
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is notorious for its resistance to various treatment modalities. The genetic heterogeneity of PDAC, coupled with the presence of a desmoplastic stroma within the tumor microenvironment (TME), contributes to an unfavorable prognosis. The mechanisms and consequences of interactions among different cell types, along with spatial variations influencing cellular function, potentially play a role in the pathogenesis of PDAC. Understanding the diverse compositions of the TME and elucidating the functions of microscopic neighborhoods may contribute to understanding the immune microenvironment status in pancreatic cancer. As we delve into the spatial biology of the microscopic neighborhoods within the TME, aiding in deciphering the factors that orchestrate this intricate ecosystem. This overview delineates the fundamental constituents and the structural arrangement of the PDAC microenvironment, highlighting their impact on cancer cell biology.
Highlights
-
• Cancer cells’ intrinsic traits shape the microenvironment, causing dynamic changes in genetic and phenotypic features as the tumor grows and spreads.
-
• Exploring how cellular components and molecular features impact cancer biology warrants further investigation into the intricate interactions within the TME
-
• The spatial relationships between individual cellular and acellular components in PDAC hold the potential to provide novel insights into the dynamic and complex functions of PDAC desmoplasia,
-
• Intratumoral sub-TMEs exhibit distinct regional immune compositions, and typical spatial subregion-TLS highlight the spatial heterogeneity of the TME

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Pancreatic ductal adenocarcinoma (PDAC) stands out as an extremely aggressive cancer with significant mortality, ranking 12th in incidence and 7th in mortality globally [1, 2]. PDAC is notorious for its strong tendency to metastasize, with approximately 80% of newly diagnosed patients presenting at an advanced stage [3]. Unfortunately, the response rate to single-agent chemotherapy in advanced patients is unsatisfactory, leading to an overall 5-year survival rate of less than 10% [1,2,3]. Despite ongoing efforts, therapeutic options for PDAC remain limited, and chemotherapy stands as the most effective treatment modality [4]. While certain subgroups of patients may exhibit promising responses to chemotherapy, the development of therapeutic resistance poses a considerable obstacle to achieving long-term survival [3, 5]. Currently, there is no effective first-line targeted therapy except chemotherapy [4, 6]. However, the diverse frequencies of different KRAS missense mutations in PDAC have spurred investigations into targeted drugs tailored to specific KRAS mutation sites [5]. Nonetheless, only a subset of PDAC patients experiences favorable outcomes from cytotoxic chemotherapy, molecular-targeted agents, and immune-based therapies [6, 7]. Patients harboring BRCA1/2 mutations experienced marginal benefits from the concurrent administration of poly (ADP-ribose) polymerase (PARP) inhibitors and platinum-based chemotherapeutic agents [8]. Yet, finding effective protocols for PDAC remains a challenging task [9].
The principal constituents of the PDAC stroma predominantly create an immunosuppressive microenvironment dominated by extracellular matrix (ECM) proteins, tumor vasculature, fibroblasts, and suppressive immune cell infiltrations [6, 10]. Extensive preclinical and clinical investigations have thoroughly examined the bidirectional interactions occurring between transformed epithelial cells and the encompassing microenvironment [3, 5, 6]. It is noteworthy that the TME can vary among patients with the same cancer, and different cancer types can exhibit distinct TME [4, 11, 12]. Furthermore, according to the analysis of cellular interaction from the primary PDAC tumor and metastatic tumors, it further indicates that the absence of interactions between tumor cells and immune cells in metastatic tissues contributes to the establishment of an immunosuppressive microenvironment [13]. As anatomical site-dependent transcription may be provoked by local cues when hematopoietic immune cells reach the tissue, or it might already be epigenetically programmed during tissue development [14].
As the tumor advances to a more malignant stage, pancreatic ductal cells no longer constitute the primary element in the tumor microenvironment. Cancer-associated fibroblasts (CAFs) play a pivotal role in shaping the microenvironment of PDAC, as CAFs serve as central hubs within the stromal compartments of PDAC [15]. CAFs notably contribute by synthesizing the extracellular matrix (ECM) and engaging with cancer and immune cells [16,17,18]. Moreover, immune cells migrate within the tumor, indicating the reaction of the body’s immune system to the evolving tumorous conditions [19]. As malignant progression occurs, morphologically and compositionally diverse sub-TMEs may represent unique ecosystems within tumors [20,21,22]. Additionally, leukocyte subpopulations show differential enrichment between PDACs and across various tissue regions [10]. Quantifying the degree of leukocyte infiltration in PDAC offers an additional means of categorizing tumors into subtypes characterized by hypoinflammation, myeloid enrichment, and lymphoid enrichment [10]. The development of intratumoral and inter-tumoral heterogeneity is a hallmark of malignant TME progression [23, 24].
Furthermore, comprehensive insights obtained from atlases with multiple parameters are anticipated to steer future inquiries into the fundamental biological mechanisms that drive malignant transformation. Conventional therapies, such as chemotherapy and radiotherapy, bring about alterations in the TME, thus influencing their therapeutic impacts indirectly on cancer cells and either augmenting or disrupting responses [25, 26]. Nonetheless, it’s crucial to acknowledge that both adaptive and intrinsic resistance can serve as obstacles to therapies targeting the TME [5, 27]. Additionally, the immunosuppressive microenvironment poses significant challenges to the development of immune-based therapies [6]. The critical factors governing tumor growth, metastatic potential, primary resistance, and poor long-term survival remain to be fully elucidated [3, 6, 28].
2 Cancer cell-intrinsic characteristic drive microenvironment formation
The spatial evolution of cancer is significantly influenced by cancer cell-intrinsic characteristics, which play a pivotal role in tumor growth, invasion, metastasis, and response to therapy [6, 29]. Genomic instability often continues following cancer dissemination, leading to continuous, parallel, and sometimes convergent evolution across various metastases [30, 31]. As the tumor grows and spreads within its microenvironment, dynamic changes occur in the genetic and phenotypic features of cancer cells [29]. In primary tumors, mutations in driver genes typically provide a survival advantage, stemming from the genetic instability of clonal and subclonal tumor cells, which underpin the evolution of the tumor and its spatiotemporal heterogeneity [30, 31]. Tumorigenesis results from the combined and cumulative dysregulation of various genetic and non-genetic processes [31]. Due to the inherent genetic instability within the tumor genome, many tumorigenic events occur randomly throughout the progression of the disease [5]. Throughout the development of PDAC, the infiltration of immune cells exerts complex influences on the disease’s pathogenesis and progression, with varying immune selection pressures shaping the evolution of cancer cell clones [32]. These random occurrences are crucial for the development of heterogeneous immune microenvironments, which vary in both spatial and temporal dimensions. Concurrently, this genetic diversity influences the antigenic profile of the tumors. These alterations occur during the process of carcinogenesis [5]. Meanwhile, there already reported the TME applies selective pressure on the clonal evolution of lung tumors [33], indicating detailed spatial information is crucial to grasp how cell interactions and physical barriers affect tumor evolution.
The progression of PDAC is driven by various genomic and transcriptomic changes, including the loss of tumor suppressor genes and activation of oncogenes [3, 22]. Molecular phenomena, encompassing somatic mutations, chromosomal rearrangements, variations in copy numbers, and modifications in epigenetic patterns, play a role in fostering tumor diversity and advancing the transition from pancreatic intraepithelial neoplasia (PanIN) to PDAC [5, 34]. Genetic alterations and epigenetic anomalies within tumor cells can transform the TME, which were evidenced by the loss of SETD2 in tumor cells activates BMP2 signaling through an abnormal increase of H3K27Ac, which causes CAFs to differentiate into a lipid-rich phenotype. These lipid-abundant CAFs subsequently promote tumor growth by supplying lipids for mitochondrial oxidative phosphorylation through the ABCA8a transporter [35]. Among the various factors that regulate the TME, it is important to consider the multifactorial nature of TME modulation, especially the dual influence of both tumor intrinsic properties and the microenvironment in shaping each other [3, 34]. Notably, KRAS, TP53, CDKN2A, and SMAD4 are among the most frequently mutated genes in PDAC, with approximately 90% of PDAC cases having an oncogenic KRAS mutation. Moreover, oncogenic factors like KRAS mutations and the presence of p53 mutations have been demonstrated to play a role in the evolutionary changes within the stromal environment of PDAC [36, 37]. These mutations are associated with the activation of multiple downstream immunosuppressive pathways [38, 39] (Fig. 1). Oncogenic KRAS signaling, which is non-cell autonomous transforms pancreatic fibroblasts by triggering an inflammatory gene expression program. Consequently, these fibroblasts turn into centers of extracellular signaling and the primary producers of cytokines that drive the polarization of protumorigenic macrophages [40]. A comprehensive whole-genome analysis further unveiled that molecular subtypes are associated with distinct copy number aberrations in genes, including but not limited to mutant KRAS and GATA6 [41]. Recently, a classification system based on the characteristics of cancer cells and the TME has delineated five distinct PDAC subtypes: stroma-activated, desmoplastic, pure classical, pure basal-like, and immune classical subtypes [42]. These subtypes reflect the heterogeneity and complexity of PDAC, influenced by various factors within both cancer cells and the surrounding microenvironment [43]. Currently, the stratification in pancreatic cancer is basic and does not guide clinical management or therapy development. A refined TME classification system for PDAC, utilizing advanced techniques such as single-nucleus RNA sequencing and digital spatial profiling (DSP), provides profound molecular and cellular insights. This system delineates PDAC into three unique multicellular communities, each defined by distinct combinations of malignant, fibroblast, and immune subtypes: classical, squamoid-basaloid, and treatment-enriched. Such a classification may establish a framework for stratifying clinical trials and designing targeted therapies aimed at specific cellular interactions and phenotypes within the tumor microenvironment [44].

Cancer cell-intrinsic characteristic drive microenvironment formation in PDAC. Oncogenic factors are linked to the initiation of several downstream pathways that exert immunosuppressive effects, contributing to the evolutionary transformations observed in the milieu of PDAC
The magnitude of PDAC intrinsic characteristics includes genetic alterations, metabolic reprogramming, epigenetic alterations, and signal release, critical determinants that help malignant cells escape from an effective adaptive immune response [4, 5, 38]. In preclinical studies involving mice with Kras mutations, early pancreatic tumor progression can be accelerated by tissue damage, along with the release of the cellular cytokine interleukin 33 (IL-33). Moreover, recombinant IL-33 has been shown to induce chromatin dysregulation and expedite Kras-driven development of PanIN, underscoring the gene-environment interactions and epigenetic programs that initiate tumorigenesis [45].
No specific intrinsic or extrinsic biomarker guidelines have been validated for selecting patients. Through integrated genomic analysis, mutated genes that are involved in acquired immune suppression have been identified in certain tumors, variations in the molecular progression of pancreatic cancer subtypes, and unveil new potential targets for therapeutic intervention [38, 43]. Analysis of PDAC genomic data suggests correlations with histopathological characteristics, but it is crucial to note that only a fraction of pancreatic cancers exhibit immunocompetence. As an illustration, various subgroups of tumor cells exhibit distinctive traits related to proliferation, KRAS signaling, cell stress, and epithelial-to-mesenchymal transition. This differentiation is achieved by delineating mutations and copy number changes that set apart these tumor populations from normal and transitional cells, like acinar-to-ductal metaplasia and PanIN. Furthermore, employing pathology-guided deconvolution of spatial transcriptomic data enabled the identification of tumor and transitional subgroups marked by distinctive histological characteristics [46].
In genetically engineered mouse models, PDAC cells with oncogenic Kras signaling express granulocyte-macrophage colony-stimulating factor (GM-CSF), causing the suppression of antigen-specific T cells and the promotion of immunosuppressive myeloid-derived suppressor cell recruitment [6, 47]. Furthermore, the oncogenic Kras-induced GM-CSF accelerates the process of pancreatic neoplasia [6, 47, 48] (Fig. 1). TP53 mutations or deletions within cancer cells participate in establishing an environment that suppresses the immune response, leading to a heightened accumulation of cells originating from the bone marrow [10]. Certain PDACs, particularly those harboring TP53 alterations, exhibit modest elevations in neutrophil/eosinophil density [10]. Changes in the intrinsic characteristics of cancer cells can lead to modifications in secreted factors, cell surface receptors or ligands, extracellular vesicle (EV) content and abundance, as well as nutrient utilization [6, 29, 49]. Moreover, fibroblast reprogramming begins in the initial stages of carcinogenesis, driven by Janus kinase (JAK)/signal transducer and activator of transcription 3 (STAT3) signaling, which activate inflammatory gene expression program, encompassing various cytokines that enhance the tumor-promoting activities of myeloid cells in the pancreas. Therefore, investigating strategies to block fibroblast reprogramming during carcinogenesis could be crucial to prevent, and potentially reverse, KRAS-driven carcinogenesis [40]. The KRASG12D mutation occurs in about half of all PDAC.In a mouse model, the specific and effective inhibitors targeting KRAS mutations could halt the growth of early-stage PDAC, enhance the presence of CD8 + effector T cells within the tumor, diminish myeloid cell infiltration, and alter the behavior of cancer-associated fibroblasts [50]. Therefore, examining the tumor immune microenvironment through the lens of mutated genes associated with immune suppression reveals key mechanisms of tumor progression. As inhibitors targeting KRAS advance into clinical trials, it is crucial to comprehend the biology of KRAS- mutated genes and identify biomarkers that can predict therapeutic outcomes.
3 Overview of the tumor microenvironment in PDAC
The stroma of PDAC displays a notably significant mesenchymal section, setting it apart from other solid tumors [3, 51]. The pancreatic tumor microenvironment comprises numerous fibroblast populations, a compact extracellular matrix, and an underdeveloped vascular network, all of which collectively act as a barrier, protecting cancer cells from an effective cytotoxic immune response [4, 6, 52]. With the tumor’s growth, it brings about a range of physiological mechanisms, encompassing tumor cell proliferation, angiogenesis, and the influx of immune cells [6]. Interactions between cancer cells and immune cells tightly govern the infiltration of immune cells into the tumor site [6, 34]. Cancer cells have the ability to manipulate the microenvironment, promoting the localization of tumor-promoting immune cells while evading anti-tumor immune responses [34, 52]. Consequently, the microenvironment surrounding cancer cells plays a crucial role in dictating tumor growth, metastatic potential, and treatment resistance [3]. The crosstalk between immune cells and extracellular matrix components can uncover mechanisms of cancer pathogenesis and therapeutic resistance [53, 54]. Significant progress has been made in comprehending the stroma of PDAC, while no stroma-targeting treatments have yet been approved, many potential therapies are in the stages of preclinical or clinical development [54]. Additionally, the potential use of tissue biomarkers to identify the targeted stromal components may enhance patient selection and increase the success rates of clinical trials (Fig. 2).

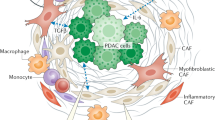
The crosstalk between various principal components of the intratumoral microenvironment in PDAC. Within the microenvironment of pancreatic cancer, immunosuppressive cells contribute to the evasion of immune surveillance by through the expression or production of diverse factors, including IL-35, IL-6, IL-10, and TGFβ,which collectively inhibit CD8 + T cells
Effector immune cells, such as CD8 + or CD4 + T cells, mount responses against tumor cells [3]. The TME of PDAC exhibits a predominant presence of infiltrating CD4 + T cells, while surprisingly, only a small number of patients’ tumor samples showed infiltration of CD8 + T cells [7]. PDAC cancer cells release pro-inflammatory cytokines and chemokines, recruiting innate and adaptive immune cells to the tumor site and inducing an inflammatory response with immune cell infiltration [11] (Fig. 2). Considering the diverse influence of various leukocyte subsets on tumor biology in PDAC, the tumor microenvironment is enriched with distinct subsets of tumor-infiltrating leukocytes, including T cells, B cells, macrophages, and myeloid cells, which encompass granulocytes, neutrophils, eosinophils, and other cell types [10, 34] (Table 1).
3.1 CD8 + T-cell
Within the PDAC microenvironment, a variety of CD8 + T-cell states can be observed, including intratumoral exhausted phenotypes with dysfunctional characteristics [10, 55]. Significantly, CD8 + T effector cells showed a noteworthy decrease in abundance within the tumor compared to the adjacent normal tissue [10]. Despite the presence of CD8 + effector T cells within the PDAC TME, they develop an exhausted and dysfunctional phenotype, suggesting that their cytotoxic potential is likely compromised [56]. Furthermore, peripheral CD8 T cells exhibited almost undetectable levels of LAG3 expression, while the majority of them expressed LAG3 within the TME. This suggests that CD8 T cell dysfunction is predominantly prevalent at the tumor site [56]. Additionally, immune exclusion, caused by physical barriers or other mechanisms restricting the ability of immune cells to reach and attack cancer cells, seems to dominate the progression of the disease [10, 57]. PDAC cells and the tumor microenvironment have the capability to produce diverse immunosuppressive factors, including interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), which exacerbate the inhibition of immune cell activity [3, 58] (Fig. 2). The presence of tumor-infiltrating CD8 + T cells is associated with a response to immunotherapy, while the CD8 + T cell-mediated anti-tumor immune responses appear to be under constraints in PDAC [59, 60]. However, the infiltration pattern of T cells in PDAC tumors isn’t widespread; instead, the spatial arrangement is concentrated and restricted to distinct clusters neighboring tumor cells, within the stromal context, or as integral components of tumor-associated lymphoid structures (TLS) [10] (Fig. 3). The optimal activation of CD8 + T cells required the expression of CD137, which was associated with activated effector T cell signatures and correlated with increased overall survival (OS) [61].

Spatial heterogeneity of TME and landscape of typical spatial subregion-TLS. Cancer-associated fibroblasts (CAFs) are critical components of the tumor stroma, which include myofibroblast CAFs (myCAFs), immunogenic CAFs (iCAFs), and antigen-presenting CAFs (apCAFs), contributing to extracellular matrix production. TLS contains other cell populations, such as dendritic cells (DCs), CD21 + follicular dendritic cells (FDCs), B cells, CD68 + macrophages, follicular reticular cells and CD83 + DCs
3.2 CD4 + T cells
CD4 + helper T cells have a significant impact on various immune cells, particularly in enhancing CD8 + T cell responses [62] (Fig. 2). The Th1 subtype of CD4 + T cells, in particular, plays an antitumor role by supporting antitumor cytotoxic CD8 + cells and B cells, as well as directly contributing to cancer cell death through the production of interferon-gamma (IFN-γ) and tumour necrosis factor-α(TNF-α) [63]. Conversely, the Th2 subtype releases anti-inflammatory signaling molecules. Mounting evidence suggests that CD4 + T cells could have a pivotal impact on shaping the effectiveness of ICB [62, 63]. Moreover, apart from antigen-presenting signals from DCs cells, MHC class II-expressing cancer-associated fibroblasts (CAFs) can also present antigens to CD4 + T cells [64] (Fig. 2). CD4 + T cells and their associated cells have the potential to modulate the immune response in pancreatic cancer [65]. CD4 + T cells are also enlisted and contribute to the advancement of the disease through the release of IL-17 [66]. The trajectory investigation unveiled that primary naive CD4 + T cells predominantly transform into Treg and CD4 + CD52 + subtypes within the context of PDAC. Furthermore, CD4 + CD52 + cells located at tumor sites exhibited elevated levels of CCL2, CCL5, and CCL20, which act as attractants for Tregs and macrophages to the TME [56] (Fig. 2). The spatial investigation provided further evidence for the presence of fatigued CD4 + T and regulatory T cells (Tregs) characteristics within the TME, especially highlighting a heightened occurrence of CD4 T cells expressing TIGIT both in the vicinity of and within the tumor mass [56].
A subset of CD4 + T cells known as Tregs holds significant immunosuppressive properties and plays a pivotal role in upholding immune equilibrium. They act as gatekeepers, regulating the immune response. Tregs exert their suppressive effects on effective antitumor immunity through various mechanisms, which can vary depending on the specific context and cues within the tumor microenvironment [67]. For instance, studies on mouse models have demonstrated that Treg cells can diminish the effectiveness of vaccines [68]. During the progression of pancreatic cancer, through the attenuation of mesothelial characteristics and the adoption of fibroblastic traits, mesothelial cells undergo a conversion into antigen-presenting cancer-associated fibroblasts (apCAFs), which were driven by interleukin-1 and TGF-β [16]. Notably, apCAFs directly interact with and induce naive CD4 + T cells to differentiate into Tregs in an antigen-specific manner [16] (Fig. 2). Depletion of Tregs prompted the development of inflammatory fibroblast subtypes, which in turn facilitated the infiltration of myeloid cells via CCR1, unveiling unforeseen communication between Tregs and fibroblasts [69]. Investigating Tregs-targeted cancer therapies poses challenges due to the critical role of Tregs in preventing auto-immunity.
3.3 B cells
B lymphocytes are crucial in coordinating humoral immunity. Furthermore, B cells are capable of infiltrating PDAC lesions, though their presence is notably less frequent in comparison to T cells [70] (Fig. 2). They can also be found within intratumoral TLS, where they promote T cell activation through antigen presentation (Fig. 2). When it comes to cancer, B cells have the ability to elicit anti-tumor effects by engaging in antibody-dependent cytotoxicity and triggering complement activation [71]. Among tumor-infiltrating B cell subsets, some promote the proliferation of cancer cells through the secretion of IL-35 [72]. Besides, B cells can also contribute to inflammation and immunosuppression by secreting anti-inflammatory and pro-angiogenic mediators, and activating complement, ultimately supporting tumor growth. B cells are key components and initiators of TLS, recruiting T cells, dendritic cells (DCs), and other cells to establish important positions within tumors (Fig. 3). TLS has been associated with a positive prognosis in various tumors [22, 73, 74]. B cells were observed scattered throughout the PDAC TME but decreased in number near tumor epithelial cells.Additionally, B cells within the PDAC exhibit a regulatory and immunosuppressive phenotype [56]. Based on single-cell RNA sequence data, the cell-cell communication analysis indicated that B cells expressing TGFB1 exhibited a tendency to establish more intimate interactions with CD8 + T cells and macrophages compared to other cell types present within the TME [56]. Besides the lower abundance of naive B cells in PDAC, the tumor-infiltrating naive B cells in PDAC displayed significantly elevated TGFB1 expression levels [56]. The specific locations and functions of TLSs will be discussed further in the following sections.
Different infiltration patterns and tumor microenvironments can induce B cell differentiation along different pathways, thereby affecting their overall effects. B cells’ antitumor function primarily relies on the secretion of antibodies against tumor-associated antigens (TAAs) and providing costimulatory signals to TAA-specific CD4 + T cells, thereby activating T cells (Fig. 2). B cells’ antibodies can directly kill tumor cells through antibody-dependent cytotoxicity (ADCC) and phagocytosis or activate T cell immune responses by presenting TAAs to T cells through DC cells. Additionally, B cells are capable of functioning as proficient antigen-presenting cells (APCs) to independently activate T cells [75,76,77].
3.4 MDSCs
Heterogeneous bone marrow cell populations, including dendritic cells, macrophages, granulocytes, early myeloid progenitor cells, and neutrophils, collectively referred to as myeloid-derived suppressor cells (MDSCs), possess robust immunosuppressive capacities [11, 78] (Fig. 2). The myeloid cell populations within the PDAC tumor microenvironment primarily lean towards subsets that facilitate the progression of the tumor [56]. Within the TME, myeloid cells exhibit considerable spatial and transcriptional diversity, and STAT3 plays a crucial role in reprogramming monocytes [25, 79]. Via paracrine signaling or direct interactions between cells, MDSCs can elicit suppressive impacts on T cells, NK cells, B cells, and DCs. Additionally, they promote tumor cell survival by producing various factors [11, 80, 81]. Furthermore, MDSCs can induce the generation of regulatory T cells (Tregs) in a cell-cell dependent manner, and the crosstalk between MDSCs and Tregs contributes to the immunosuppressive environment in PDAC [82]. CXCR2 signaling in the myeloid-derived suppressor cells plays a crucial role in promoting pancreatic tumorigenesis and is essential for pancreatic cancer metastasis [83]. The presence of MDSCs in the TME is regulated by EGFR/MAPK-dependent mechanisms that influence PD-L1 expression on tumor cells, facilitating immune evasion [84]. Since MDSCs display variations in their phenotypic, morphological, and functional characteristics, it is crucial to investigate their mechanisms and precisely understand their roles in disease-related pathophysiology, including tumor progression [85]. The CD14 + monocyte population showed elevated gene expression levels related to angiogenesis and immunoregulation, indicating their potential tumor-promoting role [56]. Therapeutically, the immunosuppressive activity of MDSCs can be redirected. In autochthonous PDAC, depleting granulocytic MDSCs (Gr-MDSCs) promotes the accumulation of intratumoral activated CD8 + T cells and induces tumor apoptosis, while also remodeling the tumor stroma [81].
3.5 TAMs
Tumor-associated macrophages (TAMs) display significant diversity in their source, originating from various origins like tissue-resident macrophages derived from the yolk sac or macrophages infiltrating from the bone marrow [86, 87]. Moreover, within tumors, multiple subsets of TAMs coexist. TAMs facilitate immune suppression and represent a highly plastic population of immune cells with both tumor-promoting and anti-tumor functions [88]. Macrophages can polarize into either M1 or M2 phenotypes, leading to the induction of inflammation or immune suppression, respectively. For instance, TAMs in a hypoxic environment release higher levels of interleukins IL-6 and IL-10, elevate PD-1 expression on T cells, and increase the production of CCL17 and CCL22, promoting the induction of Tregs, which contribute to the establishment of an immunosuppressive environment [87, 89] (Fig. 2). The pro-tumorigenic functions of TAMs include promoting angiogenesis, immunosuppression, extracellular matrix transformation, metastasis formation, and resistance to chemotherapeutic and checkpoint blockade immunotherapies [90, 91]. While TAMs can also counteract cancer progression by directly mediating phagocytosis of cancer cells or cytotoxic tumor killing [89].Macrophages surrounding pancreatic cancer cells undergo a loss of their ability for T cell priming while fostering a microenvironment that supports tumor growth [56]. Recently, an inflammatory circuit involving the interplay between cancer cells and macrophages expressing interleukin-1β (IL-1β + TAMs), aided by prostaglandin E2 (PGE2) and tumor necrosis factor (TNF), revealed physical proximity to IL-1β + TAMs was linked to the inflammatory reprogramming of cells in PDAC [92]. Macrophage-centered therapeutic strategies to change the functional subclasses hold great antitumor potential.
3.6 CAFs
Cancer-associated fibroblasts (CAFs) are critical components of the tumor stroma. Within the TME of PDAC, CAFs constitute a significant portion, accounting for as much as 85% of the tumor volume. This poses challenges for anti-cancer therapy, as it impedes the effective delivery of treatments to target tumor tissue. CAFs include myofibroblast CAFs (myCAFs), immunogenic CAFs (iCAFs), and antigen-presenting CAFs (apCAFs), contributing to extracellular matrix production, immunosuppression, vascular remodeling, tumor proliferation, and metastasis (Fig. 3) [93, 94]. Numerous studies have indicated the presence of two main broadly characterized subgroups, known as myCAFs and iCAFs, exhibiting variances in transcriptomic profiles, spatial distribution, and functional attributes [95]. Indications suggest that the characterization of phenotypes will provide insights into the functional variety of CAFs. Specifically, FAP + CAFs have been identified as facilitators of tumor growth and significant contributors to the immunosuppressive characteristics of the TME [96].
CAFs exhibit multiple functionally phenotypic heterogeneities, contributing to the complexity of their biological functions during PDAC progression [97, 98]. CAFs exhibit pleiotropic and opposing functions in TME [54]. Under certain conditions, CAFs may also impede tumor progression [98]. The spatial heterogeneity of intratumoral CAFs indicates a paradigm shift in their biology, where distinct subtypes such as myofibroblastic and inflammatory CAFs distinctly contribute to PDAC progression [99]. Additionally, CAFs can influence the heterogeneity of PDAC cancer cells [5, 21].
The impact of CAFs on modifying the immune TME has been extensively investigated [54]. A fundamental function of cancer-associated fibroblasts (CAFs) lies in their creation and modification of the ECM, leading to changes in the mechanical characteristics of the ECM and impacting the actions of cancer cells and immune cells [97]. MHC class II-expressing CAFs (apCAFs) directly induce naive CD4 + T cells to differentiate into Tregs, thereby inhibiting the proliferation of cytotoxic CD8 + T cells [64]. CAFs exert an impact on angiogenesis and possess a potent ability to modulate the immune response, thereby contributing to immune evasion in the context of cancer [98]. CAFs also secrete immunomodulatory factors, including CXCL8, CXCL10, TGF-β, IL-6, IL-10, and others, that regulate the innate immune response [58, 97, 100] (Fig. 2). Moreover, CAFs promote immune suppression by influencing the activities of TAMs and MDSCs within the TME [11, 101]. Furthermore, CAFs in triple-negative breast cancer (TNBC) can also express immune checkpoint molecules like PD-L1 [102]. However, it remains uncertain whether CAFs in PDAC express PD-L1.
CAFs are derived from mesenchymal stem cells, and bone marrow (BM)-derived macrophages can convert into CAFs within tumors [103]. CAFs are supposed to play an immunosuppressive role through the production of extracellular matrix components and the secretion of immunosuppressive factors such as CXCL12 and FAP [101]. While CAFs promote cancer progression, metastasis, and resistance against chemo- and immunotherapies, they can also restrict tumor invasion, suggesting potential opportunities to balance their protumorigenic and antitumorigenic effects that require further validation [97, 104].
Sub-TMEs can be characterized by fibroblast plasticity and the regional interactions of CAFs with immune cells, involving various subtypes, differentiation states, and treatment responses. This signifies heterogeneity within the TME is not coincidental but rather indicates essential functional units of the tissue [21]. Thus, the interactions and spatial heterogeneity of CAFs impact cell function and may contribute to PDAC pathogenesis [98]. In summary, CAFs exert their influence on the immunotherapy response through various mechanisms, ultimately creating an immunosuppressive microenvironment. Understanding CAF heterogeneity and their interactions with other components of the TME could potentially enhance the efficacy of immunotherapy [105]. Consequently, targeting CAFs has emerged as a promising strategy to enhance the overall therapeutic response to immunotherapy.
3.7 ECM
The extracellular matrix (ECM) contains a network of fibrous proteins, such as collagens, fibronectin, laminins, elastin, proteoglycans/glycosaminoglycans, and several other glycoproteins [51]. Interestingly, there are similarities in matrix changes between PDAC and pancreatitis. Notably, among the most prominent ECM proteins in PDAC are fibrillar collagens, particularly collagen I and III [106]. The ECM surrounding cancer cells not only acts as a scaffold for tumor cell organization but also plays a crucial role in transmitting mechanical signals to activate various immunosuppressive pathways [106].
Cancer-associated fibroblasts (CAFs) play a significant role in the deposition of fibrillar collagens within the ECM. Deletion of myofibroblast-derived collagen 1 in PDAC results in upregulated Cxcl5 in cancer cells via SOX9, leading to the suppression of CD8 + T cells and the recruitment of myeloid-derived suppressor cells, thus accelerating PDAC initiation in mouse models and impairing overall survival [107]. Moreover, embryonically derived TAMs tend to exhibit a pro-fibrotic transcriptional profile [86]. The ECM directly interacts with CAFs, tumors, and immune cells through integrins, mainly mediating adhesion signaling pathways, which can drive metastasis and hinder treatment efficacy [108]. Integrins also activate tumor immune escape through the TGFβ-SOX4 pathway, limiting T cell-mediated tumor killing [109, 110]. TGF-β signaling cascades, activated by integrins, have been implicated in reducing the tumor-killing capacity of CD8 + T cells in various cancers [111]. Furthermore, TGFβ contributes to immunosuppression and fibrosis [112], thereby impairing the efficacy of immunotherapy [113]. However, targeting fibrosis can reverse immunosuppression in the TME, as shown by effective modulation of the tumor microenvironment using TGF-β-derived peptide vaccination in animal models of PDAC [27, 111].
ECM proteins can increase intratumoral pressure, hindering the effective delivery of drugs to PDAC cancer cells [99]. Blocking TGFβ signaling can restore the ECM and remodel the tumor vasculature, thereby increasing chemotherapy efficacy [112]. Moreover, the matrix components of the stromal compartment adjacent to cancer cells seem particularly similar between primary and metastatic PDAC lesions [114], suggesting that metastatic lesions acquire typical behavior of primary tumors by recruiting fibroblasts and leukocytes, resulting in a dense desmoplastic TME [115]. Hyaluronan, another ECM component, has been found to have a detrimental effect, showing a negative association with survival [114, 116]. Despite attempts to use PEGylated hyaluronidase (PEGPH20) in combination with chemotherapy to degrade hyaluronic acid, it has not shown to provide a survival advantage to patients [117].
3.8 Vasculature
The tumor vasculature in PDAC is a crucial component of the tumor microenvironment and plays a significant role in the disease’s aggressiveness, metastasis, treatment response, and resistance to treatments [28, 118,119,120]. Angiogenesis in PDAC is highly disorganized and irregular; therefore, the new blood vessels are structurally abnormal, leading to a chaotic and leaky vascular network [28, 121]. The blood vessels in PDAC often lack pericytes and smooth muscle cells, which normally provide structural support and stability to blood vessels. This deficiency contributes to the fragile and leaky nature of the tumor vasculature, facilitating the spread of the disease to distant sites [28, 118]. The scarcity of functional vasculature near pancreatic cancer tumor cells is well-established [122]. Due to the abnormal vasculature, resulting in persistent and severe hypoxia within the TME [118, 120]. Insufficient vascularization leads to a microenvironment distinguished by factors such as low pH, hypoxia, modified metabolism, and the evasion of immune responses [28, 118, 123].
CAFs-induced desmoplastic reactions impact tumor vascularization by producing various pro-angiogenic factors such as VEGF-A, FGF2, and CXCL12 to recruit and activate endothelial progenitor cells and myeloid cells with pro-angiogenic potential, indirectly coordinating tumor angiogenesis [105, 124, 125]. Hypoxia-inducible factors (HIF) drive maintenance of redox homeostasis, activation of autophagy, and epigenetic regulation [120, 123, 126]. Hypoxia synergized with cancer cell-derived cytokines lead fibroblasts to convert to inflammatory CAFs (iCAF) in a HIF1α-dependent manner [127]. Stromal HIF2 regulates immune suppression through recruiting M2 macrophages and Tregs, as well as tumor fibrosis [128]. The abnormal tumor vasculature hampers the infiltration and activity of immune cells and poses challenges for delivering chemotherapy and other therapeutics effectively to the tumor; therefore, hypoxia promotes the selection of more aggressive cancer cells and creates an immunosuppressive TME contributing to therapeutic resistance [118, 120, 129].
Understanding the complexities of the tumor vasculature in PDAC is crucial for developing targeted therapies, while several anti-angiogenesis agents have faced setbacks in late-stage clinical trials for PDAC [99]. The degradation of fibrosis in PDAC is associated with increased vascularity [125]. Promoting tumor vessel normalization as a strategy to enhance drug delivery to tumors and sensitize them to ICB/chemotherapy renews the potential for an anti-vasculature approach in PDAC [130,131,132]. As research progresses, advancements in our understanding of the tumor vasculature’s intricacies in PDAC may lead to novel and more effective therapeutic interventions.
4 Spatial heterogeneity and landscape
Investigation of multicellular formations revealed that the variations in phenotype within tumors were localized to specific regions or lesions; however, multicellular structures distinguishing patients with different clinical outcomes [133]. For instance, IL-1β + TAMs are in proximity to CD31 + VEGFR2 + endothelial cells in PDAC [92]. Furthermore, fibroblasts constitute the majority of the tumor volume in PDAC and exhibit significant transcriptional heterogeneity, with distinct functions and spatial relationships [134]. CAFs extracted from different subregions exhibit distinct functional and phenotypic characteristics [21]. The spatial distribution of CAFs subsets within PDAC shows considerable heterogeneity across tissues and tumors [134]. These spatial dynamics of CAFs can influence PDAC cancer cell heterogeneity, indicating intrinsic aggressiveness of PDAC [134] (Fig. 3). α-SMA + CAFs are mainly localized close to well-differentiated cancer cell nests, while fibroblast activation protein (FAP+) CAFs, known for secreting IL6 and other inflammatory mediators, tend to be found juxtaposed to poorly differentiated tumor regions, highlighting CAF heterogeneity in PDAC biology depending on their proximity to cancer cells [96, 100] (Fig. 3). Anti-fibrotic strategies in PDAC aim to counteract the desmoplastic reaction and improve the penetration of chemotherapy into the tumor. Meanwhile, depletion of FAP (+) CAFs or targeting of CXCL12 sensitizes PDAC to ICB in pre-clinical models [96, 100]. Recent findings suggest that the expression of CD105 can be used to categorize CAFs into two functionally distinct subtypes: tumor permissive CD105 + CAFs and tumor-suppressive CD105-CAFs. CD105-CAFs exhibit a restraining effect dependent on functional adaptive immunity, indicating coordinated relationships between immune cell subsets and mesenchymal cells [134] (Fig. 3).
myCAFs are located close to neoplastic cells and are believed to be responsible for extracellular matrix deposition. In research conducted by Moncada et al., primary pancreatic tumors were subjected to multimodal intersection analysis. The study revealed focused enrichments and unique co-enrichments of subpopulations like ductal cells, macrophages, dendritic cells, and cancer cells, in relation to other cell types, within specific spatial regions [135] (Fig. 3). Moreover, the researchers identified instances where inflammatory fibroblasts and cancer cells expressing a stress-response gene module were colocalized. This approach, which delineates the arrangement of subpopulations as defined by single-cell RNA sequencing, has the potential to be expanded for uncovering inherent interactions within intricate tissue structures [135]. The different CAF subpopulations can have an impact on immunomodulation and mechanic responsiveness, and ICB therapy can lead to shifts in CAFs subpopulation distributions, affecting tumor growth [93]. Based on the studies reviewed herein, it becomes clear that subTMEs seem to originate from overarching differentiation stages of intricate CAFs collectives, exhibiting synchronized phenotypes, behaviors, and functions [21]. The characteristics of fibroblasts and ECMs play a crucial role in shaping the biochemical and physical barriers that lead to the exclusion of T cells from the TME [64, 107]. As a result, T cells in human PDAC are often found to exhibit a patchy distribution [136]. For instance, studies have demonstrated that T cells are less abundant in the regions of the tumor containing cancer cells compared to the stroma lacking cancer cells [137]. Research on the spatial architecture of the TME concerning immune targets is relatively limited. The spatial relationships between individual cellular and acellular components in PDAC hold the potential to provide novel insights into the dynamic and complex functions of PDAC desmoplasia [20]. An assessment of the functional properties of the identified immune subsets is warranted.
5 Typical spatial subregion-TLS
The cellular composition of the TME shows T cells dispersed throughout and B cells tending to form aggregates. Moreover, the spatial dynamics of CAFs delineating the functions of different CAF subpopulations [138]. Spatial analyses performed by multiplexed immunohistochemical revealing highly inter- and intrapatient spatial heterogeneity of leukocyte infiltrate [10, 55]. For example, in various human malignancies, the presence of TLS has been linked to positive patient prognoses [139, 140], and favorable responses to ICB [22, 73]. This suggests that TLS may have a beneficial impact on anti-tumor immune responses. TLS can be found either within the tumor (intratumorally) or around the tumor (peritumorally), moreover TLS maturation exhibited prognostic significance in the intratumoral TLS (+) group, whereas it did not show the same prognostic value in the peritumoral TLS (+) group [141]. Intratumoral TLS exhibited notable heterogeneity in their location, cellular composition, and spatial organization across various types of cancers [141]. Typically, TLS is composed of B cells forming the core, surrounded by T cells, with B cells and T cells being the main constituents [140, 142](Fig. 3). Within the T cell population of TLS, there is a higher likelihood of follicular helper T cells (Tfh) presence, but Th1, cytotoxic CD8 + T cells, or Treg cells may also be present [143,144,145]. (Fig. 3). Additionally, TLS contains other cell populations, such as dendritic cells (DCs), including CD21 + follicular dendritic cells (FDCs), crucial for germinal center formation and affinity maturation of B cells, and CD83 + DCs, mainly localized in the T cell zone (also known as DC-LAMP + DCs) [73, 146,147,148,149]. Some CD68 + macrophages may also be found in TLS, possibly involved in clearing apoptotic cells [150]. Apart from the cellular components described above, numerous stromal cells play a role similar to follicular reticular cells (FRCs) in secondary lymphoid tissues (spleen and lymph nodes), anchoring TLS to sites of chronic inflammation [151]. An essential circuit, known as the high post -endothelial venule (HEV), is also required to provide lymphocytes with access to TLS [148] (Fig. 3).
The cellular components and functional status of the TME are influenced by various factors, including the origin of tumorigenesis, intrinsic characteristics of cancer cells, stage of tumorigenesis, and patient characteristics [3]. An intriguing observation is the existence of a “deserted TME state,” characterized by strong enrichment of ECMs that supports tumor differentiation, while there is a “reactive state” characterized by an abundance of cellular stress response gene sets that promote tumor progression [21]. Additionally, these findings indicate the existence of separate and possibly autonomous regional patterns within the TME. The intratumoral heterogeneity, particularly in PDAC, poses a significant challenge in attributing clear functions to the TME [10, 21]. The immune infiltrate within the tumor is not evenly distributed; instead, it varies significantly among distinct areas based on regional epithelial phenotypes [10, 56]. Convincing evidence supports the idea that intratumoral sub-TMEs exhibit significant differences in composition, representing distinct regional immune milieus [21, 152].Subregions of the TME distinct from the composition of TLS have also been identified in our laboratory through multiplex immunofluorescence staining findings (Fig. 4), but their biological significance remains elucidated. Despite these observations, the exact mechanisms underlying TLS formation in tumors remain inadequately understood.

Subregions of the tumor microenvironment that differ from the composition of TLS. (A) Representative mIHC images showing distinct spatial patterns of cellular organization units discovered by our team (not published). (B) Voronoi diagrams of TLS identified. (C) Segmentation pattern diagram of cells identified by AI software. (D) Voronoi diagrams of another subregion. (E) Composition ratio of TLS. (F) Composition ratio of another subregion
6 Conclusion and perspectives
The PDAC tumor immune microenvironment (TiME) displayed a low-immunogenic ecosystem characterized by substantial heterogeneity both within and between tumors [55]. Immunotherapy often fails in PDAC primarily due to a lack of antigen-experienced T effector cells. The stromal and immune compartments of the TME are pivotal in determining the progression and therapeutic response of the disease. To date, no immune-based biomarker has been approved for clinical use in PDAC. A meta-analysis using immunohistochemistry revealed that high levels of CD4 and CD8 T-lymphocytes correlate with better disease-free survival, whereas high levels of CD163 are associated with poorer overall survival. The presence of CD3, CD20, FoxP3, and CD68 cells, as well as PD-L1 expression, did not show significant prognostic relevance [153]. Studies have shown that only tumors with both a high count of neoantigens and significant CD8 + T-cell infiltration are linked to prolonged survival in patients [154]. Moreover, overexpression of fibroblast activation protein (FAP) in the tumor microenvironment is associated with worse overall and disease-free survival in pancreatic ductal adenocarcinoma, suggesting its role in disease progression and metastasis [17]. Additionally, survival outcomes were significantly correlated with the expression levels of specific markers for EMT + cancer cells, activated CAFs, and endothelial cells [94]. It has become evident that treatments significantly influence the spatial and temporal heterogeneity within tumors, which subsequently affects the extent and persistence of the response [26, 155]. A deep understanding of the immunosuppressive milieu is essential for identifying biomarkers that could improve patient stratification.
Recently, advancements in multiplex imaging have deepened our understanding of the immune heterogeneity and dual pro-tumor and anti-tumor roles of the immune elements in PDAC [10]. They can enhance our ability to categorize the phenotypic and spatial immune profiles of PDAC. Besides by applying machine learning (ML) techniques to investigate a highly multiplexed, spatial proteomic dataset derived from single-cell analyses in human pancreatic cancer aid in molecular cancer immunology applications [156]. Additionally, the spatial perspective presents novel opportunities to comprehend the ever-changing crosstalk between the immune system and tumor cells, introducing an extra dimension of spatial information to the study of the TME.
A spatial perspective enhances understanding of the dynamic interactions between the immune system and tumor cells, adding a layer of spatial complexity to TME studies and potentially improving the classification of PDAC’s phenotypic and spatial immune profiles [10, 56]. Spatial computational analysis has highlighted the prognostic importance of CD8 + cell density at the tumor center and its proximity to tumor cells, significantly impacting survival rates [57]. Additionally, increased CD8 + T cell presence within lymphoid clusters correlates with better survival outcomes [55]. Post-neoadjuvant chemotherapy, M1-polarized macrophages situated near tumor cells have been linked to improved pathological responses and survival outcomes [26]. However, a higher ratio of Tregs to total T cells during treatment is associated with shorter survival [157]. Additionally, CD68 + TAMs were notably closer to tumor cells in PDACs with liver recurrences, underscoring the prognostic importance of both the density and spatial distribution of immune cell infiltrates [158]. CCL5 and CXCL10 are consistently linked to robust antitumor immunity and the regulation of CD8 + T cells, suggesting that therapeutically stimulating the production of CCL5 and CXCL10 in recurrent PDACs might enhance T cell recruitment [158]. Distinct recurrence patterns in PDAC correlate with unique, spatially structured inflammatory and stromal responses. These responses are propelled by tumor-host interactions, generating distinct selective pressures that likely influence the tissue-specific spread of PDAC cells [158]. A precise comprehension of the spatial and temporal dynamics of response or resistance to intratumoral heterogeneity in the tumor microenvironment is crucial not only for identifying predictive biomarkers, but also for defining new therapeutic targets.
Understanding the complex genetic landscape and molecular attributes of PDAC offers crucial insights into how intrinsic factors shape the tumor immune microenvironment. Research utilized single-cell RNA sequencing technology to analyze the transcriptomes of individual cells derived from dissociated primary tumors or metastatic biopsies from patients with PDAC, revealing the composition of tumor cells emerged as a critical determinant in classifying PDAC subtypes [94]. Comprehending the intricate genetic makeup and molecular features of PDAC can offer valuable insights into how intrinsic factors influence the evolution of the TME [29, 46]. Yet, the link between tumor genetics and immune response remains a subject of debate, warranting further investigation to clarify this connection. Genomic studies aimed at identifying distinct molecular subtypes in PDAC are crucial for developing more effective translational therapies. The drivers of this heterogeneity and the full definition of tumor sections with spatial resolution are still not completely understood.
The interplay of internal and external factors shapes the diversity of PDAC subtypes, which could guide efforts to reprogram the disease towards more treatable forms. Nevertheless, the stromal and immune compartments of the tumor microenvironment (TME) play crucial roles in dictating the course of disease progression and response to therapy. A more thorough comprehension of PDAC’s immune contexture and its influencing factors may assist in identifying further predictive biomarkers and therapeutic targets, and also shedding light on the mechanisms of tumor recurrence.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- PDAC:
-
Pancreatic ductal adenocarcinoma
- TME:
-
Tumor microenvironment
- ECM:
-
Extracellular matrix
- PanIN:
-
Pancreatic intraepithelial neoplasia
- IL-33:
-
Cellular cytokine interleukin 33
- EV:
-
Extracellular vesicle
- ICB:
-
Immune checkpoint blockades
- Tregs:
-
Regulatory T cells
- MDSCs:
-
Myeloid-Derived Suppressor Cells
- CAFs:
-
Cancer-Associated Fibroblasts
- TLS:
-
Tumor-associated lymphoid structures
- FRCs:
-
Follicular reticular cells
- HEV:
-
High post -endothelial venule
- ADCC:
-
Antibody-dependent cytotoxicity
- APCs:
-
Antigen-presenting cells
- Teff:
-
Antigen-experienced T effector cells
- DSP:
-
Digital spatial profiling
- IFN-γ:
-
Interferon-gamma
- TNF-α:
-
Tumour necrosis factor-α
- FAP:
-
Fibroblast activation protein
References
P. Rawla, T. Sunkara, V. Gaduputi, Epidemiology of pancreatic Cancer: global trends, etiology and risk factors. World J. Oncol. 10(1), 10–27 (2019). https://doi.org/10.14740/wjon1166
H. Sung, J. Ferlay, R.L. Siegel et al., Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71(3), 209–249 (2021). https://doi.org/10.3322/caac.21660
J.D. Mizrahi, R. Surana, J.W. Valle et al., Pancreatic cancer. Lancet. 395(10242), 2008–2020 (2020). https://doi.org/10.1016/s0140-6736(20)30974-0
N.A. Ullman, P.R. Burchard, R.F. Dunne et al., Immunologic strategies in pancreatic Cancer: making Cold tumors Hot. J. Clin. Oncol. 40(24), 2789–2805 (2022). https://doi.org/10.1200/jco.21.02616
A.A. Connor, S. Gallinger, Pancreatic cancer evolution and heterogeneity: integrating omics and clinical data. Nat Rev Cancer. 22(3): 131– 42 (2022). https://doi.org/10.1038/s41568-021-00418-1
C.J. Halbrook, C.A. Lyssiotis, M. Pasca di Magliano et al., Pancreatic cancer: advances and challenges. Cell. 186(8), 1729–1754 (2023). https://doi.org/10.1016/j.cell.2023.02.014
L.J. Padrón, D.M. Maurer, M.H. O’hara et al., Sotigalimab and/or nivolumab with chemotherapy in first-line metastatic pancreatic cancer: clinical and immunologic analyses from the randomized phase 2 PRINCE trial. Nat. Med. 28(6), 1167–1177 (2022). https://doi.org/10.1038/s41591-022-01829-9
M.M. Wattenberg, D. Asch, S. Yu et al., Platinum response characteristics of patients with pancreatic ductal adenocarcinoma and a germline BRCA1, BRCA2 or PALB2 mutation. Br. J. Cancer. 122(3), 333–339 (2020). https://doi.org/10.1038/s41416-019-0582-7
Z.A. Wainberg, H.S. Hochster, E.J. Kim et al., Open-label, phase I study of Nivolumab Combined with nab-Paclitaxel Plus Gemcitabine in Advanced Pancreatic Cancer. Clin. Cancer Res. 26(18), 4814–4822 (2020). https://doi.org/10.1158/1078-0432.Ccr-20-0099
S.M. Liudahl, C.B. Betts, S. Sivagnanam et al., Leukocyte heterogeneity in pancreatic ductal adenocarcinoma: phenotypic and spatial features Associated with clinical outcome. Cancer Discov. 11(8), 2014–2031 (2021). https://doi.org/10.1158/2159-8290.Cd-20-0841
B. Uzunparmak, I.H. Sahin, Pancreatic cancer microenvironment: a current dilemma. Clin. Transl Med. 8(1), 2 (2019). https://doi.org/10.1186/s40169-019-0221-1
J. Watt, H.M. Kocher, The desmoplastic stroma of pancreatic cancer is a barrier to immune cell infiltration. Oncoimmunology. 2(12), e26788 (2013). https://doi.org/10.4161/onci.26788
S. Zhang, W. Fang, S. Zhou et al., Single cell transcriptomic analyses implicate an immunosuppressive tumor microenvironment in pancreatic cancer liver metastasis. Nat. Commun. 14(1), 5123 (2023). https://doi.org/10.1038/s41467-023-40727-7
T. Krausgruber, N. Fortelny, V. Fife-Gernedl et al., Structural cells are key regulators of organ-specific immune responses. Nature. 583(7815), 296–302 (2020). https://doi.org/10.1038/s41586-020-2424-4
C. Neuzillet, A. Tijeras-Raballand, C. Ragulan et al., Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 248(1), 51–65 (2019). https://doi.org/10.1002/path.5224
H. Huang, Z. Wang, Y. Zhang et al., Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell. 40(6): 656– 73.e7 (2022). https://doi.org/10.1016/j.ccell.2022.04.011
A. Lo, C.P. Li, E.L. Buza et al., Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight. 2(19) (2017). https://doi.org/10.1172/jci.insight.92232
X. Liu, J. Xu, B. Zhang et al., The reciprocal regulation between host tissue and immune cells in pancreatic ductal adenocarcinoma: new insights and therapeutic implications. Mol. Cancer. 18(1), 184 (2019). https://doi.org/10.1186/s12943-019-1117-9
K. Chen, Q. Wang, M. Li et al., Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. EBioMedicine. 66, 103315 (2021). https://doi.org/10.1016/j.ebiom.2021.103315
J.L. Carstens, P. Correa de Sampaio, D. Yang et al., Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 8, 15095 (2017). https://doi.org/10.1038/ncomms15095
B.T. Grünwald, A. Devisme, G. Andrieux et al., Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell. 184(22), 5577–92e18 (2021). https://doi.org/10.1016/j.cell.2021.09.022
B.A. Helmink, S.M. Reddy, J. Gao et al., B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 577(7791): 549– 55 (2020). https://doi.org/10.1038/s41586-019-1922-8
Z.R. Qian, D.A. Rubinson, J.A. Nowak et al., Association of alterations in main driver genes with outcomes of patients with resected pancreatic ductal adenocarcinoma. JAMA Oncol. 4(3), e173420 (2018). https://doi.org/10.1001/jamaoncol.2017.3420
M. Peng, Y. Ying, Z. Zhang et al., Reshaping the pancreatic Cancer microenvironment at different stages with chemotherapy. Cancers (Basel). 15(9) (2023). https://doi.org/10.3390/cancers15092448
C. Mota Reyes, S. Teller, A. Muckenhuber et al., Neoadjuvant therapy remodels the pancreatic Cancer Microenvironment via Depletion of Protumorigenic Immune cells. Clin. Cancer Res. 26(1), 220–231 (2020). https://doi.org/10.1158/1078-0432.Ccr-19-1864
A. Dias Costa, S.A. Väyrynen, A. Chawla et al., Neoadjuvant Chemotherapy is Associated with altered Immune Cell Infiltration and an anti-tumorigenic microenvironment in Resected Pancreatic Cancer. Clin. Cancer Res. 28(23), 5167–5179 (2022). https://doi.org/10.1158/1078-0432.Ccr-22-1125
M.H. Andersen, Novel immune modulatory vaccines targeting TGFβ. Cell. Mol. Immunol. 20(5), 551–553 (2023). https://doi.org/10.1038/s41423-023-01000-5
S. Li, H.X. Xu, C.T. Wu et al., Angiogenesis in pancreatic cancer: current research status and clinical implications. Angiogenesis. 22(1), 15–36 (2019). https://doi.org/10.1007/s10456-018-9645-2
de K.E. Visser, J.A. Joyce, The evolving tumor microenvironment: from cancer initiation to metastatic outgrowth. Cancer Cell. 41(3), 374–403 (2023). https://doi.org/10.1016/j.ccell.2023.02.016
P.J. Campbell, S. Yachida, L.J. Mudie et al., The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 467(7319), 1109–1113 (2010). https://doi.org/10.1038/nature09460
S. Yachida, S. Jones, I. Bozic et al., Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 467(7319), 1114–1117 (2010). https://doi.org/10.1038/nature09515
H. Sakamoto, M.A. Attiyeh, J.M. Gerold et al., The Evolutionary origins of recurrent pancreatic Cancer. Cancer Discov. 10(6), 792–805 (2020). https://doi.org/10.1158/2159-8290.Cd-19-1508
K.S.S. Enfield, E. Colliver, C.S.Y. Lee et al., Spatial Architecture of myeloid and T cells orchestrates Immune Evasion and Clinical Outcome in Lung Cancer. Cancer Discov. (2024). https://doi.org/10.1158/2159-8290.Cd-23-1380
E. Hessmann, S.M. Buchholz, I.E. Demir et al., Microenvironmental determinants of pancreatic Cancer. Physiol. Rev. 100(4), 1707–1751 (2020). https://doi.org/10.1152/physrev.00042.2019
N. Niu, X. Shen, Z. Wang et al., Tumor cell-intrinsic epigenetic dysregulation shapes cancer-associated fibroblasts heterogeneity to metabolically support pancreatic cancer. Cancer Cell. 42(5): 869– 84.e9 (2024). https://doi.org/10.1016/j.ccell.2024.03.005
C.J. Tape, S. Ling, M. Dimitriadi et al., Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell. 65(4): 910– 20 (2016). https://doi.org/10.1016/j.cell.2016.03.029
C. Vennin, P. Mélénec, R. Rouet et al., CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 10(1), 3637 (2019). https://doi.org/10.1038/s41467-019-10968-6
P. Bailey, D.K. Chang, K. Nones et al., Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 531(7592), 47–52 (2016). https://doi.org/10.1038/nature16965
V.T. Smit, A.J. Boot, A.M. Smits et al., KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 16(16), 7773–7782 (1988). https://doi.org/10.1093/nar/16.16.7773
A. Velez-Delgado, K.L. Donahue, K.L. Brown et al., Extrinsic KRAS Signaling shapes the pancreatic microenvironment through fibroblast reprogramming. Cell. Mol. Gastroenterol. Hepatol. 13(6), 1673–1699 (2022). https://doi.org/10.1016/j.jcmgh.2022.02.016
M. Chan-Seng-Yue, J.C. Kim, G.W. Wilson et al., Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 52(2), 231–240 (2020). https://doi.org/10.1038/s41588-019-0566-9
F. Puleo, R. Nicolle, Y. Blum et al., Stratification of pancreatic ductal adenocarcinomas based on Tumor and Microenvironment features. Gastroenterology. 155(6), 1999–2013e3 (2018). https://doi.org/10.1053/j.gastro.2018.08.033
W.Q. Wang, L. Liu, H.X. Xu et al., Infiltrating immune cells and gene mutations in pancreatic ductal adenocarcinoma. Br. J. Surg. 103(9), 1189–1199 (2016). https://doi.org/10.1002/bjs.10187
W.L. Hwang, K.A. Jagadeesh, J.A. Guo et al., Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat. Genet. 54(8), 1178–1191 (2022). https://doi.org/10.1038/s41588-022-01134-8
D. Alonso-Curbelo, Y.J. Ho, C. Burdziak et al., A gene-environment-induced epigenetic program initiates tumorigenesis. Nature. 590(7847), 642–648 (2021). https://doi.org/10.1038/s41586-020-03147-x
D. Cui Zhou, R.G. Jayasinghe, S. Chen et al., Spatially restricted drivers and transitional cell populations cooperate with the microenvironment in untreated and chemo-resistant pancreatic cancer. Nat. Genet. 54(9), 1390–1405 (2022). https://doi.org/10.1038/s41588-022-01157-1
L.J. Bayne, G.L. Beatty, N. Jhala et al., Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 21(6), 822–835 (2012). https://doi.org/10.1016/j.ccr.2012.04.025
Y. Pylayeva-Gupta, K.E. Lee, C.H. Hajdu et al., Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 21(6), 836–847 (2012). https://doi.org/10.1016/j.ccr.2012.04.024
E.E. Montalvo-Javé, N. Nuño-Lámbarri, G.N. López-Sánchez et al., Pancreatic Cancer: genetic conditions and epigenetic alterations. J. Gastrointest. Surg. 27(5), 1001–1010 (2023). https://doi.org/10.1007/s11605-022-05553-0
K.K. Mahadevan, K.M. Mcandrews, V.S. Lebleu et al., KRAS(G12D) inhibition reprograms the microenvironment of early and advanced pancreatic cancer to promote FAS-mediated killing by CD8(+) T cells. Cancer Cell. 41(9), 1606–20e8 (2023). https://doi.org/10.1016/j.ccell.2023.07.002
A.D. Theocharis, S.S. Skandalis, C. Gialeli et al., Extracellular matrix structure. Adv. Drug Deliv Rev. 97, 4–27 (2016). https://doi.org/10.1016/j.addr.2015.11.001
W.J. Ho, E.M. Jaffee, L. Zheng, The tumour microenvironment in pancreatic cancer - clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 17(9), 527–540 (2020). https://doi.org/10.1038/s41571-020-0363-5
D. Lv, Y. Fei, H. Chen et al., Crosstalk between T lymphocyte and extracellular matrix in tumor microenvironment. Front. Immunol. 15, 1340702 (2024). https://doi.org/10.3389/fimmu.2024.1340702
X. Mao, J. Xu, W. Wang et al., Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol. Cancer. 20(1), 131 (2021). https://doi.org/10.1186/s12943-021-01428-1
H. Mi, S. Sivagnanam, C.B. Betts et al., Quantitative spatial profiling of Immune populations in pancreatic ductal adenocarcinoma reveals Tumor Microenvironment Heterogeneity and Prognostic biomarkers. Cancer Res. 82(23), 4359–4372 (2022). https://doi.org/10.1158/0008-5472.Can-22-1190
S. Yousuf, M. Qiu, L. Von Voith et al., Spatially resolved Multi-omics single-cell analyses inform mechanisms of Immune Dysfunction in Pancreatic Cancer. Gastroenterology. (2023). https://doi.org/10.1053/j.gastro.2023.05.036
Y. Masugi, T. Abe, A. Ueno et al., Characterization of spatial distribution of tumor-infiltrating CD8(+) T cells refines their prognostic utility for pancreatic cancer survival. Mod. Pathol. 32(10), 1495–1507 (2019). https://doi.org/10.1038/s41379-019-0291-z
M.H. Sherman, G.L. Beatty, Tumor Microenvironment in Pancreatic Cancer Pathogenesis and Therapeutic Resistance. Annu. Rev. Pathol. 18, 123–148 (2023). https://doi.org/10.1146/annurev-pathmechdis-031621-024600
P.C. Tumeh, C.L. Harview, J.H. Yearley et al., PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 515(7528), 568–571 (2014). https://doi.org/10.1038/nature13954
A.X. Zhu, A.R. Abbas, De M.R. Galarreta et al., Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat. Med. 28(8), 1599–1611 (2022). https://doi.org/10.1038/s41591-022-01868-2
K. Li, J.A. Tandurella, J. Gai et al., Multi-omic analyses of changes in the tumor microenvironment of pancreatic adenocarcinoma following neoadjuvant treatment with anti-PD-1 therapy. Cancer Cell. 40(11), 1374–91e7 (2022). https://doi.org/10.1016/j.ccell.2022.10.001
M. Ruterbusch, K.B. Pruner, L. Shehata et al., In vivo CD4(+) T cell differentiation and function: revisiting the Th1/Th2 paradigm. Annu. Rev. Immunol. 38, 705–725 (2020). https://doi.org/10.1146/annurev-immunol-103019-085803
A. Takeuchi, T. Saito, CD4 CTL, a cytotoxic subset of CD4(+) T cells, their differentiation and function. Front. Immunol. 8, 194 (2017). https://doi.org/10.3389/fimmu.2017.00194
E. Elyada, M. Bolisetty, P. Laise et al., Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals Antigen-Presenting Cancer-Associated fibroblasts. Cancer Discov. 9(8), 1102–1123 (2019). https://doi.org/10.1158/2159-8290.Cd-19-0094
T. Komura, Y. Sakai, K. Harada et al., Inflammatory features of pancreatic cancer highlighted by monocytes/macrophages and CD4 + T cells with clinical impact. Cancer Sci. 106(6), 672–686 (2015). https://doi.org/10.1111/cas.12663
Y. Zhang, W. Yan, E. Mathew et al., CD4 + T lymphocyte ablation prevents pancreatic carcinogenesis in mice. Cancer Immunol Res. 2(5): 423– 35 (2014). https://doi.org/10.1158/2326-6066.Cir-14-0016-t
C. Mota Reyes, E. Demir, K. Çifcibaşi et al., Regulatory T cells in pancreatic Cancer: of mice and men. Cancers (Basel). 14(19) (2022). https://doi.org/10.3390/cancers14194582
B.P. Keenan, Y. Saenger, M.I. Kafrouni et al., A Listeria vaccine and depletion of T-regulatory cells activate immunity against early stage pancreatic intraepithelial neoplasms and prolong survival of mice. Gastroenterology. 146(7), 1784–94e6 (2014). https://doi.org/10.1053/j.gastro.2014.02.055
Y. Zhang, J. Lazarus, N.G. Steele et al., Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 10(3): 422– 39 (2020). https://doi.org/10.1158/2159-8290.Cd-19-0958
A.H. Ko, A.C. Jordan, E. Tooker et al., Dual targeting of Mesothelin and CD19 with Chimeric Antigen Receptor-Modified T Cells in patients with metastatic pancreatic Cancer. Mol. Ther. 28(11), 2367–2378 (2020). https://doi.org/10.1016/j.ymthe.2020.07.017
M.C.A. Wouters, B.H. Nelson, Prognostic significance of Tumor-infiltrating B cells and plasma cells in Human Cancer. Clin. Cancer Res. 24(24), 6125–6135 (2018). https://doi.org/10.1158/1078-0432.Ccr-18-1481
Y. Pylayeva-Gupta, S. Das, J.S. Handler et al., IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Discov. 6(3): 247– 55 (2016). https://doi.org/10.1158/2159-8290.Cd-15-0843
R. Cabrita, M. Lauss, A. Sanna et al., Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 577(7791), 561–565 (2020). https://doi.org/10.1038/s41586-019-1914-8
F. Petitprez, De A. Reyniès, E.Z. Keung et al., B cells are associated with survival and immunotherapy response in sarcoma. Nature. 577(7791): 556– 60 (2020). https://doi.org/10.1038/s41586-019-1906-8
C. Cui, J. Wang, E. Fagerberg et al., Neoantigen-driven B cell and CD4 T follicular helper cell collaboration promotes anti-tumor CD8 T cell responses. Cell. 184(25), 6101–18e13 (2021). https://doi.org/10.1016/j.cell.2021.11.007
R.A.M. Rossetti, N.P.C. Lorenzi, K. Yokochi et al., B lymphocytes can be activated to act as antigen presenting cells to promote anti-tumor responses. PLoS One. 13(7), e0199034 (2018). https://doi.org/10.1371/journal.pone.0199034
S. Hong, Z. Zhang, H. Liu et al., B cells are the Dominant Antigen-presenting cells that activate naive CD4(+) T cells upon immunization with a Virus-Derived Nanoparticle Antigen. Immunity. 49(4), 695–708e4 (2018). https://doi.org/10.1016/j.immuni.2018.08.012
J. Deng, J.B. Fleming, Inflammation and myeloid cells in Cancer Progression and Metastasis. Front. Cell. Dev. Biol. 9, 759691 (2021). https://doi.org/10.3389/fcell.2021.759691
R. Trovato, A. Fiore, S. Sartori et al., Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J. Immunother Cancer. 7(1), 255 (2019). https://doi.org/10.1186/s40425-019-0734-6
J.Q. Fan, M.F. Wang, H.L. Chen et al., Current advances and outlooks in immunotherapy for pancreatic ductal adenocarcinoma. Mol. Cancer. 19(1), 32 (2020). https://doi.org/10.1186/s12943-020-01151-3
I.M. Stromnes, J.S. Brockenbrough, K. Izeradjene et al., Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 63(11), 1769–1781 (2014). https://doi.org/10.1136/gutjnl-2013-306271
C. Siret, A. Collignon, F. Silvy et al., Deciphering the Crosstalk between Myeloid-Derived Suppressor Cells and Regulatory T Cells in pancreatic ductal adenocarcinoma. Front. Immunol. 10, 3070 (2019). https://doi.org/10.3389/fimmu.2019.03070
C.W. Steele, S.A. Karim, J.D.G. Leach et al., CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. 29(6), 832–845 (2016). https://doi.org/10.1016/j.ccell.2016.04.014
Y. Zhang, A. Velez-Delgado, E. Mathew et al., Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut. 66(1), 124–136 (2017). https://doi.org/10.1136/gutjnl-2016-312078
A. Thyagarajan, M.S.A. Alshehri, K.L.R. Miller et al., Myeloid-derived suppressor cells and pancreatic Cancer: implications in Novel Therapeutic approaches. Cancers (Basel). 11(11) (2019). https://doi.org/10.3390/cancers11111627
Y. Zhu, J.M. Herndon, D.K. Sojka et al., Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity. 47(2): 323– 38.e6 (2017). https://doi.org/10.1016/j.immuni.2017.07.014
T. Lazarov, S. Juarez-Carreño, N. Cox et al., Physiology and diseases of tissue-resident macrophages. Nature. 618(7966), 698–707 (2023). https://doi.org/10.1038/s41586-023-06002-x
Y. Zhu, B.L. Knolhoff, M.A. Meyer et al., CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 74(18), 5057–5069 (2014). https://doi.org/10.1158/0008-5472.Can-13-3723
A. Mantovani, P. Allavena, F. Marchesi et al., Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 21(11), 799–820 (2022). https://doi.org/10.1038/s41573-022-00520-5
A.R. Poh, M. Ernst, Tumor-Associated macrophages in Pancreatic Ductal Adenocarcinoma: Therapeutic opportunities and Clinical challenges. Cancers (Basel). 13(12) (2021). https://doi.org/10.3390/cancers13122860
S. Chen, A. Saeed, Q. Liu et al., Macrophages in immunoregulation and therapeutics. Signal. Transduct. Target. Ther. 8(1), 207 (2023). https://doi.org/10.1038/s41392-023-01452-1
N. Caronni, LA F. Terza, F.M. Vittoria et al., IL-1β(+) macrophages fuel pathogenic inflammation in pancreatic cancer. