
Abstract
Cold tolerance is the primary limiting factor affecting the quality and productivity of chrysanthemum (Dendranthema morifolium). However, few studies have investigated the molecular mechanisms underlying the low temperature response in chrysanthemum. In the present study, we used RNA-Seq technology to compare the transcript profiles of chrysanthemum leaves exposed to two different temperatures (20 and −8 °C). A total of 13.54 Gb of clean reads were assembled into 71,971 unigenes with an average length of 694 bp, and 33,282 unigenes were annotated identified from five well-known protein databases. Of these, 9579, 24,252, 7123, 22,554, and 32,891 unigenes were separately identified in the COG, GO, KEGG, Swiss-Prot, and NCBI databases, respectively. Differentially expressed genes (1592 upregulated and 718 downregulated) were identified between the control (CK) and low temperature treatment (T) groups. KEGG pathway enrichment analysis identified 20 significantly different pathways. Many genes encoding important transcription factors (e.g. CBF/DREB, bHLH, MYC, and ZAT) as well as proteins (e.g. CCX, CBP, CML, and MAPK) involved in cold signal transduction were up- or down-regulated in the low temperature treatment group. Genes involved in ABA signal transduction and biosynthesis of unsaturated fatty acids were also identified. The expression profiles of these genes were analyzed during the cold treatment stage. These results provide important information for further studies on gene discovery in chrysanthemum and suggest a potential molecular mechanism for the response to low temperature in this plant species.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Chrysanthemum (Dendranthema morifolium) is a popular ornamental plant worldwide (Silva et al. 2013). Because the growing market requires cold-tolerant and perpetual-flowering chrysanthemum cultivars, improving cold tolerance is an important goal for breeders. Low environmental temperatures can easily damage chrysanthemum plants, which can cause significant losses in many temperate crops (Chinnusamy et al. 2007; Janská et al. 2010; Yang et al. 2005). In Arabidopsis, there are multiple and complex cold-response pathways, and many genes involved in the response to low temperature have been identified (Janská et al. 2010; Yamaguchi-Shinozaki and Shinozaki 2006); however, systematic investigations of the molecular basis of low temperature tolerance in chrysanthemum are lacking.
Cold signal transduction involves a series of physiological, biochemical, and molecular reactions in many plants (Yamaguchi-Shinozaki and Shinozaki 2006). Firstly, the cold stress signal is perceived by plants and the physical state of the membrane plays a role in this cold perception (Beck et al. 2004, 2007). Changes in the plasma membrane lead to an increase in the cytosolic Ca2+ ion concentration (Komatsu et al. 2007). Certain protein kinases related Ca2+ influx is recognized as key regulators of plasma membrane change (Saijo et al. 2000). Mitogen activated protein kinase (MAPK) functions in low temperature signal transduction and regulates low temperature tolerance (Yang et al. 2011). Three candidate low temperature response genes (ZmMAPKKK, ZmCLC-D and ZmRLK) were shown to be dysregulated under conditions of chilling stress in maize (Yang et al. 2011). Secondly, cold-regulated gene expression is regulated by different pathways that can be ABA-dependent or ABA-independent (Beck et al. 2007). Three candidate genes (CBFs, DREB1s, and COR) belonging to the ethylene responsive element binding protein family are regulated after receiving the cold stress signal (Stockinger et al. 1997). ICE1 encodes a MYC-like basic helix-loop-helix protein that activates the expression of many downstream genes, leading to a significantly enhanced tolerance to chilling (Chinnusamy et al. 2003; Stockinger et al. 1997). Thirdly, many metabolites including some with protective effects against cold stress and reactive oxygen species (ROS) are photosynthetically regulated (Heidarvand and Amiri 2010), and the proteins associated with these metabolites including certain cold associated and inducible proteins such as dehydrins (DHNs) (Close 1996; Kosová et al. 2007; Location 2006), antifreeze proteins (AFPs) (Guy 1999; Marilyn et al. 1997), heat shock proteins (HSPs) (Banzet et al. 1998; Guy 1999), cold-shock domain proteins (CSDPs) (Guy 1999; Sommerville 1999), and enzymes such as alternative oxidases and desaturases, are differentially expressed in response to cold stress
In our previous study, we found that − 8 °C was the half lethal temperature in the autumn chrysanthemum, and that 20 °C was the optimal temperature for autumn chrysanthemum cultivation. We measured physiological indexes such as membrane fatty and unsaturated fatty acids in autumn chrysanthemum (Li et al. 2013). However, the molecular mechanism of the autumn chrysanthemum response to cold temperature remains unclear. In the present study, we used RNA-Seq technology to characterize the transcriptomes of the autumn chrysanthemum cultivar ‘Jin Long Teng Fei’ in response to the half lethal temperature. We aimed to identify candidate genes to improve cold tolerance in chrysanthemum and increase the genetic resources available for hardy breeding strategies for chrysanthemum.
