
Abstract
This study aims to explore the functions and mechanisms of long noncoding RNA small nucleolar RNA host gene 5 (SNHG5) in chronic constriction injury (CCI)-induced neuropathic pain (NP). An NP rat model was established using the CCI method and the NP severity was evaluated by paw withdrawal threshold (PWT) and paw withdrawal latency (PWL). The expression of SNHG5, CDK9, and SCN9A was quantified in rat dorsal root ganglion, in addition to the detections of apoptosis, pathological changes, neuron number, and the co-localization of Nav1.7 and cleaved caspase-3 with NeuN. In ND7/23 cells, the apoptosis and lactate dehydrogenase concentration were assessed, as well as the relationship between SNHG5, CDK9, and SCN9A. In the dorsal root ganglion of CCI-treated rats, SNHG5 and SCN9A were upregulated and downregulation of SNHG5 suppressed SCN9A expression, increased the PWT and PWL, blocked neuroinflammation and neuronal apoptosis, and alleviated NP. Mechanistically, SNHG5 recruited CDK9 to enhance SCN9A-encoded Nav1.7 expression and promoted peripheral neuronal apoptosis and injury. In addition, SCN9A overexpression nullified the alleviative effects of SNHG5 deficiency on NP and neuron loss in CCI rats. In conclusion, SNHG5 promotes SCN9A-encoded Nav1.7 expression by recruiting CDK9, thereby facilitating neuron loss and NP after spinal nerve injury, which may offer a promising target for the management of NP.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuropathic pain (NP) is a complex and severe pain disorder arising from a lesion or disease influencing the somatosensory system [1]. Antidepressant drugs and Ca2+ channel α2δ ligands are commonly used for NP treatment, yet caution should be used when administering these drugs to older people due to their adverse effects such as sedation and altered mental status [2, 3]. Owing to the adverse effects and poor efficacy of conventional therapies in clinical practice, deepening the knowledge of the mechanisms behind NP is necessary for identifying more effective pharmacological treatment options [4].
The mechanisms of NP are multifaceted and engage with structural and functional changes in nociceptive pathways from the injured site of peripheral nerves to the dorsal root ganglion (DRG) and spinal cord [5]. Peripheral sensitization appears when primary afferent nociceptive neurons are increasingly sensitive to external mechanical or thermal stimuli at the lesion site, which is controlled by proinflammatory cytokines and other agents [6]. Spontaneous activities of primary afferents, mediated by inflammatory mediators, result in central sensitization, which is essential for the development and maintenance of NP [7]. Voltage-gated sodium channels (VGSCs or Navs), including Nav1.1–1.9, are strongly expressed in peripheral sensory neurons [8]. Nav1.7, encoded by SCN9A gene, is abundant in the primary afferent nociceptive and sympathetic neurons and is responsible for the production of action potentials and the transduction of pain signals [9]. Inhibition of Nav1.7 in mice was found to relieve spinal nerve injury-induced NP and formalin-induced inflammatory pain [10]. These findings indicate that Nav1.7 is of enormous importance for the transmission of neuroinflammation-mediated pain signals, and exploration of Nav1.7-related mechanisms may hold great promise for developing novel therapeutic strategies for NP.
A number of long noncoding RNAs (lncRNAs) have been identified in DRGs, spinal cord, and other pain-related regions of animals and humans, and these lncRNAs affect NP development and maintenance by mediating pain-related genes and elevating neuronal excitability in primary sensory neurons of DRG [11]. The lncRNA small nucleolar RNA host gene 5 (SNHG5) was upregulated in mouse DRGs in response to experimental NP [12] and knockdown of SNHG5 blunted NP development and suppressed activation of astrocytes and microglia in mouse spinal cord [13]. The lncRNA X inactivate-specific transcript boosted inflammatory pain through upregulation of Nav1.7 by sequestering microRNA (miR)-146a [14]. These reports suggest that lncRNA SNHG5 may contribute to NP development by mediating Nav1.7. In addition, cyclin-dependent kinase-9 (CDK9) is an elongation factor that phosphorylates RNA polymerase II (RNAPII) to advance transcription elongation of genes [15]. CDK9 expression was elevated by nerve injury to increase RNAPII phosphorylation in the promoter of metabotropic glutamate receptor subtype 5 (mGluR5), an excitatory G protein-coupled receptor that regulates neurotransmitter signals, thereby promoting mGluR5 transcription in the dorsal horn for NP development [16]. Bioinformatics prediction in the present study showed an intriguing interaction between SNHG5, CDK9, and SCN9A. Therefore, we hypothesized that SNHG5 may recruit CDK9 to the promoter of SCN9A and regulate the expression of Nav1.7 channel protein in NP, which may contribute to the understanding of Nav1.7-related mechanisms in NP and provide promising therapeutic targets for the disease.
Materials and methods
NP rat model
Thirty-six male adult Sprague–Dawley rats (200 ~ 230 g) were reared at 25 °C with sufficient food and water and a 12-h light/dark cycle for 1 week of acclimatization, after which 12 rats were randomly grouped into sham and chronic constriction injury (CCI) groups with six rats in each group. As previous methods described [17], an NP rat model was induced with CCI under sodium pentobarbital anesthesia (40 mg/kg, 4390–16-3, Merck) via intraperitoneal injection. The sciatic nerve was exposed in a sterile environment, disassociated from surrounding tissues, and ligated with 4–0 thread at 1 mm intervals. The sciatic nerve of sham-operated animals was exposed and disassociated but not ligated. Fourteen days after the operation, the animals were sacrificed and their L5 DRGs were collected and stored at − 80 °C.
Short hairpin RNA (shRNA) lentiviruses knocking down SNHG5 (sh-SNHG5) vectors were designed and synthesized according to the target sequence of SNHG5 (Table 1). sh-SNHG5, SCN9A overexpression lentiviruses (LV-SCN9A), and their negative controls (NCs) (sh-NC and LV-NC) were purchased from Systems Biosciences (Mountain View, USA) and used for animal experiments. The other 24 rats were arranged into sh-SNHG5 (intrathecal injection with sh-SNHG5 before CCI modeling), sh-NC (intrathecal injection with sh-NC before CCI modeling), sh-SNHG5 + LV-SCN9A (intrathecal injection with sh-SNHG5 and LV-SCN9A before CCI modeling), and sh-SNHG5 + LV-NC (intrathecal injection with sh-SNHG5 and LV-NC before CCI modeling) groups (n = 6 rats per group). Hamilton syringes equipped with 30-gauge needles were placed into the subarachnoid space of the lumbar spine L4–L6, and the hind limbs of the rats were anesthetized with 2% lidocaine. Following experiments were carried out 24 h after catheter implantation. Three days before the modeling, corresponding lentiviruses were intrathecally injected into the rats through the catheters to reduce SNHG5 expression or elevate SCN9A level. The intrathecal injection was performed once a day for consecutive three days (10 µL/day, lentivirus titer of 2 × 108 pfu/mL). After the intrathecal injection, the rats were subjected to CCI modeling as above. The rats were euthanatized on day 14 after the CCI procedure and their DRGs were preserved for the following experiments. The animal experimentation was approved by the ethics committee of First Affiliated Hospital of Fujian Medical University (No. IACUC FJMU 2023–0038). All animal experiments in this work were conducted in accordance with the guidelines for the care and use of laboratory animals formulated by the Chinese Institutional Animal Protection and Use Committee.
