L, Fischer RL et al (2009) Genome-wide demethylation of Arabidopsis endosperm. Science 324:1451–1454
Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Iglesias FM, Cerdán PD (2016) Maintaining epigenetic inheritance during DNA replication in plants. Front Plant Sci 7:38
Jacobsen SE, Meyerowitz EM (1997) Hypermethylated SUPERMAN epigenetic alleles in Arabidopsis. Science 277:1100–1103
Kaeppler SM, Kaeppler HF, Rhee Y (2000) Epigenetic aspects of somaclonal variation in plants. Plant Mol Biol 43:179–188
Kaeppler SM, Phillips RL (1993) Tissue culture-induced DNA methylation variation in maize. Proc Natl Acad Sci USA 90:8773–8776
Kankel MW, Ramsey DE, Stokes TL, Flowers SK, Haag JR, Jeddeloh JA, Riddle NC, Verbsky ML, Richards EJ (2003) Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 163:1109–1122
Khraiwesh B, Arif MA, Seumel GI, Ossowski S, Weigel D, Reski R, Frank W (2010) Transcriptional control of gene expression by microRNAs. Cell 140:111–122
Krueger F, Andrews SR (2011) Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics 27:1571–1572
Kubis S, Baldwin A, Patel R, Razzaq A, Dupree P, Lilley K et al (2003) The Arabidopsis ppi1 mutant is specifically defective in the expression, chloroplast import, and accumulation of photosynthetic proteins. Plant Cell 15:1859–1871
Laird PW (2010) Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet 11:191–203
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Larkin PJ, Scowcroft W (1981) Somaclonal variation–a novel source of variability from cell cultures for plant improvement. Theor Appl Genet 60:197–214
Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11:204–220
Li LC, Dahiya R (2002) MethPrimer: designing primers for methylation PCRs. Bioinformatics 18:1427–1431
Libeau P, Durandet M, Granier F, Marquis C, Berthomé R, Renou JP et al (2011) Gene expression profiling of Arabidopsis meiocytes. Plant Biol 13:784–793
Lim CJ, Yang KA, Hong JK, Choi JS, Yun DJ, Hong JC et al (2006) Gene expression profiles during heat acclimation in Arabidopsis thaliana suspension-culture cells. J Plant Res 119:373–383
Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR (2008) Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133:523–536
Liu Y, Yan J, Qin Q, Zhang J, Chen Y, Zhao L et al (2020) Type one protein phosphatases (TOPPs) contribute to the plant defense response in Arabidopsis. J Integr Plant Biol 62:360–377
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408
Manning K, Tor M, Poole M, Hong Y, Thompson AJ, King GJ et al (2006) A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat Genet 38:948–952
Maruta T, Yoshimoto T, Ito D, Ogawa T, Tamoi M, Yoshimura K et al (2012) An Arabidopsis FAD pyrophosphohydrolase, AtNUDX23, is involved in flavin homeostasis. Plant Cell Physiol 53:1106–1116
Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD et al (2010) Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466:253–257
Mihara H, Suzuki N, Muhammad JS, Nanjo S, Ando T, Fujinami H et al (2017) Transient receptor potential vanilloid 4 (TRPV 4) silencing in Helicobacter pylori-infected human gastric epithelium. Helicobacter 22:e12361
Miura K, Agetsuma M, Kitano H, Yoshimura A, Matsuoka M, Jacobsen SE et al (2009) A metastable DWARF1 epigenetic mutant affecting plant stature in rice. Proc Natl Acad Sci USA 106:11218–11223
Miguel C, Marum L (2011) An epigenetic view of plant cells cultured in vitro: somaclonal variation and beyond. J Exp Bot 62:3713–3725
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue culture. Physiol Plant 15:473–497
Müller E, Brown PTH, Hartke S, Lörz H (1990) DNA variation in tissue-culture-derived rice plants. Theor Appl Genet 80:673–679
Nagata T, Niyada E, Fujimoto N, Nagasaki Y, Noto K, Miyanoiri Y et al (2010) Solution structures of the trihelix DNA-binding domains of the wild-type and a phosphomimetic mutant of Arabidopsis GT-1: mechanism for an increase in DNA-binding affinity through phosphorylation. Proteins Struct Funct Bioinform 78:3033–3047
Niederhuth CE, Schmitz RJ (2014) Covering your bases: inheritance of DNA methylation in plant genomes. Mol Plant 7:472–480
Park Y, Wu H (2016) Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics 32:1446–1453
