
Abstract
Background
Although monogenic diabetes accounts for a small proportion of diabetes cases, accurate diagnosis may significantly change treatment. This study aimed to contribute to knowledge about the genotype-phenotype relationship in monogenic diabetes.
Methods
This study used data from a tertiary centre in Turkey. Genetic analysis outcomes for 36 patients were evaluated. The panel included 23 genes related to maturity-onset diabetes of the young (MODY), neonatal diabetes, and some genes related to hyperglycemic hypoglycemia. The next-generation sequencing method was used after DNA isolation from the peripheral blood.
Results
Mutations were identified in 19 (52.8%) of 36 patients. Of the 19 mutations, 7 (36.8%) were new mutations. A total of 20 cases met the MODY clinical criteria, and mutations were identified in 11 (55%) of them. In total, nine patients had more than one mutation. Mutations were identified on the ABCC8 (n = 7), PDX1 (n = 6), GLIS3 (n = 6), ZFP57 (n = 5), GCK (n = 4), HNF1A (n = 3), GLUD (n = 3), and HNF4A, KLF11, NKX2-2, and INSR genes (n = 1 each).
Conclusion
Our findings highlight a broad clinical and genetic spectrum of MODY, and genetic analysis may provide a better understanding of diabetes and improve the individualised treatment approach.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nearly 90% of diabetics have type 2 diabetes mellitus (DM) and 5–10% have type 1 DM. Type 1 and type 2 DM have no single genetic cause. Variations causing an increase in risk, and some reducing risk, were identified, and a multi-genetic effect was reported. However, a genetic variation may be identified as the true cause of DM in 2–5% of diabetics [1,2,3,4]. Monogenic DM cases comprise maturity-onset diabetes of the young (MODY), neonatal DM (NDM), and some rare diabetic syndromes. The most common is MODY. NDM is rare, as reported in 1 per 90,000 live births in Europe [5]. Monogenic diabetic forms should be suspected in cases without classic type 2 DM or type 1 DM clinical findings or diabetics with intense family history, and genetic studies should be planned.
NDM is generally defined as DM emerging in the first 6 months after birth. Temporary or permanent types may be present. The most common causes of NDM are KCNJ11, ABCC8, and INS heterozygous gene mutations. Homozygote INS mutations may create a clinical course very similar to type 1 DM [6]. Some other genes had also been identified to cause NDM. NDM has a lower heritability rate than MODY [7].
The term “MODY” was first used in 1975 [8]. However, mutations responsible for this DM form were revealed in the 1990s [9]. With the use of new-generation sequencing devices in recent years, recognising genes related to disease has increased. Finally, 14 MODY-related genes were identified. However, not all the 14 forms of ‘MODY’ are indeed true MODY.
Whether some rare MODY types like MODY 7,8,9,11 are indeed MODY is debatable [10].
Especially for diabetics under 45 years old, the frequency of MODY reaches up to 5%, and 80% of these cases were misdiagnosed as type 1 DM or type 2 DM [11]. Genetic confirmation of the diagnosis of MODY may lead to managing more appropriate treatment approaches. While some monogenic DM forms respond to sulfonylurea treatment very well, newer drugs such as DPP4 and SGLT2 inhibitors are also being studied to manage MODY [10]. Identification of new MODY mutations and reporting of their clinical data contribute to a better understanding of the MODY phenotypes and may provide developing more improved therapeutic approaches.
In this article, we present analysis results of a gene panel including 23 genes and the clinical characteristics of cases. This study aimed to contribute to knowledge about the genotype-phenotype relationship in monogenic DM.
Methods
This single-centre study included 36 patients whose gene analysis was performed between January 2018 and December 2019. Analyses were performed in a University Medical Genetic laboratory with the new-generation sequencing method after DNA isolation from the peripheral blood. Results were assessed with Ion reporter v. 5.6 and IGV software. The panel included genes related to MODY and NDM and some genes related to hyperinsulinemic hypoglycemia.
The pathogenicity of the mutations was classified according to the American College of Medical Genetics and Genomics criteria [12]. The Clinvar, HGMD, and Varsome databases were also used to assess mutations. In silico analysis was also performed using SIFT, MutationTaster, Human Splicing Finder, Mutation Assessor, and Polyphen2software. GERP score, DANN score, and GnomAD frequency were also employed for the assessment of variations.