Behavioral testing
Behavioral studies measuring pain thresholds to mechanical and thermal stimuli were performed without any use of analgesic drugs before the CCI (day 0) and on days 1, 3, 5, 7, and 14 after the CCI procedure according to the procedure developed by Li et al. [18]. The animals were examined for their development of NP behaviors, which was used to identify the NP rat model. Behavioral experiments were performed from 9:00 am to 4:00 pm on sham-operated and CCI rats by a researcher blind to the experimentation, in what is known as a double-blind method.
Paw withdrawal mechanic threshold (PWT) was measured using von Frey tests. The rats were placed in a glass box with metal mesh floor and stimulated with von Frey hair for 5 ~ 6 s with an interval of 5 min.
Paw withdrawal thermal latency (PWL) was tested using radiant heat stimulation (Plantar Analgesia Meter; IITC Life Science, USA). When paw withdrawal occurred, the timer was stopped and the time was automatically recorded. The cutoff time was 20 s.
Hematoxylin and eosin staining
Rat DRG paraffin sections were dewaxed and rehydrated with xylene and alcohol gradient before 2 min of hematoxylin staining (C0105S, Beyotime) and 2 s of hydrochloric acid alcohol differentiation. After washing, the sections were stained with eosin (Beyotime) for 2 min, dried, and photographed from five random fields. Three sections were collected from each sample to analyze the pathological changes in the DRGs.
Nissl staining
DRG tissues were fixed in 4% paraformaldehyde (PFA) (P0099, Beyotime), dehydrated, and embedded before preparation of 4-μm-thick sections, dewaxing, and hydration. Methylene Blue stain (M8030-10, Solarbio) was dropped onto the sections for 10 min of waiting period and Nissl differentiation was performed for 1–3 min until Nissl bodies were clearly observed by microscopy. Ammonium molybdate solution was added to the sections for 3–5 min and the sections were rinsed with distilled water, dehydrated in absolute alcohol, and sealed in neutral balsam. The images of neurons were captured under an optical microscope and processed with the software provided by Media Cybernetics (Rockville, MD, USA).
Immunofluorescence
First, immunofluorescence was used to analyze NeuN level in rat DRGs. DRGs were fixed in 4% PFA, and 4-μm-thick DRG sections were sealed with 5% conventional goat serum (SL038, Solarbio) and 0.3% Triton X-100 (P1080, Solarbio) [in phosphate buffered solution (0.01 M PBS, PH = 7.4)], incubated with a NeuN antibody (ab104224, Abcam, Cambridge, MA, USA) at 4 °C overnight and a secondary antibody at room temperature for 2 h, and imaged under an Olympus BX53 microscope (Japan). Three sections of five samplers were randomly selected from each sample.
Immunohistochemistry
DRGs were fixed for 48 h in 4% PFA and prepared into 4-μm-thick paraffin sections. Following baking for 20 min and dewaxing in xylene, the sections were washed once in distilled water and thrice in PBS, added with 3% H2O2 for 10 min of waiting period at room temperature, and washed thrice with PBS before antigen repair. Afterwards, the sections were sealed with goat serum for 20 min at room temperature, incubated with anti-CDK9 (ab239364, Abcam) and biotin-labeled goat anti-rabbit IgG (A0216, 1:200, Beyotime) at room temperature for 2 h. After washing and color development with DAB, the sections were stained with hematoxylin for 3 min, dehydrated, permeabilized, sealed, and imaged.
TUNEL
Apoptosis in DRGs was tested using a TUNEL kit (11,684,795,910, Roche, Basel, Switzerland). After dewaxing, alcohol hydration, and washing in PBS, DRG Sects. (4 μm) were maintained with proteinase K (39,450–01-6, Merck) for 20 min, rinsed with PBS, and sealed for 10 min before incubation with 500 μl TUNEL reaction solution (50 μl TdT + 450 μl fluorescein-conjugated dUTP solutions) at 37 °C for 1 h in a dark and wet box. The sections were imaged with a fluorescence microscope for counting of apoptotic cells. The nuclei of apoptotic cells showed red and that of stained cells showed blue. Image J software was used to count cells (cell apoptosis index = red cells/total cells × 100%).
Enzyme linked immunosorbent assay (ELISA)
The levels of tumor necrosis factor (TNF)-α (ab208348, Abcam), interleukin (IL)-1β (ab100768, Abcam), IL-6 (ab100712, Abcam), and IL-10 (ab255729, Abcam) in rat DRGs were measured using ELISA kits. A microplate reader was used to evaluate the optical density at 450 nm to calculate their concentrations.
Cell culture and transfection
ND7/23 cells, a rat DRG neuroblastoma cell line (#92,090,903, Sigma-Aldrich, St. Louis, MO, USA) (R-ND7/23), were cultured in high-glucose-DMEM growth medium (12,430,054, ThermoFisher Scientific, San Jose, CA, USA) supplemented with 10% fetal bovine serum (10,091,148, ThermoFisher Scientific), 50 U/mL penicillin, and 50 µg/mL streptomycin (15,070,063, ThermoFisher Scientific) at 37 °C with 5% CO2/95% air. Small interfering RNA (siRNA) sequences silencing SNHG5, CDK9, and SCN9A (si-SNHG5, si-CDK9, and si-SCN9A, respectively), SNHG5 and CDK9 overexpression plasmids (oe-SNHG5 and oe-CDK9, respectively), and their NCs were provided by GenePharma (Shanghai, China) and delivered into ND7/23 cells using Lipofectamine 2000 (11,668,027, Invitrogen, Carlsbad, CA, USA). The final concentrations of the si-RNAs and plasmids were 100 nM and 2 µg, respectively. The sequences of siRNAs are shown in Table 2.