2 Materials and methods
2.1 Plant material and cold treatment
Plant material from the autumn chrysanthemum cultivar ‘Jin Long Teng Fei’ was collected at Henan Agricultural University. In our previous study, the half lethal temperature of autumn chrysanthemum was − 8 °C, and the optimal temperature was 20 °C (Li et al. 2013); therefore, we choose a treatment of − 8 °C for 10 min. Eighteen plants were separated into two groups, a control group (CK) with nine plants grown and a low temperature treatment group (T) with nine plants that were all initially grown at 20 °C for 1 w, and then followed by a treatment at 8 °C for 2 h and −8 °C for 10 min in the T group. For each group, nine leaves were sampled from three biological replicates (e.g. three plants were one biological replicate, with three leaves taken from each plant). Both groups (CK and T) were immediately frozen below −80 °C until RNA extraction. A total RNA from each replicate was extracted, and three total RNA were mixed in total 30 μg in equal amounts. Each group had a mixed RNA from three replicates that was used for sequencing.
2.2 RNA extraction, library construction, and sequencing
Total RNA was extracted from two groups (CK and T) and subjected to purity, concentration, and integrality testing. Qualified RNA (20 μg for each sample) was selected for library construction. Sequencing libraries were generated using NEBNext®Ultra™ RNA Library Prep Kit for Illumina®(NEB, USA) following manufacturer’s recommendations and index codes were added to attribute sequences to each sample. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5X). First strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase(RNase H-). Second strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of 3’ ends of DNA fragments, NEBNext Adaptor with hairpin loop structure were ligated to prepare for hybridization. In order to select cDNA fragments of preferentially 200–250 bp in length, the library fragments were purified with AMPure XP system (Beckman Coulter, Beverly, USA). Then 3 μg USER Enzyme (NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37 °C for 15 min followed by 5 min at 95 °C before PCR. Then PCR was performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers and Index (X) Primer. At last, PCR products were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2500 system.
2.3 Sequence assembly and annotation
RNA sequencing was performed by the Biomarker Biotechnology Corporation (Beijing, China). Raw data were checked according to the quality score and low-quality data were filtered and removed. High quality clean data were acquired and used to assemble unigenes using Trinity software (Stefan et al. 2008). Firstly, sequence reads were broken into shorter reads (K-mer), followed by extension of K-mers to contigs. Secondly, components were assembled to search the pair-end overlaps, and then De Bruijin maps were generated to acquire transcripts (Grabherr et al. 2011). Finally, unigenes were generated from the transcripts.
Unigene annotation was performed by BLAST with an E-value < 10−5 (Altschul et al. 1997). Five databases were used to annotate unigenes, including the National Center for Biotechnology Information (NCBI), non-redundant protein (Nr), Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), Gene Ontology (GO), and Cluster of Orthologous Groups of proteins (COG).