In this study, the following 23 genes were investigated: HNF1A, GCK, HNF4A, PDX1, HNF1B, NEUROD1, KLF11, ZFP57, PAX4, INS, BLK, ABCC8, KCNJ11, RFX6, HADH, SLC16A1, FOXP3, G6PC2, NEUROG3, GLIS3, NKX2-2, GLUD1, and INSR.
The clinical history and laboratory results of the patients were obtained from the hospital records. MODY’s clinical diagnosis was defined as follows: DM diagnosis at a young age, positive family history (autosomal dominant inheritance observed in at least two or three generations), absence of C-peptide negativity, and lack of β-cell autoimmunity [13].
Statistical data analysis used SPSS version 23.0 software (IBM Corp., Armonk, NY, USA). The mean and standard deviation values were used for descriptive statistics.
Results
Thirty-six patients referred for the genetic study were assessed in this study. Of all the participants, 25 were female (69.5%), and 11 were male (30.5%). The mean age was 31.4 ± 15.5 years.
Thirty patients had analysis requested with suspicion of monogenic DM, two paediatric patients had hypoglycemia suspicion, and the reason for analysis could not be identified in four patients.
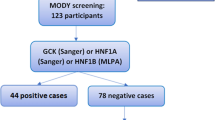
Nineteen different variants were detected in 19 of 36 cases. Nine patients had more than one variant. Seven variants were novel. Thirteen of the mutations were interpreted as variant of uncertain significance, likely pathogenic or pathogenic. A total of 20 cases met the MODY clinical criteria, and mutations were identified in 11 (55%) of them. Four mutation-positive cases with diabetes did not meet all of the MODY criteria. The other four patients with the mutation also did not fully meet the DM diagnostic criteria. Of the 16 patients with negative mutation analysis, nine met the clinical criteria for MODY and were considered to have mutation-negative MODY (Figure 1). One patient with hyperinsulinemic hypoglycemia was identified to have a mutation of the HADH gene, but he was not evaluated in the analyses of this study.

Distribution of analyzed cases according to mutation status and diagnosis
Mutations were present on HNF1A in three patients, HNF4A in one, KLF11 in one, PDX1 in six, ABCC8 in seven, GLIS3 in six, XFP57 in five, GCK in four, GLUD1 in three, NKX2-2 in one, and INSR in one. The clinical features of the cases are shown in Table 1, and database analyses of the detected mutations are shown in Table 2.
Glucokinase (GCK) mutation
Four patients had GCK mutations. One of the patients was 28 years old and was diagnosed with DM at age 13. The HbA1c value was 6.8% with diet alone. The two other patients were a mother and daughter in family B. Both were not using pharmacologic agents for DM treatment. The mother had HbA1c value of 6.9%, while the daughter had 7.1%. The fourth patient had GCK and ABCC8 mutations. The patient was 4 years old and diagnosed with DM at age 2. HbA1c value was 6.4% with diet alone. There was no family history of DM.
HNF1A mutation
Three patients had HNF1A mutation. One of the patients with HNF1A mutation had anti-GAD positivity, and the C-peptide was at unmeasurably low levels, so she was diagnosed with type 1 DM. The other two patients with HNF1A mutation were a mother and a daughter. The daughter was 33 years old and had no DM with an HbA1c of 5.1%. The mother was 64 years old without DM.
Family A
Family A was identified to have multiple gene mutations. Genetic analysis was performed for seven family members, and six were identified to have mutations. The proband was 23 years old and diagnosed with DM 4 years ago. Her last HbA1c was 5.3% with diet alone. The proband’s mother was 46 years old and had DM for 15 years with HbA1c of 6.8% under both insulin and oral anti-diabetic (OAD) treatment. The mother had diabetic polyneuropathy and coronary artery disease. A sibling of the proband, diagnosed at age 16, had DM for 5 years. The HbA1c value was 6.5% with insulin and OAD treatment. Another sibling was diagnosed at age 17 and had DM for 8 years, and his HbA1c value was 4.8% with insulin treatment alone. The maternal aunt of the proband was 42 years old and had DM. Another maternal aunt of the proband was 40 years old without a DM diagnosis.
KLF11 mutation
A new mutation was identified on the KLF11 gene in a 57-year-old woman diagnosed with DM at age 10. Her last HbA1c value was 8.0% with OAD and insulin treatment. She had diabetic proliferative retinopathy and diabetic polyneuropathy. There was a family history of MODY with typical three generations.