RNA extraction
Nucleo-cytoplasmic separation was completed based on the instructions of a nucleo-cytoplasmic separation kit (AM1921, Thermo Fisher, San Jose, CA, USA). Reverse transcription fluorescence–quantitative polymerase chain reaction (RT–qPCR) was used to determine the lncRNA SNHG5 RNA expression in the nucleus and cytoplasm, with GAPDH and U6 serving as positive controls.
Flow cytometry analysis
Neuronal apoptosis at early and late stages was evaluated by flow cytometry analysis. ND7/23 cells (2 × 106) were added to 1 mL PBS and centrifuged at 1500 rpm for 3 min. After washing twice, the cells were resuspended in 300 µL pre-cold 1 × Binding Buffer and incubated at room temperature in the dark with 3 µL Annexin V-FITC and 5 µL PI (AP101-100-kit, MULTI SCIENCES, Hangzhou, Zhejiang, China) for 10 min. Afterwards, the cells were mixed with 200 µL pre-cold 1 × Binding Buffer and evaluated by a flow cytometer (NovoCyte 2060R, ACEA Biosciences Inc., Hangzhou, China).
Lactate dehydrogenase (LDH) test
LDH content in ND7/23 cell supernatant was tested with an LDH cytotoxicity assay kit (C0016, Beyotime). A microplate reader was used to detect the optical density of LDH at 490 nm.
RNA pull-down assay
RNA–protein interaction sequencing (RPIseq) (http://pridb.gdcb.iastate.edu/RPISeq/about.php) is a family of machine learning classifiers for predicting RNA–protein interactions using only sequence information. The interaction between the lncRNA SNHG5 and CDK9 was predicted by RPIseq and an RNA pull-down assay was completed according to the instructions of a Pierce™ Magnetic RNA–Protein Pull-Down kit (Thermo Fisher Scientific, MA, USA). Biotinylated lncRNA SNHG5 or NC was incubated with ND7/23 cell lysate at 25 °C for 2 h and the lncRNA SNHG5/CDK9 complexes were captured by streptavidin-conjugated immunomagnetic beads at 25 °C for 1 h. The complexes were eluted by incubation with proteinase K at 25 °C for 2 h and the protein levels in the complexes were evaluated by western blotting.
RNA immunoprecipitation (RIP)
RIP assay was completed based on the instructions of the Magna RIP Kit (17–701, EMD Millipore, Billerica, MA, USA). ND7/23 cells at a density of 2 × 106 cells/mL were fixed in 4% PFA and lysed with RIPA buffer. The cell lysate was incubated with anti-CDK9 (ab239364, Abcam) or anti-IgG (#5946, CST, Danvers, MA, USA) at 4 °C, after which the lysate was added with proteinase K-contained beads and RNA was extracted with Trizol. RNA purification was performed with a RNeasy Mini Kit (QIAGEN), and RNA expression was quantified by RT–qPCR.
Chromatin immunoprecipitation (ChIP)
ChIP assay was performed using an EZ-ChIP™ Kit (17–371, Millipore). Following 36 h of cell culture, ND7/23 cells were fixed with 4% PFA, neutralized with glycine, and collected for centrifugation. The cell pellet was suspended in PMSF-contained cell lysis buffer and centrifuged, with the supernatant discarded. DNA fragmentation was performed in an ice bath using ultrasonication. The cell supernatant was divided into two parts: 10% supernatant was used as the control and the other 90% was incubated with magnetic bead-conjugated anti-CDK9 or anti-IgG. After immunoprecipitation, the DNA bound to CDK9 protein was eluted and purified with a DNA purification kit (Beyotime) before qPCR analysis. In short, the principle of ChIP assay was as follows: in the living cells, after the DNA–protein complex was fixed by PFA, DNA fragmentation was conducted in an ice bath using ultrasonication. Next, these fragments were enriched and precipitated by specific antigen–antibody binding reactions. The DNA bound to CDK9 protein was eluted and purified. Finally, qPCR assay was used to detect the sequence information of target DNA.
Dual-luciferase reporter assay
The binding sites between CDK9 and SCN9A were screened in the hTFtarget database (http://bioinfo.life.hust.edu.cn/hTFtarget#!/) [19], and wild (SCN9A-WT: CAAAGAGAAACAGGAA) and mutated (SCN9A-MUT: AAAAAAAAAAAAAAAA) sequences were designed, generated, and inserted into luciferase reporter vectors (pGL3-Basic, Promega, Madison, WI, USA). The vectors were transfected into ND7/23 cells with oe-CDK9, si-SNHG5 + oe-CDK9, oe-SNHG5 + si-CDK9, or oe-NC, with the dose of 30 nM. The activities of Firefly luciferase and Renilla luciferase were evaluated by a luciferase detector (Promega) to quantitatively reflect the regulation of CDK9 on target genes. The interaction sites between CDK9 and target gene 3 'UTR were further determined by site-specific mutation method. Relative luciferase activity was defined as the ratio of Firefly luciferase activity to Renilla luciferase activity (the internal control: Renilla luciferase activity). Three independent experiments were set.
RT–qPCR
Total RNA was isolated from rat DRGs or ND7/23 cells by Trizol (15,596,018, Thermo Fisher Scientific) and converted into cDNA by a Reverse Transcription System (A3500, Promega). Gene expression was measured on a LightCycler 480 instrument (Roche) under the conditions provided by the SYBR Green PCR kit (DRR820A, TaKaRa, Otsu, Japan) (predenaturation at 95 °C for 5 min, 40 cycles of denaturation at 95 °C for 10 s, annealing at 60 °C for 10 s, and extension at 72 °C for 20 s). Relative RNA expression of target genes was quantified by the 2−∆∆Ct method (see Table 3 for primer sequences).
Western blotting
L5 DRGs and ND7/23 cells were treated with RIPA buffer (P0013B, Beyotime) (containing 1% PMSF) and centrifuged at a low speed. Afterwards, protein concentration was determined with the BCA method (P0010, Beyotime). Premixed supernatant was added with a fivefold volume of protein buffer (P0015L, Beyotime), boiled at 100 °C for denaturation, and preserved at − 20 °C. Denatured protein was unfrozen on ice, and 20 μL of protein underwent sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto a PVDF membrane (IPVH00010, Merck) using the semi-wet method before 1 h of sealing in 5% nonfat milk at room temperature. After washing, incubation with a primary antibody against CDK9 (ab239364, Abcam), Nav1.7 (ab65167, Abcam), Bax (ab32503, Abcam), Bcl-2(ab32124, Abcam), cleaved-caspase-3 (#9654, CST), and β-actin (ab8227, Abcam) was performed at 4 °C overnight, followed by TBST washing and incubation with a TBST-diluted secondary antibody at room temperature for 1 h. After color development with ECL, the results were identified and analyzed in a gel scanning analyzer.