2.4 Expression calculation and identification of differentially expressed unigenes
Unigene expression was calculated using expectation–maximization (RSEM) software (Li and Dewey 2011). RPKMs (reads per kilobases per million reads) were used to evaluate the expression of the unigenes (Li and Dewey 2011). Differentially expressed genes (DEGs) were identified using the Benjamini–Hochberg method (Benjamini and Hochberg 1995) with a false discovery rate (FDR) of < 0.01 and a fold change > 2 (Hu et al. 2010). The identified DEGs were annotated and enrichment analysis was performed with topGO (Alexa and Rahnenfuhrer 2007) and KEGG terms (Kanehisa and Goto 2000).
2.5 Real-time quantitative PCR
Real-time quantitative PCR was performed to verify the DEG results obtained by RNA-Seq. A total of 2 μg of RNA were extracted from the CK and T groups after quality and integrity testing. The PrimeScript RT Kit (Takara, China) was used to synthesize the first strand cDNA. The tests were performed using the Applied Biosystems 7500 Real Time PCR system with a 20-μl reaction volume containing 1 μl template, 300 mM each primer, and 10 μl SYBR Premix Ex Taq II (Takara, China). The reaction conditions were 30 s at 95 °C, 40 cycles of 5 s at 95 °C and 34 s at 60 °C. Three replicates were performed for each experiment and relative expression was calculated using the 2−ΔΔCt method with 18 s ribosomal RNA as the reference gene.
3 Results
3.1 Sequence assembly
A total of 13.54 Gb clean data were obtained after data cleaning and quality checks. The control (CK) and low temperature treatment groups (T) yielded 34,971,435 and 32,734,410 clean reads, respectively. The Q30 values exceeded 89% for all samples (Table 1).
To increase the depth of sequencing, CK and T were combined for assembly by Trinity. A total of 376,073,043 bp of clean reads were assembled into 8,120,672 contigs (Table 2). Then, components were constructed using the paired-end overlaps of the contigs, and 238,436 transcripts (N50 = 1336 bp; mean length = 923.41 bp) were assembled by De Bruijn mapping. Finally, a total of 71,917 high-quality unigene sequences (N50 = 1181 bp; mean length = 694.49 bp) were obtained (Table 2). Thus, high quality clean data were provided for further analysis.
3.2 Gene annotation
The Nr protein, Swiss-Prot, COG, GO, and KEGG databases were used to annotate 33,282 unigenes (46.27%) out of 71,917 unigenes with an E-value threshold of < le-5, indicating that 53.73% (38,936 unigenes) had no annotation in any of the databases due to the lack of genomic and transcriptomic information on chrysanthemum (Table 2).
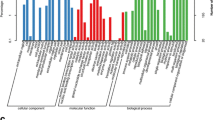
GO provides three ontologies (cellular components, biological processes, and molecular functions) to analyze genes (Conesa et al. 2005). Based on the blast results, 24,252 unigenes were annotated and covered 56 GO functional categories. Figure 1 shows that the ‘cell part’ (14,984 unigenes) term in cellular components, the ‘catalytic activity’ (12,594 unigenes) term in molecular functions, and the ‘metabolic process’ (16,249 unigenes) term in biological processes were the most dominant for the three ontologies. A high number of unigenes belonging to ‘cell’ (14,812 unigenes), ‘organelle’ (12,638 unigenes), ‘binding’ (11,555 unigenes), and ‘cellular process’ (15,085 unigenes) were also identified within the sequencing results. In addition, A low number of unigenes from the ‘extracellular matrix part’ (6 unigenes), ‘channel regulator activity’ (1 unigenes), and ‘carbon utilization’ (5 unigenes) categories were identified in each of the three GO ontologies.