NKX2-2 mutation
A new mutation was identified on the NKX2-2 gene in a 43-year-old patient diagnosed with DM 6 years ago. The HbA1c value was 7.5% with OAD treatment alone. There was a typical family history of MODY.
HNF4A mutation
One patient had HNF4A mutation. Simultaneous GLIS3 mutation was also identified. The patient was 23 years old and diagnosed with DM 7 years ago. The HbA1c value was 10.1% with OAD treatment alone. There was a typical family history of MODY.
ABCC8 mutation
All seven patients with the ABCC8 mutation also had a mutation in another gene. Three different mutations were identified for the ABCC8 gene, and two of these were new mutations.
INR mutation
A new mutation was identified for the INR gene in a 4-year-old patient with DM diagnosis at age 2. She was receiving basal and bolus insulin treatment, and the last HbA1c value was 7.7%. The mother or father had no DM.
GLIS3 mutation
One patient had a new mutation on the GLIS3 gene. However, this patient was considered type 1 DM based on clinical findings and undetectable C-peptide levels.
Mutation-negative MODY
Nine patients did not have mutations but with clinical features that met the MODY criteria. The mean age was 40 ± 11.5 years, and the mean age at diagnosis was 29.4 ± 7.9 years. The mean of the last HbA1c value was 10.0% ± 3.1%, and the mean body mass index was 26.6 ± 3.0 kg/m2.
Discussion
Our study investigated the results of a panel containing genes that might cause monogenic DM from a single centre in Turkey. Of the 36 patients investigated, 19 different mutations were detected, 7 of which were novel. A total of 20 patients fully met the clinical criteria for MODY, and 11(%55) of these patients had mutations. A recent study in Turkey reported a mutation identification rate of 65% in 43 children [14]. However, the mutation identification rate may vary according to the inclusion criteria for the analysis. Expanding the criteria used to request genetic analysis may decrease the positivity rates to 10–20% [13]. A recent multi-centre study identified a lower mutation positivity rate of 17.6% in 204 cases from Mediterranean countries [15].
Although genetic studies on monogenic DM have increased, novel genes could not be identified except for the neonatal period. However, the allelic spectrum of the known genes continues to expand. Some of the new variants detected do not meet all of the classic MODY criteria, adding a new perspective to MODY [16]. These variants suggest that monogenic diabetes, and some other types of diabetes may share a common genetic spectrum. We also identified variants in four diabetic cases that did not fully meet the MODY criteria. However, these variants may be benign or de novo mutations, as well as they can also be considered to support this new perspective. One patient with HNF1A mutation was interpreted as type 1 DM because of C-peptide negativity and anti-GAD antibody positivity. One patient with GLIS3 mutation had intense insulin requirements and low C-peptide level. Another pediatric case with INSR mutation was accepted as type 1 DM because of a lack of family history and clinical findings. One patient had ABCC8 and GCK mutation; however, indeterminate MODY was recognised due to a lack of family history.
Of the mutations identified in our study, four were GCK gene mutations. GCK mutations cause MODY type 2 and are associated with mild DM. Elevated fasting blood sugar can be detected since birth; however, no significant progression is expected over the years [9]. Pharmacological treatment for those with GCK mutations may not help in lowering blood sugar [18]. The diagnoses were made during childhood in this study, and the serum glucose levels were not very high. Patients had mild progression of DM and were managed with diet and lifestyle changes. Two patients (mother and daughter; P7, P8) had mild DM findings despite having c.91A>T non-sense mutations. Similarly, those with heterozygous non-sense or frameshift mutations of the GCK gene may have mild progression of clinical DM [18].
P12, with family history and mild serum glucose elevation, meet the typical MODY type 2 diagnosis. The identified GCK c.895G>C variant was previously reported as pathological.
P15 was the case that identified the GCK and ABCC8 mutations together mentioned above but was considered indeterminate MODY due to the absence of family history. The identified GCK c.895G>C variant was previously reported as pathological. The ABCC8 c.1259T>G variant was interpreted as pathogenic in most in silico analyses, but this patient’s clinical course was consistent with MODY type 2.
HNF1A mutation was identified in three patients. HNF1A mutations cause the frequently encountered MODY type 3 [19]. One of these patients (P17) had been interpreted as type 1 DM due to C-peptide negativity and positive anti-GAD antibody. This patient had HNF1A c.1108-27C>T mutation in the intronic region, which was considered benign. The other two patients were a mother and daughter (P9, P10) with HNF1A c.1108-27C>T intronic region mutation. One of these patients had pre-DM emerging only with weight gain. These cases did not fully meet the DM diagnostic criteria; analysis was requested because of a family history of severe DM. These two cases were considered in the non-DM group, and this mutation was interpreted as benign because of the lack of obvious DM diagnosis.
We identified the KLF11 gene mutation, a rare cause of MODY, in P11. KLF11 regulates PDX1 transcription in pancreatic beta cells [9]. Our 57-year-old patient had been diagnosed with DM at age ten and did not have adequate blood sugar control despite both OAD and intensive insulin treatment. She also had diabetic neuropathy and proliferative retinopathy. Very few cases were related to KLF-11 mutation [20]. A study screening patients with obesity reported that one patient had a KLF11 variant. The 16-year-old patient was reported to have hyperlipidemia accompanied by mild DM progression [21]. Considering that our patient had complicated DM, KLF11 mutations might be associated with severe disease progression.
In family A, mutations were identified in six of seven patients analysed, and five had DM (P1, P2, P3, P5, P6). Interpreting the phenotype-genotype correlation was difficult because multiple mutations were identified in this family. This family may be considered PDX1 mutation associated with MODY type 4, but family analysis showed that patient–healthy individual segregation was partly consistent with the PDX1 c.246T>C variant. This mutation was assessed as VUS. All six family members with the mutation had c.246T>C synonymous variant on the PDX1 gene. The PDX1 gene is an important transcription factor gene for pancreas development and beta-cell maturation. MODY type 4 families are scarce. Generally, affected family members are diagnosed at a young age, and the majority may require insulin [10]. Autoantibodies against PDX1 may also cause type 1 DM [22]. The identified PDX1 variant was not found in the Clinvar database, and it was assessed as likely benign in the Varsome database. However, it was predicted to affect the splice region in the Human Splicing Finder database.
The ABCC8 c.2116+39T>A variant identified in five members of the family A (P1, P2, P4, P5, and P6) was in the intronic region and not reported in the Clinvar database. This gene codes sulfonylurea receptor 1 (SUR1) subunits in the ATP-susceptible potassium channel and thus plays a role in regulating insulin secretion [23]. ABCC8 mutations were first associated with MODY type 12 in 2012, and it may also cause NDM [24]. MODY type 12 families may be obese and overweight. They respond well to sulfonylurea treatment [25]. In this study, the family members had average weight. Additionally, it was predicted not to affect the splice region in the Human Splicing Finder database, and this mutation was assessed as VUS.
The GLIS3 c.1056G>C variant is also identified in four family A members. GLIS3 is a member of the GLIS family of Krüppel-like zinc finger transcription factors. It is dominantly expressed in the pancreas, thyroid, and kidneys. Mutations in GLIS3 cause an NDM syndrome characterised by congenital hypothyroidism and polycystic kidney. Variants are reported at high rates among those with type 1 DM and type 2 DM. It works with the PDX1 gene in insulin gene transcription control [26]. However, this GLIS3 gene mutation was not detected in P2, who has moderate DM. Clinvar database reported this mutation in a neonate with DM and congenital hypothyroidism and interpreted as VUS. This variant was not predicted to affect the splice region in the Human Splicing Finder database, and this mutation was assessed as VUS.
Another variant identified in family A was a c.1103A>T missense variant on the ZFP57 gene. The ZFP57 gene codes a transcription factor necessary to adequately sustain methylation during early embryonic development. ZFP57 mutations are reported to be associated with multiple locus-imprinting disorders in transient NDM [27]. However, the phenotype effect of this gene on the juvenile and adult periods is unknown. The ZFP57 c.1103A>T variant was reported as VUS in the Varsome database and was predicted to affect the splicing region in the Human Splicing Finder database. However, this variant was not detected in the proband but was detected in the non-diabetic P4. It was interpreted as benign in most in silico analyses.
Additionally, two of the five family A members with DM were identified to have GLUD1 c.200T>C variant. GLUD1 ensures the synthesis of glutamate dehydrogenase, a mitochondrial matrix enzyme included in the tricarboxylic acid cycle. Mutations were associated with familial hyperinsulinemic hypoglycemia [28]. Most in silico analyses assessed this mutation as pathogenic.
In family A, possible mutations on other unexamined genes may be associated with DM development. Multiple mutations may also have an additive effect on DM progression [29].
HNF4A c.724G>A mutation was identified in P14. HNF4A encodes a nuclear transcription factor. Pathogenic mutations are associated with MODY type 1. HNF4A mutations comprised 7.5% of MODY cases in a new study from Mediterranean countries, including Turkey [15]. MODY type 1 cases were generally diagnosed in the young adult period, progressed over time, and may require multiple OAD or insulin treatments [30]. P14 was diagnosed in the adolescent period and had high HbA1c levels despite intense treatment. The identified mutation was interpreted as pathogenic in most in silico analyses. This patient also had c.1585C>G missense mutation on the GLIS3 gene. This variant was interpreted as pathological in most in silico analyses.
P16 had GLIS3 c.893C>A variant. This variant is generally interpreted as VUS, while it was assessed as benign in the Clinvar database. This patient was diagnosed with type 1 DM due to intense insulin treatment, C-peptide negativity, and lack of family history.
P13 was identified to have c.*73G>A intronic region mutation on the NKX2-2 gene. This gene is a transcription factor gene related to pancreas development. Mutations are associated with NDM [31]. This patient met the MODY clinical criteria; however, he was diagnosed with DM in adulthood. This variant is interpreted as LB; however, it is in the 3′ untranslated region and may have acted by altering the gene’s RNA stability and expression level.
P18, without DM, had ABCC8 c.1332+4delC intronic region deletion. Some cases are assessed as benign and VUS in the Clinvar database. According to the Human Splicing Finder prediction algorithm, this mutation is predicted to affect splicing. However, it was not considered to affect DM development because P18 had no DM. This patient was also identified to have GLUD1 c.1568 G>A missense mutation. A case of hyperammonemia hyperinsulinemia syndrome was reported in the Clinvar database. However, the clinical findings of this patient were not consistent with hypoglycemia.
P19 was identified to have INSR c.1777G>T missense mutation. INSR gene mutations may cause severe insulin-resistant syndromes and hyperinsulinemic hypoglycemia [32]. However, no clinical case has been reported. P19 was diagnosed with DM at age 2 years and has used insulin since then. Her mother and father did not have DM. Clinical findings were not consistent with insulin resistance. The C-peptide level was slightly low, but autoantibody tests could not be obtained. The diagnosis was not definite for this patient, but she was considered to have type 1 DM.
Although we analysed all 23 MODY-related genes in all cases, our major limitation was the limited number of cases from a single centre. Another limitation is that mutations in the deep intronic regions of the regulatory sequences and large copy changes could not be detected due to the limitation of our method.
Conclusion
This study analysed data from a single centre in Turkey and aimed to contribute to the phenotype-genotype correlation in monogenic DM. We detected some new mutations and reviewed the clinical findings of previously known mutations. We found that a mutation can be detected in only half of clinically diagnosed MODY patients, and some mutation carriers do not have all of the classic MODY traits. These findings highlight a broad clinical and genetic spectrum of monogenic DM. Detecting new genes and new mutations is critical for a better understanding this form of DM and may improve the individualised treatment approach.
Data Availability
The data are available from the corresponding authors.
References
Irgens HU, Molnes J, Johansson BB, et al. Prevalence of monogenic diabetes in the population-based Norwegian Childhood Diabetes Registry. Diabetologia. 2013;56(7):1512–9. https://doi.org/10.1007/s00125-013-2916-y.
Fendler W, Borowiec M, Baranowska-Jazwiecka A, et al. Prevalence of monogenic diabetes amongst Polish children after a nationwide genetic screening campaign. Diabetologia. 2012;55(10):2631–5. https://doi.org/10.1007/s00125-012-2621-2.
Shepherd M, Shields B, Hammersley S, et al. Systematic population screening, using biomarkers and genetic testing, identifies 2.5% of the UK pediatric diabetes population with monogenic diabetes. Diabetes Care. 2016;39(11):1879–88. https://doi.org/10.2337/dc16-0645.
Gat-Yablonski G, Shalitin S, Phillip M. Maturity onset diabetes of the young–review. Pediatr Endocrinol Rev PER. 2006;3(Suppl 3):514–20.
Iafusco D, Massa O, Pasquino B, et al. Minimal incidence of neonatal/infancy onset diabetes in Italy is 1:90,000 live births. Acta Diabetol. 2012;49(5):405–8. https://doi.org/10.1007/s00592-011-0331-8.
Al-Agha AE, Ahmad IAA, Basnawi FAA, Al-Nasser EYF. A recessive mutation in the insulin gene in neonatal diabetes. Turk J Endocrinol Metab. 2015;19(1):25–7. https://doi.org/10.4274/tjem.2326.
Lemelman MB, Letourneau L, Greeley SAW. Neonatal diabetes mellitus: an update on diagnosis and management. Clin Perinatol. 2018;45(1):41–59. https://doi.org/10.1016/j.clp.2017.10.006.
Tattersall RB, Fajans SS. A difference between the inheritance of classical juvenile-onset and maturity-onset type diabetes of young people. Diabetes. 1975;24(1):44–53. https://doi.org/10.2337/diab.24.1.44.
Anık A, Çatlı G, Abacı A, Böber E. Maturity-onset diabetes of the young (MODY): an update. J Pediatr Endocrinol Metab JPEM. 2015;28(3–4):251–63. https://doi.org/10.1515/jpem-2014-0384.
Aarthy R, Aston-Mourney K, Mikocka-Walus A, et al. Clinical features, complications and treatment of rarer forms of maturity-onset diabetes of the young (MODY) - A review. J Diabetes Complicat. 2021;35(1): 107640. https://doi.org/10.1016/j.jdiacomp.2020.107640.
Thanabalasingham G, Pal A, Selwood MP, et al. Systematic assessment of etiology in adults with a clinical diagnosis of young-onset type 2 diabetes is a successful strategy for identifying maturity-onset diabetes of the young. Diabetes Care. 2012;35(6):1206–12. https://doi.org/10.2337/dc11-1243.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K. Rehm HL Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
Owen KR. Monogenic diabetes: old and new approaches to diagnosis. Clin Med Lond Engl. 2013;13(3):278–81. https://doi.org/10.7861/clinmedicine.13-3-278.
Ağladıoğlu SY, Aycan Z, Çetinkaya S, et al. Maturity onset diabetes of youth (MODY) in Turkish children: sequence analysis of 11 causative genes by next generation sequencing. J Pediatr Endocrinol Metab JPEM. 2016;29(4):487–96. https://doi.org/10.1515/jpem-2015-0039.
Vaxillaire M, Bonnefond A, Liatis S, et al. Monogenic diabetes characteristics in a transnational multicenter study from Mediterranean countries. Diabetes Res Clin Pract. 2020;171:108553. https://doi.org/10.1016/j.diabres.2020.108553.
Owen KR. Monogenic diabetes in adults: what are the new developments? Curr Opin Genet Dev. 2018;50:103–10. https://doi.org/10.1016/j.gde.2018.04.006.
Tattersall R. Maturity-onset diabetes of the young: a clinical history. Diabet Med J Br Diabet Assoc. 1998;15(1):11–4. https://doi.org/10.1002/(SICI)1096-9136(199801)15:1%3c11::AID-DIA561%3e3.0.CO;2-0.
Ping Xiao Y, Hua XuX, Lan Fang Y, et al. GCK mutations in Chinese MODY2 patients: a family pedigree report and review of Chinese literature. J Pediatr Endocrinol Metab JPEM. 2016;29(8):959–64. https://doi.org/10.1515/jpem-2015-0354.
Valkovicova T, Skopkova M, Stanik J, Gasperikova D. Novel insights into genetics and clinics of the HNF1A-MODY. Endocr Regul. 2019;53(2):110–34. https://doi.org/10.2478/enr-2019-0013.
Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V, et al. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci U S A. 2005;102(13):4807–12. https://doi.org/10.1073/pnas.0409177102.
Kleinberger JW, Copeland KC, Gandica RG, et al. Monogenic diabetes in overweight and obese youth diagnosed with type 2 diabetes: the TODAY clinical trial. Genet Med Off J Am Coll Med Genet. 2018;20(6):583–90. https://doi.org/10.1038/gim.2017.150.
Li S-W, Koya V, Li Y, et al. Pancreatic duodenal homeobox 1 protein is a novel beta-cell-specific autoantigen for type I diabetes. Lab Investig J Tech Methods Pathol. 2010;90(1):31–9. https://doi.org/10.1038/labinvest.2009.116.
Patch AM, Flanagan SE, Boustred C, Hattersley AT, Ellard S. Mutations in the ABCC8 gene encoding the SUR1 subunit of the KATP channel cause transient neonatal diabetes, permanent neonatal diabetes or permanent diabetes diagnosed outside the neonatal period. Diabetes Obes Metab. 2007;9(Suppl 2):28–39. https://doi.org/10.1111/j.1463-1326.2007.00772.x.
Bowman P, Flanagan SE, Edghill EL, et al. Heterozygous ABCC8 mutations are a cause of MODY. Diabetologia. 2012;55(1):123–7. https://doi.org/10.1007/s00125-011-2319-x.
Lin L, Quan H, Chen K, Chen D, Lin D, Fang T. ABCC8-related maturity-onset diabetes of the young (MODY12): a report of a Chinese family. Front Endocrinol. 2020;11:645. https://doi.org/10.3389/fendo.2020.00645.
Wen X, Yang Y. Emerging roles of GLIS3 in neonatal diabetes, type 1 and type 2 diabetes. J Mol Endocrinol. 2017;58(2):R73–85. https://doi.org/10.1530/JME-16-0232.
Touati A, Errea-Dorronsoro J, Nouri S, et al. Transient neonatal diabetes mellitus and hypomethylation at additional imprinted loci: novel ZFP57 mutation and review on the literature. Acta Diabetol. 2019;56(3):301–7. https://doi.org/10.1007/s00592-018-1239-3.
Ninković D, Sarnavka V, Bašnec A, et al. Hyperinsulinism-hyperammonemia syndrome: a de novo mutation of the GLUD1 gene in twins and a review of the literature. J Pediatr Endocrinol Metab JPEM. 2016;29(9):1083–8. https://doi.org/10.1515/jpem-2016-0086.
Forlani G, Zucchini S, Rocco AD, et al. Double heterozygous mutations involving both HNF1A/MODY3 and HNF4A/MODY1 genes: a case report. Diabetes Care. 2010;33(11). Accessed January 16, 2021. https://cyberleninka.org/article/n/1249234
Warncke K, Kummer S, Raile K, et al. Frequency and characteristics of MODY 1 (HNF4A Mutation) and MODY 5 (HNF1B Mutation): analysis from the DPV database. J Clin Endocrinol Metab. 2019;104(3):845–55. https://doi.org/10.1210/jc.2018-01696.
Auerbach A, Cohen A, Ofek Shlomai N, et al. NKX2-2 mutation causes congenital diabetes and infantile obesity with paradoxical glucose-induced ghrelin secretion. J Clin Endocrinol Metab. 2020;105(11). https://doi.org/10.1210/clinem/dgaa563
Sethi A, Foulds N, Ehtisham S, et al. Heterozygous insulin receptor (INSR) mutation associated with neonatal hyperinsulinemic hypoglycaemia and familial diabetes mellitus: case series. J Clin Res Pediatr Endocrinol. 2020;12(4):420–6. https://doi.org/10.4274/jcrpe.g.,alenos.2019.2019.0106.
Acknowledgements
Preparation for publication of this article is supported by the Society of Endocrinology and Metabolism of Turkey.
Funding
No funding was received for conducting this study.
Author information
Authors and Affiliations
Contributions
E.K.,G.G.O and U.A. collected the data. E.K, E.S.S and U.A. prepared Tables and Figures. E.K, F.S and E.S.S designed the study, interpreted the data, and wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Statement of ethics
Ethics permission was granted by Canakkale 18 Mart University Clinical Research Ethics Committee dated 11.11.2020, decision no. 2020-13. This research is strictly complying with the guidelines for human studies and was conducted.
Conflict of interest
The authors declare no competing interests.
Ethical considerations
Ethics permission was granted by University Clinical Research Ethics Committee dated 11.11.2020, decision no. 2020-13.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Karakilic, E., Saygili, E.S., Silan, F. et al. New results for monogenic diabetes with analysis of causative genes using next-generation sequencing: a tertiary centre experience from Turkey. Int J Diabetes Dev Ctries 42, 703–712 (2022). https://doi.org/10.1007/s13410-021-01027-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13410-021-01027-2