Statistical analysis
Data analysis was performed by GraphPad Prism 8.0 version and the results are mean ± standard deviation. Statistically significant differences between two groups were identified by t test. When multiple groups of data were compared, one-way or two-way analysis of variance was used instead, followed by Tukey’s multiple comparisons test. Statistical significance was set at p < 0.05.
Results
LncRNA SNHG5 expressed strongly in CCI-induced NP rats
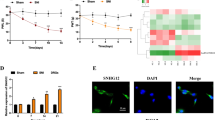
The severity of NP in CCI-induced rats was evaluated by PWT and PWL. Rats in the CCI group showed decreased PWT (p < 0.001) and PWL (p < 0.01) beginning 3 days after the surgery (Fig. 1A), suggesting that NP was successfully induced in CCI-treated rats.

CCI-induced NP rats had higher SNHG5 expression. A PWT (g) and PWL (g) were determined at days 0, 1, 3, 7, and 14 after operation. B Immunofluorescence was used to visualize the expression of NeuN in rat DRGs. C H&E staining was used to show the histological structures of rat DRGs (the nucleus appears blue and the cytoplasm appears red under the light microscope) and Nissl staining to observe the number of neurons in the DRGs (the neuron appears lavender blue and the nucleus appears purple blue under the light microscope). D SNHG5 expression in rat DRGs was evaluated by RT–qPCR. E ELISA assayed the expression of TNF-α, IL-1β, IL-6, and IL-10 in rat DRGs. F TUNEL tested the apoptotic rate in rat DRGs. N = 6. Data were shown as mean ± standard deviation. Statistically significant differences between two groups were identified by t test. When multiple groups of data were compared, one-way or two-way analysis of variance was used instead, followed by Tukey’s multiple comparisons test. *P < 0.05 vs. sham group. CCI, chronic constriction injury; DRG dorsal root ganglion; PWL paw withdrawal mechanic threshold; PWL paw withdrawal thermal latency
The expression of NeuN, a marker for neurons, was visualized by immunofluorescence. Compared with the sham group, the CCI group showed lower expression of NeuN in rat DRGs (Fig. 1B). H&E staining results indicated that sham-operated rats had normal histological structures in their DRGs with the cells regularly arranged; CCI-induced rats had irregular cell arrangement and larger intracellular space, in addition to neuron shrinkage and loss shown by Nissl staining, compared with sham-operated rats (Fig. 1C).
Next, the expression of the lncRNA SNHG5 was determined by RT–qPCR in rat DRGs. SNHG5 was upregulated by 1.82-fold in CCI rats (Fig. 1D; p < 0.01). Moreover, ELISA results indicated increased levels of TNF-α (2.46-fold), IL-1β (2.14-fold), and IL-6 (3.41-fold), as well as decreased IL-10 (0.44-fold), in the DRGs of CCI rats (Fig. 1E). Shown by TUNEL staining, the apoptotic rate in DRGs was enhanced in the CCI group compared to that in the sham group (Fig. 1F; 24.36 ± 2.52 vs. 3.72 ± 0.62, p < 0.001). These results suggested that the lncRNA SNHG5 might be associated with CCI-induced NP and neuron loss.
Downregulating the lncRNA SNHG5 attenuated CCI-induced neuron loss and NP in rats
To validate the role of SNHG5 in NP, we first evaluated the efficacy of sh-SNHG5 lentiviruses. RT–qPCR results showed that sh-SNHG5-1 knocked down SNHG5 expression most significantly and, therefore, was used in subsequent experiments (Fig. 2A). CCI-induced rats were injected with sh-SNHG5 and the expression of SNHG5 in rat DRGs was determined by RT–qPCR. Compared with the sh-NC group, the sh-SNHG5 group showed significantly reduced SNHG5 expression (Fig. 2B; 0.52-fold, p < 0.01).

Knockdown of SNHG5 blocked CCI-induced NP and neuron loss in rats. A Efficacy of sh-SNHG5 lentiviruses was tested by RT–qPCR. B Expression of SNHG5 in DRGs of rats injected with sh-SNHG5 was assayed. C PWT (g) and PWL (g) of rats injected with sh-SNHG5 were determined at days 0, 1, 3, 7, and 14 after operation. D Immunofluorescence was used to visualize the expression of NeuN in rat DRGs. E H&E staining was used to show the histological structures of rat DRGs and Nissl staining to observe the number of neurons in the DRGs. F ELISA assayed the expression of TNF-α, IL-1β, IL-6, and IL-10 in rat DRGs. G TUNEL tested the apoptotic rate in rat DRGs. N = 6. Data were shown as mean ± standard deviation. Statistically significant differences between two groups were identified by t test. When multiple groups of data were compared, one-way or two-way analysis of variance was used instead, followed by Tukey’s multiple comparisons test. *P < 0.05 vs. sh-NC group. CCI chronic constriction injury; DRG dorsal root ganglion; PWL paw withdrawal mechanic threshold; PWL paw withdrawal thermal latency
Knockdown of SNHG5 enhanced the PWT (p < 0.01) and PWL (p < 0.01) of CCI-treated rats (Fig. 2C) and boosted the expression of NeuN in the DRGs of CCI-induced rats (Fig. 2D). H&E and Nissl staining results showed alleviated tissue injury and neuron loss in CCI-induced rats in response to SNHG5 knockdown (Fig. 2E). Moreover, the sh-SNHG5 group showed lower levels of pro-inflammatory factors TNF-α (0.45-fold), IL-1β (0.43-fold), and IL-6 (0.41-fold), and higher IL-10 expression (1.96-fold) in rat DRGs (Fig. 2F). Compared to the sh-NC group, the sh-SNHG5 group also showed a decrease in the apoptotic rate of DRGs (Fig. 2G; 22.85 ± 4.65 vs. 9.05 ± 2.06, p < 0.001). Overall, knockdown of SNHG5 blocked CCI-induced NP and neuron loss in rats.
LncRNA SNHG5 interacted with CDK9 in ND7/23 cells
The focus of this study was shifted to the regulatory mechanisms of SNHG5 in NP. First, the results of nucleo-cytoplasmic separation showed that SNHG5 mainly localized in the nuclei of ND7/23 cells (Fig. 3A), which suggested that this lncRNA may play a role in transcriptional regulation. According to a previous report, SCN9A-encoded Nav1.7 channel protein is of great importance for nociception and chronic hyperalgesia and involved in pain behaviors, such as inflammatory pain and NP [20]. Shown by RT–qPCR and western blotting, SCN9A mRNA expression (1.76-fold) and Nav1.7 protein level (1.62-fold) were upregulated in the CCI group than in the sham group, and SCN9A mRNA expression (0.69-fold) and Nav1.7 protein level (0.66-fold) in the sh-SNHG5 group were downregulated compared with those in the sh-NC group (Fig. 3B, C). These data indicated that SNHG5 may regulate the transcription of SCN9A.