Map of GO functional categories. Red lines indicate the most dominant category in the ontologies. Blue lines indicate the least dominant category in the ontologies. (Color figure online)
COG provides a method to search genes by eukaryotic COG classification or ID, which was useful to identify homologous proteins. Based on the results, 9579 unigenes were annotated covering 25 KOG categories (Fig. 2). The most dominant categories were ‘general function prediction only’ (2664, 27.81%) followed by ‘signal transduction mechanisms’ (1259, 13.14%), ‘transcription’ (1433, 14.96%), and ‘replication, recombination and repair’ (1569, 16.38%). The least dominant categories were ‘extracellular structures’ (0, 0%), ‘nuclear structure’ (4, 0%), and ‘cell motility’ (13, 0.14%).

Map of Cog function classifications. Red rectangles indicate high frequency categories. Blue rectangles indicate low frequency categories. (Color figure online)
KEGG allows the identification of potential pathways associated with the annotated genes in a network. A total of 7123 unigenes were annotated in the database covering 116 pathways. The dominant pathways were ‘metabolic pathway’ (1925 unigenes) and ‘biosynthesis of secondary metabolites’ (845 unigenes). In addition, 277 unigenes were mapped to ‘plant hormone signal transduction’, 68 unigenes to ‘phosphatidylinositol signaling system’, 55 unigenes to ‘biosynthesis of unsaturated fatty acids’, and 22 unigenes to ‘circadian rhythm-plant’.
3.3 Comparison of transcriptomes between the control group (CK) and the cold treated group (T) in chrysanthemum
A search for DEGs from the database with the proper threshold value (FDR < 0.01 and FC ≥ 2) identified 2310 unigenes, with 1592 upregulated and 718 downregulated genes between CK and T. Similarly, the GO, COG, and KEGG databases were used to annotate DEGs, in which we identified 1833 unigenes, indicating that 477 DEGs (20%) had no match in any of these databases.
The GO annotation showed that 1354 DEGs were enriched in 56 groups as follows: 16 in the ‘cellular component’ group, 16 in the ‘molecular function’ group, and 24 in the ‘biological process’ category. Figure 3 shows the functional categories of DEGs compared to those in all annotated unigenes. A greater difference between DEGs and all annotated unigenes was associated with a higher probability of the categories identified. In the ‘metabolic process’ category, the most significantly different terms were ‘structural molecule activity’, ‘nutrient reservoir activity’, and ‘nucleic acid binding transcription factor activity’. In the ‘biological process’ category, the most significantly different terms were ‘cell killing’ and ‘biological adhesion’. Finally, in the ‘cellular component’ category the most significantly different terms were ‘membrane-enclosed lumen’, ‘extracellular region part’, and ‘extracellular matrix’. Enrichment analysis of DEGs annotated on GO was performed by topGO. Ten terms were selected as the top10 categories with significant enrichment on GO (Table 3).

Map of GO functional categories of DEGs. Red cylinders indicate the unigene number in GO classification of all annotated unigenes. Blue cylinders indicate unigene number in GO classification of DEGs. (Color figure online)
KEGG pathway analysis showed that 336 out of 1836 DEGs were assigned to 50 pathways. Figure 4 shows the dominant pathways, which were as follows: plant hormone signal transduction (28), phenylpropanoid biosynthesis (23), photosynthesis-antenna proteins (21), plant-pathogen interaction (21), phenylalanine metabolism (19), and starch and sucrose metabolism (18). Enrichment analysis of KEGG pathways was performed to search for significant pathways. The top 20 significant pathways are shown in Fig. 5, including photosysthesis-antenna proteins, phenylpropanoid biosynthesis, phenylalanine metabolism, flavonoid biosynthesis, biosynthesis of unsaturated fatty acids, nitrogen metabolism, plant hormone signal transduction, and stilbenoid, diarylheptanoid and gingerol biosynthesis. These findings indicated that a diverse range of metabolic processes were activated by low temperature treatment in chrysanthemum.