Park JS, Park JH, Park YD (2019) Construction of pseudomolecule sequences of Brassica rapa ssp. pekinensis inbred line CT001 and analysis of spontaneous mutations derived via sexual propagation. PloS One 14(9):e0222283
Quadrana L, Almeida J, Asis R, Duffy T, Dominguez PG, Bermudez L et al (2014) Natural occurring epialleles determine vitamin E accumulation in tomato fruits. Nat Commun 5:3027
Schmitz RJ, He Y, Valdés-López O, Khan SM, Joshi T, Urich MA et al (2013) Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res 23:1663–1674
Seymour DK, Becker C (2017) The causes and consequences of DNA methylome variation in plants. Curr Opin Plant Biol 36:56–63
Shen Y, Zhang J, Liu Y, Liu S, Liu Z, Duan Z et al (2018) DNA methylation footprints during soybean domestication and improvement. Genome Biol 19:1–14
Smulders MJM, De Klerk GJ (2011) Epigenetics in plant tissue culture. Plant Growth Regul 63:137–146
Soppe WJJ, Jacobsen SE, Alonso-Blanco C, Jackson JP, Kakutani T, Koornneef M et al (2000) The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol Cell 6:791–802
Stroud H, Greenberg MV, Feng S, Bernatavichute YV, Jacobsen SE (2013a) Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152:352–364
Stroud H, Ding B, Simon SA, Feng S, Bellizzi M, Pellegrini M et al. (2013b) Plants regenerated from tissue culture contain stable epigenome changes in rice. eLife 2:e00354
Stelpflug SC, Eichten SR, Hermanson PJ, Springer NM, Kaeppler SM (2014) Consistent and heritable alterations of DNA methylation are induced by tissue culture in maize. Genetics 198:209–218
Sun J, Jiang H, Xu Y, Li H, Wu X, Xie Q, Li C (2007) The CCCH-type zinc finger proteins AtSZF1 and AtSZF2 regulate salt stress responses in Arabidopsis. Plant Cell Physiol 48:1148–1158
Taiko KT, Saze H, Kakutani T (2015) DNA methylation within transcribed regions. Plant Physiol 168:1219–1225
Teixeira FK, Colot V (2009) Gene body DNA methylation in plants: a means to an end or an end to a means? EMBO J 28:997–998
Uthup TK, Ravindran M, Bini K, Thakurdas S (2011) Divergent DNA methylation patterns associated with abiotic stress in Hevea brasiliensis. Mol Plant 4:996–1013
Vaattovaara A, Brandt B, Rajaraman S, Safronov O, Veidenberg A, Luklová M, Wrzaczek M (2019) Mechanistic insights into the evolution of DUF26-containing proteins in land plants. Commun Biol 2:1–18
Weeda S, Zhang N, Zhao X, Ndip G, Guo Y, Buck GA, Ren S (2014) Arabidopsis transcriptome analysis reveals key roles of melatonin in plant defense systems. PLoS ONE 9:e93462
Xie Z, Johansen LK, Gustafson AM, Kasschau KD, Lellis AD, Zilberman D, Jacobsen SE, Carrington JC (2004) Genetic and functional diversification of small RNA pathways in plants. PLoS Biol 2:e104
Xing MQ, Zhang YJ, Zhou SR, Hu WY, Wu XT, Ye YJ et al (2015) Global analysis reveals the crucial roles of DNA methylation during rice seed development. Plant Physiol 168:1417–1432
Yang X, Han H, Carvalho DDD, Lay FD, Jones P, Liang G (2014) Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 26:577–590
Zhang H, Lang Z, Zhu JK (2018) Dynamics and Function of DNA Methylation in Plants. Nat Rev Mol Cell Biol 19:489–506
Zilberman D, Henikoff S (2007) Genome-wide analysis of DNA methylation patterns. Development 134:3959–3965
Acknowledgements
This work was carried out with the support of the “Cooperative Research Program for Agriculture Science and Technology Development (Project No. PJ01324601)”, Rural Development Administration, Republic of Korea.
Author information
Authors and Affiliations
Contributions
HM Lee and JS Park performed the majority of the experiment and data analysis, and developed the regenerated lines. HM Lee and JS Park contributed equally to this work. YH Shin contributed to experimental implementation. YD Park designed the experiment and analyzed data. All authors contributed to and corrected the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Inhwa Yeam.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lee, HM., Park, JS., Shin, YH. et al. Alterations in DNA methylation patterns in regenerated Chinese cabbage (Brassica rapa ssp. pekinensis) plants derived from tissue culture. Hortic. Environ. Biotechnol. 62, 605–618 (2021). https://doi.org/10.1007/s13580-020-00310-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13580-020-00310-1