Interaction between SNHG5 and CDK9 in ND7/23 cells. A Localization of SNHG5 was identified by nucleo-cytoplasm separation. B, C Expression of SCN9A in rat DRGs was depicted by RT–qPCR and western blotting. D, E Binding of SNHG5 to CDK9 was verified by RNA pull-down (D) and RIP (E) assays. F, G RT–qPCR and western blotting were used to quantify the expression of CDK9 and SCN9A in ND7/23 cells transfected with oe-SNHG5 or si-SNHG5. H, I Expression of CDK9 in rat DRGs was tested by RT–qPCR (H) and immunohistochemistry (I). N = 6 in animal experiments. Cellular experiments were repeated in triplicate. Data were shown as mean ± standard deviation. Statistically significant differences between two groups were identified by t test and those among multiple groups were verified by one-way analysis of variance with Tukey’s multiple comparisons test. *P < 0.05 vs. sham, oe-NC, si-NC, bio-NC, or IgG group; #P < 0.05 vs. sh-NC group. CCI chronic constriction injury; DRG dorsal root ganglion
RPIseq analysis showed a high possibility of SNHG5 interacting with CDK9 (prediction using random forest classifier: 0.55; prediction using support vector machine classifier: 0.91). Moreover, the results of RNA pull-down assay demonstrated that CDK9 expression was elevated in the bio-SNHG5 group than in the bio-NC group (Fig. 3D). RIP results revealed that SNHG5 expression was extensively enriched by the anti-CDK9 antibody rather than anti-IgG antibody (Fig. 3E).
Next, the regulation of SCN9A expression by SNHG5 in ND7/23 cells was identified. RT–qPCR and western blot results manifested that SCN9A expression was upregulated (mRNA: 1.72-fold and protein: 1.63-fold) in response to oe-SNHG5 transfection and downregulated (mRNA: 0.45-fold and protein: 0.56-fold) after transfection with si-SNHG5, while the alterations in SNHG5 expression had no effects on CDK9 expression (Fig. 3F, G).
In addition, RT–qPCR and immunohistochemistry results indicated that CDK9 mRNA and protein expression was enhanced by 1.62-fold in the DRGs of rats treated with CCI, but downregulating SNHG5 expression did not influence CDK9 expression (Fig. 3H, I). Overall, SNHG5 interacted with CDK9.
LncRNA SHNG5 promoted Nav1.7 expression by recruiting CDK9 to the SCN9A promoter
The hTFtarget database suggested that CDK9 was a transcription factor of SCN9A (Fig. 4A). To verify the binding of CDK9 to SCN9A promoter, ND7/23 cells were transfected with oe-CDK9 or si-CDK9. Shown by RT–qPCR and western blotting, upregulation of CDK9 boosted SCN9A expression (mRNA: 1.86-fold and protein: 1.55-fold) and knockdown of CDK9 reduced SCN9A levels (mRNA: 0.41-fold and protein: 0.48-fold) in the ND7/23 cells (Fig. 4B, C). Dual luciferase reporter assay demonstrated that overexpressing CDK9 increased the luciferase intensity of SCN9A–WT and did not change that of SCN9A–MUT (Fig. 4D). ChIP–PCR analysis of CDK9 binding to the SCN9A promoter in ND7/23 cells showed that the anti-CDK9 antibody increased the enrichment of SCN9A promoter sequence in the complexes compared to the anti-IgG antibody (Fig. 4E). These findings disclosed that CDK9 positively controlled the transcription of SCN9A gene.

SNHG5 recruited CDK9 to the SCN9A promoter and thereby promoted Nav1.7 expression. A hTFtarget database predicted the binding site between CDK9 and SCN9A. B, C Expression of CDK9 and SCN9A was evaluated using RT–qPCR and western blotting after transfection with oe-CDK9 or si-CDK9. D, E Binding of CDK9 to the SCN9A promoter was determined with dual luciferase reporter (D) and ChIP (E) assays. F, G. ChIP (F) and dual-luciferase reporter (G) assays were used to probe whether SNHG5 recruits CDK9 to the SCN9A promoter after transfection with oe-SNHG5 + si-CDK9 or si-SNHG5 + oe-CDK9. Each experiment was independently repeated three times. Data were shown as mean ± standard deviation. Statistically significant differences between two groups were identified by t test and those among multiple groups were verified by one-way analysis of variance with Tukey’s multiple comparisons test. *P < 0.05 vs. sham, oe-NC, si-NC, or IgG group; #P < 0.05 vs. oe-SNHG5 or si-SNHG5 group
For the sake of validating the recruitment of CDK9 in the SCN9A promoter by SNHG5, ChIP and luciferase reporter assays were carried out. The binding of CDK9 to the SCN9A promoter was enhanced by SNHG5 overexpression, which was nullified by CDK9 knockdown; the binding between CDK9 and the SCN9A promoter was inhibited by SNHG5 knockdown, while overexpression of CDK9 blocked the negative effect of SNHG5 knockdown on the binding (Fig. 4F). Moreover, for the SCN9A–WT reporter vector, the change in luciferase activity induced by SNHG5 overexpression or knockdown was partially nullified by CDK9 knockdown or overexpression; for the SCN9A–MUT reporter vector, the alterations in CDK9 and SNHG5 expression did not influence the luciferase activity (Fig. 4G). Taken together, CDK9 transcriptionally activated SCN9A, and SNHG5 recruited CDK9 to the SCN9A promoter and thus facilitated SCN9A-encoded Nav1.7 channel protein expression in peripheral neurons.
LncRNA SNHG5 facilitated peripheral neuron injury through the CDK9/SCN9A axis
ND7/23 cells were transfected with oe-SNHG5 or oe-SNHG5 + si-CDK9/si-SCN9A to assess the influence of the SNHG5/CDK9/SCN9A axis on peripheral neurons. Compared with the oe-NC group, the oe-SNHG5 group had higher expression of SNHG5 (2.56-fold) and higher levels of SCN9A mRNA (1.88-fold) and Nav1.7 protein (1.74-fold); compared with the oe-SNHG5 group, the oe-SNHG5 + si-CDK9 group showed decreased levels of CDK9 (mRNA: 0.47-fold and protein: 0.56-fold) and SCN9A (mRNA: 0.65-fold and protein: 0.67-fold), and the oe-SNHG5 + si-SCN9A group had lower SCN9A expression (mRNA: 0.51-fold and protein: 0.57-fold) (Fig. 5A and B). Flow cytometry results showed elevated apoptotic rates in response to SNHG5 upregulation (Fig. 5C; 4.25 ± 0.82 vs. 14.31 ± 2.44), which was recapitulated by the immunoblots of Bax, cleaved caspase-3, and Bcl-2. Upregulation of SNHG5 increased the protein levels of Bax (1.92-fold) and cleaved caspase-3 (1.85-fold) and decreased Bcl-2 protein level (0.42-fold) (Fig. 5D). In addition, LDH content in ND7/23 cells overexpressing SNHG5 was increased by 2.16-fold (Fig. 5E). However, downregulating CDK9 or SCN9A offset, in part, the promoting effects of SNHG5 overexpression on the apoptosis and injury in peripheral neurons (Fig. 5C and E).

SNHG5 regulated the CDK9/SCN9A axis to promote peripheral neuronal injury. A, B Expression of SNHG5, CDK9, and SCN9A was determined by RT–qPCR and/or western blotting. C Apoptotic rate of ND7/23 cells was evaluated by flow cytometry analysis (the X-axis represents Annexin-V-FITC, and the Y-axis represents PI). D Western blotting was used to show Bax, cleaved caspase-3, Bcl-2 protein levels. E LDH test was performed to evaluate neuron injury. Data were shown as mean ± standard deviation. Statistically significant differences among multiple groups were verified by one-way analysis of variance with Tukey’s multiple comparisons test. *P < 0.05 vs. oe-NC group; #P < 0.05 vs. oe-SNHG5 group. LDH lactate dehydrogenase
LncRNA SNHG5 enhanced neuron loss and NP in CCI-induced rats by activating SCN9A through recruitment of CDK9
The effects of the SNHG5/CDK9/SCN9A axis in NP were further validated in vivo by injection of sh-SNHG5 and LV-SCN9A into CCI rats. The results of RT–qPCR and western blotting found that SCN9A mRNA expression (2.05-fold, p < 0.01) and Nav1.7 protein level (1.53-fold, p < 0.05) were boosted, while the expression of SNHG5 and CDK9 was unchanged in the sh-SNHG5 + LV-SNC9A group compared with those in the sh-SNHG5 + LV-NC group (Fig. 6A and B). Upregulation of SCN9A aggravated mechanical (p < 0.001) and thermal (p < 0.01) hyperalgesia blocked by SNHG5 knockdown (Fig. 6C). H&E and Nissl staining results suggested that, in the absence of SNHG5, SCN9A overexpression promoted tissue injury in rat DRGs and reduced the number of neurons in the DRGs (Fig. 6D). In addition, immunofluorescence results revealed the reduction in neuron number (Fig. 6E). ELISA showed that compared to the sh-SNHG5 + LV-NC group, the sh-SNHG5 + LV-SCN9A group had higher levels of TNF-α (2.16-fold), IL-1β (2.19-fold), and IL-6 (2.72-fold) and a lower level of IL-10 (0.49-fold) (Fig. 6F). TUNEL results demonstrated a promoting effect of SCN9A overexpression on neuronal apoptosis in the presence of SNHG5 knockdown (Fig. 6G; 18.65 ± 3.88 vs. 9.13 ± 2.52, p < 0.001). Overall, SNHG5 may recruit CDK9 to activate SCN9A transcription and promote Nav1.7 protein expression, thus affecting CCI-induced neuron loss and NP.

SNHG5 promoted SCN9A transcription by CDK9 to enhance neuron loss and NP in CCI rats. A, B Expression of SNHG5, CDK9, and SCN9A was determined by RT–qPCR and/or western blotting. C PWT (g) and PWL (g) of rats injected with sh-SNHG5 and LV-SCN9A were determined at days 0, 1, 3, 7, and 14 after operation. D H&E staining was used to show the histological structures of rat DRGs and Nissl staining to observe the number of neurons in the DRGs. E Immunofluorescence was used to visualize the expression of NeuN in rat DRGs. F. ELISA assayed the expression of TNF-α, IL-1β, IL-6, and IL-10 in rat DRGs. G TUNEL tested the apoptotic rate in rat DRGs. N = 6. Data were shown as mean ± standard deviation. Statistically significant differences between two groups were identified by t test. When multiple groups of data were compared, one-way or two-way analysis of variance was used instead, followed by Tukey’s multiple comparisons test. *P < 0.05 vs. sh-SNHG5 + LV–NC group. CCI chronic constriction injury; DRG dorsal root ganglion; PWL paw withdrawal mechanic threshold; PWL paw withdrawal thermal latency
Discussion
Previous NP research has identified numerous lncRNAs in pain-related regions and their expression levels were altered in affected pain pathways by peripheral inflammation and nerve injury [1, 21, 22]. In this study, we found increased SNHG5 level and the inhibitory effects of SNHG5 deficiency on CCI-induced neuron loss, hyperalgesia, and peripheral neuron injury. Mechanistic investigations showed that the lncRNA SNHG5 recruited CDK9 to the promoter of SCN9A and promoted the expression of Nav1.7 channel protein, thereby facilitating neuron injury and NP development in CCI-induced rats.
Previous inflammation research reveals an interesting association between SNHG5 and inflammation. Particularly, SNHG5 was found to play a pro- or anti-inflammatory role in various disease settings, such as chronic obstructive pulmonary disease, epilepsy, and ulcerative colitis [23,24,25]. SNHG5 has been shown to augment activation of astrocytes and microglia in spinal cord injury by directly interacting with Krüppel-like factor 4, an inflammation-associated transcription factor [26]. In periodontal inflammation, SNHG5 attenuated TNF-α-induced inflammation by inhibiting the translocation of the nuclear factor-kappa B (NF-κB) p65 subunit and inactivating the NF-κB signaling pathway [27]. Our experiments found SNHG5 was strongly expressed in rat DRGs after CCI induction. Apart from relieving inflammation, our data demonstrated that knockdown of SNHG5 alleviated neuron loss as well as mechanical and thermal hyperalgesia induced by CCI in rat DRGs. Previous mechanistic studies highlighted the functions of SNHG5 in NP through competitive endogenous RNA regulatory networks. For example, SNHG5 reduced CCI-induced NP and inflammation by upregulating the expression of calcium/calmodulin-dependent protein kinase II α via miR-142-5p [12, 13]. SNHG5 also stimulated the activation of astrocytes and microglia and promoted NP by targeting miR-154-5p which downregulated the expression of C–X–C motif chemokine 13 [12, 13]. In this work, nucleo-cytoplasm separation results revealed that SNHG5 was mainly localized in the nuclei of peripheral neurons and function experiments demonstrated a direct interaction between SNHG5 and CDK9. Rescue experiments indicated that inhibition of CDK9 attenuated SNHG5 overexpression-promoted peripheral neuronal apoptosis and cytotoxicity. Evidently, the immune and inflammatory modulatory roles of CDK9 have been confirmed in different diseases and cancers [28]. For instance, inhibition of CDK9 in nucleus pulposus cells suppressed pro-inflammatory cytokine-induced catabolism, which is considered a promising therapeutic target for managing intervertebral disk degeneration [29]. CDK9 phosphorylation was induced under hyperglycemic conditions and led to activation of the MAPK signaling pathway, which enhanced the transcription of inflammatory cytokines, including TNF-α, IL-1β, and IL-6 [30].
Although there are limited reports on the specific mechanisms of CDK9 in NP, other CDKs have been delineated to affect the commencement and maintenance of NP directly by cell cycle regulation or by transcription, epigenetic regulation, and metabolism. As a classical cell cycle-related kinase, CDK4 was upregulated after spinal cord injury, and cell cycle arrest by administration of CR8, a selective CDK inhibitor, rescued motor deficits, mechanical hyperalgesia, and neuroinflammation in mice [31]. Otherwise, CDK5 was summarized to regulate its targets (cytoplasmic proteins, integral membrane proteins, and protein complexes such as ion channels and membrane receptors) and thus enhance pain hypersensitivity in NP, either by facilitating the transmission of proteins to the plasma membrane, by enhancing their transcription and expression, by increasing the interaction between its target proteins and others and thereby promoting their function, or by promoting morphine nociceptive tolerance [32]. CDK9-dependent RNAPII phosphorylation in the mGluR5 promoter enhanced mGluR5 transcription and contributed to NP development [16]. Outside of such mechanism, we found CDK9 positively regulated SCN9A gene transcription and that SNHG5 enhanced the recruitment of CDK9 in the promoter of SCN9A to facilitate SCN9A-encoded Nav1.7 protein expression, thus regulating experimental NP-induced neuron loss and inflammation. Considering the multiple mechanisms of CDKs in NP, the regulatory mechanisms of CDK9 in NP are worthy of further exploration.
In conclusion, this study provided a potential explanation of NP maintenance, that is, the lncRNA SNHG5 interacted with CDK9 to promote Nav1.7 expression, and contributed to the understanding of the mechanisms of Nav1.7 transmitting pain signals in NP. However, experiments on animals cannot completely mimic the commencement and development of NP in humans, future investigations can be made in the clinical setting to confirm our conclusion. In addition, the commonly used cell counting method was used in this study as previously described [33], and more scientific cell counting methods can be used in future experiments to improve the accuracy of our experimental results, like stereologically unbiased method. Considering the association of the SNHG5/CDK9/SCN9A axis with inflammation, future studies can pay more attention to the relationship between this axis and proinflammatory pathways, such as the NF-κB signaling pathway, for deepening the understanding of the functions of SNHG5 in NP. Overall, this study may offer promising targets for the management of NP.
Data availability
The data sets used or analyzed during the current study are available from the corresponding author on reasonable request.
References
Zhang Q, Zhu D, Li Q. LncRNA CRNDE exacerbates neuropathic pain in chronic constriction injury-induced(CCI) rats through regulating miR-146a-5p/WNT5A pathway. Bioengineered. 2021;12:7348–59.
Attal N. Pharmacological treatments of neuropathic pain: the latest recommendations. Rev Neurol (Paris). 2019;175:46–50.
Pedowitz EJ, Abrams RMC, Simpson DM. Management of neuropathic pain in the geriatric population. Clin Geriatr Med. 2021;37:361–76.
Liu P, Cheng J, Ma S, Zhou J. Paeoniflorin attenuates chronic constriction injury-induced neuropathic pain by suppressing spinal NLRP3 inflammasome activation. Inflammopharmacology. 2020;28:1495–508.
Zhang L, Chen X, Wu L, Li Y, Wang L, Zhao X, Zhao T, Zhang L, Yan Z, Wei G. Ameliorative effects of escin on neuropathic pain induced by chronic constriction injury of sciatic nerve. J Ethnopharmacol. 2021;267: 113503.
Chu LW, Cheng KI, Chen JY, Cheng YC, Chang YC, Yeh JL, Hsu JH, Dai ZK, Wu BN. Loganin prevents chronic constriction injury-provoked neuropathic pain by reducing TNF-alpha/IL-1beta-mediated NF-kappaB activation and Schwann cell demyelination. Phytomedicine. 2020;67: 153166.
Liu ZY, Song ZW, Guo SW, He JS, Wang SY, Zhu JG, Yang HL, Liu JB. CXCL12/CXCR4 signaling contributes to neuropathic pain via central sensitization mechanisms in a rat spinal nerve ligation model. CNS Neurosci Ther. 2019;25:922–36.
Li Y, North RY, Rhines LD, Tatsui CE, Rao G, Edwards DD, Cassidy RM, Harrison DS, Johansson CA, Zhang H, Dougherty PM. DRG voltage-gated sodium channel 1.7 Is upregulated in paclitaxel-induced neuropathy in rats and in humans with neuropathic pain. J Neurosci. 2018;38:1124–36.
Moutal A, Cai S, Yu J, Stratton HJ, Chefdeville A, Gomez K, Ran D, Madura CL, Boinon L, Soto M, Zhou Y, Shan Z, Chew LA, Rodgers KE, Khanna R. Studies on CRMP2 SUMOylation-deficient transgenic mice identify sex-specific Nav1.7 regulation in the pathogenesis of chronic neuropathic pain. Pain. 2020;161:2629–51.
Niu HL, Liu YN, Xue DQ, Dong LY, Liu HJ, Wang J, Zheng YL, Zou AR, Shao LM, Wang K. Inhibition of Nav1.7 channel by a novel blocker QLS-81 for alleviation of neuropathic pain. Acta Pharmacol Sin. 2021;42:1235–47.
Wu S, Bono J, Tao YX. Long noncoding RNA (lncRNA): a target in neuropathic pain. Expert Opin Ther Targets. 2019;23:15–20.
Jin S., Tian S., Ding H., Yu Z., Li M. SNHG5 knockdown alleviates neuropathic pain induced by chronic constriction injury via sponging miR‑142‑5p and regulating the expression of CAMK2A. Mol Med Rep. 2022;26.
Chen M, Yang Y, Zhang W, Li X, Wu J, Zou X, Zeng X. Long noncoding RNA SNHG5 knockdown alleviates neuropathic pain by targeting the miR-154-5p/CXCL13 axis. Neurochem Res. 2020;45:1566–75.
Sun W, Ma M, Yu H, Yu H. Inhibition of lncRNA X inactivate-specific transcript ameliorates inflammatory pain by suppressing satellite glial cell activation and inflammation by acting as a sponge of miR-146a to inhibit Na(v) 1.7. J Cell Biochem. 2018;119:9888–98.
Singh R, Bhardwaj V, Das P, Purohit R. Natural analogues inhibiting selective cyclin-dependent kinase protein isoforms: a computational perspective. J Biomol Struct Dyn. 2020;38:5126–35.
Hsieh MC, Peng HY, Ho YC, Lai CY, Cheng JK, Chen GD, Lin TB. Transcription repressor Hes1 contributes to neuropathic pain development by modifying CDK9/RNAPII-dependent spinal mGluR5 transcription. Int J Mol Sci. 2019;20:4177.
Dong J, Xia R, Zhang Z, Xu C. lncRNA MEG3 aggravated neuropathic pain and astrocyte overaction through mediating miR-130a-5p/CXCL12/CXCR4 axis. Aging (Albany NY). 2021;13:23004–19.
Zhao L, Song C, Huang Y, Lei W, Sun J. MMP-9 regulates CX3CL1/CX3CR1 in the early phase of neuropathic pain in chronic sciatic nerve constriction injury (CCI) rats. Ann Palliat Med. 2020;9:2020–7.
Zhang Q, Liu W, Zhang HM, Xie GY, Miao YR, Xia M, Guo AY. hTFtarget: a comprehensive database for regulations of human transcription factors and their targets. Genom Proteom Bioinform. 2020;18:120–8.
Xue Y, Chidiac C, Herault Y, Gaveriaux-Ruff C. Pain behavior in SCN9A (Nav1.7) and SCN10A (Nav1.8) mutant rodent models. Neurosci Lett. 2021;753:135844.
Hu JZ, Rong ZJ, Li M, Li P, Jiang LY, Luo ZX, Duan CY, Cao Y, Lu HB. Silencing of lncRNA PKIA-AS1 attenuates spinal nerve ligation-induced neuropathic pain through epigenetic downregulation of CDK6 expression. Front Cell Neurosci. 2019;13:50.
Wang W, Min L, Qiu X, Wu X, Liu C, Ma J, Zhang D, Zhu L. Biological function of long non-coding RNA (LncRNA) Xist. Front Cell Dev Biol. 2021;9: 645647.
Shen Q, Zheng J, Wang X, Hu W, Jiang Y, Jiang Y. LncRNA SNHG5 regulates cell apoptosis and inflammation by miR-132/PTEN axis in COPD. Biomed Pharmacother. 2020;126: 110016.
Wang M, Xie Y, Shao Y, Chen Y. LncRNA snhg5 attenuates status epilepticus induced inflammation through regulating NF-kappaBeta signaling pathway. Biol Pharm Bull. 2022;45:86–93.
Zhou Z, Cao J, Liu X, Li M. Evidence for the butyrate metabolism as key pathway improving ulcerative colitis in both pediatric and adult patients. Bioengineered. 2021;12:8309–24.
Jiang ZS, Zhang JR. LncRNA SNHG5 enhances astrocytes and microglia viability via upregulating KLF4 in spinal cord injury. Int J Biol Macromol. 2018;120:66–72.
Han Y, Huang Y, Yang Q, Jia L, Zheng Y, Li W. Long non-coding RNA SNHG5 mediates periodontal inflammation through the NF-kappaB signalling pathway. J Clin Periodontol. 2022;49:1038–51.
Sundar V, Vimal S, Sai Mithlesh MS, Dutta A, Tamizhselvi R, Manickam V. Transcriptional cyclin-dependent kinases as the mediators of inflammation-a review. Gene. 2021;769: 145200.
Ni W, Zhang F, Zheng L, Wang L, Liang Y, Ding Y, Yik JHN, Haudenschild DR, Fan S, Hu Z. Cyclin-dependent kinase 9 (CDK9) inhibitor atuveciclib suppresses intervertebral disk degeneration via the inhibition of the NF-kappaB signaling pathway. Front Cell Dev Biol. 2020;8: 579658.
Yang X, Luo W, Li L, Hu X, Xu M, Wang Y, Feng J, Qian J, Guan X, Zhao Y, Liang G. CDK9 inhibition improves diabetic nephropathy by reducing inflammation in the kidneys. Toxicol Appl Pharmacol. 2021;416: 115465.
Wu J, Renn CL, Faden AI, Dorsey SG. TrkB.T1 contributes to neuropathic pain after spinal cord injury through regulation of cell cycle pathways. J Neurosci. 2013;33:12447–63.
Gomez K, Vallecillo TGM, Moutal A, Perez-Miller S, Delgado-Lezama R, Felix R, Khanna R. The role of cyclin-dependent kinase 5 in neuropathic pain. Pain. 2020;161:2674–89.
Su CJ, Zhang JT, Zhao FL, Xu DL, Pan J, Liu T. Resolvin D1/N-formyl peptide receptor 2 ameliorates paclitaxel-induced neuropathic pain through the activation of IL-10/Nrf2/HO-1 pathway in mice. Front Immunol. 2023;14:1091753.
Funding
Thanks for the grant from the Fujian Provincial Health Technology Project (Project number: 2022CXA023).
Author information
Authors and Affiliations
Contributions
WCS and CRS conceived the ideas. WCS, CRS and ZXT designed the experiments. WCS, CRS, ZXT, ZXB and LNC performed the experiments. WCS, CRS, ZXT and LNC analyzed the data. WCS, CRS and ZXT provided critical materials. WCS, CRS, LNC and ZXB wrote the manuscript. WCS and CRS supervised the study. All the authors have read and approved the final version for publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare there is no conflict of interests.
Ethical approval
All animal experiments were conducted per the protocols approved by the Institutional Animal Care and Use Committee of First Affiliated Hospital of Fujian Medical University (No. IACUC FJMU 2023–0038).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, C., Chen, R., Zhu, X. et al. Long noncoding RNA small nucleolar RNA host gene 5 facilitates neuropathic pain in spinal nerve injury by promoting SCN9A expression via CDK9. Human Cell 37, 451–464 (2024). https://doi.org/10.1007/s13577-023-01019-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13577-023-01019-w