Unigene numbers in KEGG pathways of DEGs

KEGG enrichment analysis of DEGs. Symbols indicate KEGG pathways. The x-axis indicates the value of the enrichment factor, and the y-axis indicates its significance level
3.4 Identification of genes involved in cold tolerance
Genes with homology to key regulators involved in cold signal transduction in chrysanthemum were searched. In Arabidopsis, many cold signal transduction genes were identified using molecular-genetic approaches. Some of the identified genes were involved in Ca2+ signaling pathways and the mitogen-activated protein kinase (MAPK) cascade, while others encoded important transcription factors. Genes involved in the biosynthesis of unsaturated fatty acids including calcium exchangers (CCX), calmodulin-like proteins (CML), calcium-dependent protein kinases (CDPK), CBL-interaction protein kinase (CIPK), bHLH, MYB, and CBF were also identified up or down regulated in the cold treatment. Based on the protein annotations of the chrysanthemum transcriptome sequences, a number of genes were identified up or down regulated in the cold treatment, such as homologs of the CBL-interacting protein (CIPK) (6), CDPK (6), CCX (4), calcium-binding proteins (CBP) (5), calmodulin-like protein (CML) (5), MAPK (4), WRKY (6) (transcription factor), MYB (15), AP2/ERF (6), bHLH (11), TCP (2), Dehydration-responsive element-binding protein DRBP (3), and Cys2/His2-type Zinc finger protein (ZAT) (7) (Table 4). We also found 11 genes that were similar to genes related to biosynthesis of unsaturated fatty acids.
3.5 Gene expression verified by quantitative RT-PCR
Twenty genes were selected randomly for quantitative RT-PCR to confirm the gene expression profiles from the transcriptomes. These selected genes were all annotated in databases; however, not all genes had detailed predicted gene functions. For the unpredicted genes, we listed Genebank IDs of the most similar genes from other species. The predicted gene functions of these 20 genes are list in Table 5. The templates used for qRT-PCR were those originally used for RNA-Seq. The expression patterns of the 20 genes by RT-PCR were consistent with the profiles by RNA-Seq (Fig. 6).

Expression level of 20 selected genes identified from our analysis of cold tolerance in chrysanthemum. a and b show the relative expression level of 20 selected genes assessed by RT-PCR. c and d show the RPKM value of control (CK) and cold temperature (T) treatment groups. The RPKM value had been calculated as log2 and normalized the expression data to the control values
4 Discussion
There is a paucity of research on the molecular mechanisms of cold tolerance in chrysanthemum. Our present findings improve our understanding of the biochemical pathways involved in physiological processes of cold tolerance. In the present study, approximately 67 million reads were obtained and 33,282 out of 71,917 unigenes were annotated against public databases, indicating that 38,635 unigenes (53.7%) had no current annotations. The assembled unigenes in this study were assigned to an extensive range of KEGG pathways, which indicated that various transcripts are involved in the response to low temperature in chrysanthemum.
Comparative analysis of the transcriptomes provided valuable information on candidate genes involved in cold tolerance. There were 2310 DEGs, 1836 of which were annotated. Many DEGs were associated with cold signal transduction (Table 4). The function of Ca2+ as a messenger in stress signaling is mediated by three main proteins: CDPKs, CaMs, and CBLs (Miura and Furumoto 2013; Reddy et al. 2011). In Arabidopsis, overexpression of Calmodulin (CaM) downregulates the expression of cold-responsive genes (Townley and Knight 2002). CDPKs are plant-specific genes that function as important detectors in cold signal transduction (Chinnusamy et al. 2004; Maria and Grazyna 2007). In Camellla sinensis, calmodulin and CDPK genes are involved in cold signal transduction, similar to the CBL gene (Wang et al. 2013). MAPK, which is upregulated in response to cold in Arabidopsis (Ichimura et al. 2000), is regulated by different transcription factors (Teige et al. 2004; Viswanathan and Jian-Kang 2002; Zhu et al. 2007). In the present study, homologs of CIPK (6), CDPK (6), CCX (4), CBP (5), (CML) (5), and MAPK (4) were identified among the DEGs as significantly up- or down-regulated genes in response to the cold treatment in chrysanthemum. We speculate that these genes may play similar roles in cold signal transduction in chrysanthemum as their homologs in Arabidopsis and potentially other species.
Important transcription factors that have roles in the response to cold stress were also found in the DEGs identified in our experimental treatment group. In Arabidopsis, CBF/DREB controls COR in response to cold stress (Chen et al. 2008; Stockinger et al. 1997). ICE1, which encodes a bHLH protein that binds specifically to MYC in the CBF/DREB promoter region and upregulates DREB1a/CBF3. This upregulation enhances tolerance to chilling and freezing (Chen et al. 2008). The transcription factor Zat12 interacts with 24 COS genes, of which 15 are cold-repressed genes and nine are cold-induced genes (Chinnusamy et al. 2007; Vogel et al. 2005). We identified homologs of the CBF/DREB (6), bHLH (11), MYC (15), and Zat12 (7) transcription factors in chrysanthemum that had significant transcriptional changes in treatment sample. We speculate that these genes may be involved in cold signal transduction in chrysanthemum.
Since Lyons proposed the hypothesis for the membrane phase transition of cold injury (Lyons and Raison 1971), several studies showed that the proportion and content of unstaurated fatty acids in membrane lipids are closely related to cold tolerance of plants (And and Murata 1996; Murata and Los 1997; Somerville 1995). Due to the activity of fatty acid desaturase, double bonds are introduced into saturated fatty acids in plants and become unsaturated fatty acids. Here, RNA-Seq was used to compare genes related to unsaturated fatty acids in different transcriptomes subjected to subzero and normal growing temperatures. The DEGs related to the biosynthesis of unsaturated fatty acids are shown in Table 4. Most of the associated unigenes were upregulated, which indicated that low temperature could enhance the expression of these unigenes. Only two unigenes were downregulated, FAD2-7 and delta-9 fatty acid desaturase. Delta-9 fatty acid desaturase is a soluble enzyme that catalyzes the desaturation of stearoyl-ACP, which is converted to glyceride or polyunsaturated fatty acids by the action of other enzymes (Thompson et al. 1991). In extremely cold temperatures, the desaturation products of stearic acid may be mass synthesized and lead to the negative feedback of delta-9 fatty acid desaturase, resulting in the downregulation of the unigene. Further research is required to explain the change in FAD2-7 unigene transcription. Most DEGs belonged to the FAD2 category, indicating that the FAD2 gene was abundantly expressed.
Furthermore, log2FC indicates the logarithm of the expression difference between the two group, which is reflected in the change in scale. The ω-6 fatty acid desaturase showed the greatest change in expression, suggesting that it is involved in the biosynthesis of unsaturated fatty acids under conditions of low temperature. Many unigenes (20%) encoding unknown proteins were identified, which provides valuable information. Certain unknown unigenes were significantly upregulated in the cold-treated group, suggesting that they are involved in regulating the response of chrysanthemum to cold stress. Further studies should focus on the expression of these unknown proteins in response to cold stress in chrysanthemum.
References
Alexa A, Rahnenfuhrer J (2007) Gene set enrichment analysis with topGO. Encycl Syst Biol 23:193–205
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
And IN, Murata N (1996) Chilling sensitivity in plants and cyanobacteria: the crucial contribution of membrane lipids. Annu Rev Plant Biol 47:541–568
Banzet N, Richaud C, Deveaux Y, Kazmaier M, Gagnon J, Triantaphylidès C (1998) Accumulation of small heat shock proteins, including mitochondrial HSP22, induced by oxidative stress and adaptive response in tomato cells. Plant J 13:519–527
Beck EH, Heim R, Hansen J (2004) Plant resistance to cold stress: mechanisms and environmental signals triggering frost hardening and dehardening. J Biosci 29:449–459
Beck EH, Fettig S, Knake C, Hartig K, Bhattarai T (2007) Specific and unspecific responses of plants to cold and drought stress. J Biosci 32:501–510
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc 57:289–300
Chen M, Xu ZL, Li L, Cheng X, Dong J, Wang Q, Ma Y (2008) Cold-induced modulation and functional analyses of the DRE-binding transcription factor gene, GmDREB3, in soybean (Glycine max L.). J Exp Bot 60:121–135
Chinnusamy V, Ohta M, Kanrar S, Lee BH, Hong X, Agarwal M, Zhu JK (2003) ICE1: a regulator of cold-induced transcriptome and freezing tolerance in Arabidopsis. Genes Dev 17:1043–1054
Chinnusamy V, Schumaker K, Zhu JK (2004) Molecular genetic perspectives on cross-talk and specificity in abiotic stress signalling in plants. J Exp Bot 55:225–236
Chinnusamy V, Zhu J, Zhu JK (2007) Cold stress regulation of gene expression in plants. Trends Plant Sci 12:444–451
Close TJ (1996) Dehydrins: emergence of a biochemical role of a family of plant dehydration proteins. Physiol Plant 97:795–803
Conesa A, Gotz S, Garcia-Gomez J, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Guy C (1999) Molecular responses of plants to cold shock and cold acclimation. J Mol Microbiol Biotechnol 1:231–242
Heidarvand L, Amiri RM (2010) What happens in plant molecular responses to cold stress? Acta Physiol Plant 32:419–431
Hu JX, Zhao H, Zhou HH (2010) False discovery rate control with groups. J Am Stat Assoc 105:1215–1227
Ichimura K, Mizoguchi T, Yoshida R, Takashi Y, Shinozaki K (2000) Various abiotic stresses rapidly activate Arabidopsis MAP kinases ATMPK4 and ATMPK6. Plant J Cell Mol Biol 24:655–665
Janská A, Marsík P, Zelenková S, Ovesná J (2010) Cold stress and acclimation—What is important for metabolic adjustment? Plant Biol 12:395–405
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:29–34
Komatsu S, Yang G, Khan M, Onodera H, Toki S, Yamaguchi M (2007) Over-expression of calcium-dependent protein kinase 13 and calreticulin interacting protein 1 confers cold tolerance on rice plants. Mol Genet Genom (MGG) 277:713–723
Kosová K, Vítámvás P, Prášil IT (2007) The role of dehydrins in plant response to cold. Biol Plant 51:601–617
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:93–99
Li Y, Chunhui S, Yong L, Qiusheng Y (2013) Comparison of membrane fatty acids components in leaf and root of autumn chrysanthemum cultivars under low temperature (in Chinese). Plant Physiol J 49:457–462
Location PD (2006) Plant dehydrins—tissue location, structure and function. Cell Mol Biol Lett 11:536–556
Lyons JM, Raison JK (1971) A temperature-induced phase change in mitochondrial membranes and its relationship to chilling injury in plant tissues. J Biol Chem 246:4036–4040
Maria K, Grazyna M (2007) Structure and functions of plant calcium-dependent protein kinases. Acta Biochim Pol 54:219–233
Marilyn G, Mervi A, Wai Ching H, Kaarina PM, Yu XM, Un CJ, Yang DSC (1997) Antifreeze proteins in winter rye. Physiol Plant 100:327–332
Miura K, Furumoto T (2013) Cold signaling and cold response in plants. Int J Mol Sci 14:5312–5337
Murata N, Los DA (1997) Membrane fluidity and temperature perception. Plant Physiol 115:875–879
Reddy ASN, Ali GS, Celesnik H, Day IS (2011) Coping with stresses: roles of calcium- and calcium/calmodulin-regulated gene expression. Plant Cell 23:2010–2032
Saijo Y, Hata S, Kyozuka J, Shimamoto K, Izui K (2000) Over-expression of a single Ca2+-dependent protein kinase confers both cold and salt/drought tolerance on rice plants. Plant J 23:319–327
Silva JATD, Shinoyama H, Aida R, Matsushita Y, Rajd SK, Chen F, Silva JATD, Shinoyama H, Aida R, Rajd SK (2013) Chrysanthemum biotechnology: Quo vadis? Crit Rev Plant Sci 32:21–52
Somerville C (1995) Direct tests of the role of membrane lipid composition in low-temperature-induced photoinhibition and chilling sensitivity in plants and cyanobacteria. Proc Natl Acad Sci USA 92:6215–6218
Sommerville J (1999) Activities of cold-shock domain proteins in translation control. BioEssays 21:319–325
Stefan GT, Juan Miguel GG, Javier T, Williams TD, Nagaraj SH, María José N, Montserrat R, Manuel T, Joaquín D, Ana C (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435
Stockinger EJ, Gilmour SJ, Thomashow MF (1997) Arabidopsis thaliana CBF1 encodes an AP2 domain-containing transcriptional activator that binds to the C-repeat/DRE, a cis-acting DNA regulatory element that stimulates transcription in response to low temperature and water deficit. Proc Natl Acad Sci 94:1035–1040
Teige M, Scheikl E, Gasteiger E, Eulgem T, Dóczi R, Ichimura K, Shinozaki K, Dangl J, Hirt H (2004) The MKK2 pathway mediates cold and salt stress signaling in Arabidopsis. Mol Cell 15:141–152
Thompson GA, Scherer DE, Aken FV, Kenny JW, Young HL, Shintani DK, Kridl JC, Knauf VC (1991) Primary structures of the precursor and mature forms of stearoyl-acyl carrier protein desaturase from safflower embryos and requirement of ferredoxin for enzyme activity. Proc Natl Acad Sci 88:2578–2582
Townley HE, Knight MR (2002) Calmodulin as a potential negative regulator of Arabidopsis COR gene expression. Plant Physiol 128:1169–1172
Viswanathan C, Jian-Kang Z (2002) Molecular genetic analysis of cold-regulated gene transcription. Philos Trans R Soc B Biol Sci 357:877–886
Vogel J, Zarka DBH, Fowler S, Thomashow M (2005) Roles of the CBF2 and ZAT12 transcription factors in configuring the low temperature transcriptome of Arabidopsis. Plant J 41:195–211
Wang XC, Zhao QY, Ma CL, Zhang ZH, Cao HL, Kong YM, Yue C, Hao XY, Chen L, Ma JQ (2013) Global transcriptome profiles of Camellia sinensis during cold acclimation. BMC Genom 14:1–15
Yamaguchi-Shinozaki K, Shinozaki K (2006) Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu Rev Plant Biol 57:781–803
Yang T, Zhang L, Zhang T, Hua Z, Xu S, An L (2005) Transcriptional regulation network of cold-responsive genes in higher plants. Plant Sci 169:987–995
Yang G, Zou H, Wu Y, Liu H, Yuan Y (2011) Identification and characterisation of candidate genes involved in chilling responses in maize (Zea mays L.). Plant Cell Tissue Organ Cult 106:127–141
Zhu J, Dong CH, Zhu JK (2007) Interplay between cold-responsive gene regulation, metabolism and RNA processing during plant cold acclimation. Curr Opin Plant Biol 10:290–295
Acknowledgements
The research was supported by Henan Province Science and Technology Development Program (Grant 142102110046).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lu, J., Bi, H., Zhang, A. et al. Comparative transcriptome analysis by RNA-Seq of the regulation of low temperature responses in Dendranthema morifolium. Hortic. Environ. Biotechnol. 59, 383–395 (2018). https://doi.org/10.1007/s13580-018-0042-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13580-018-0042-y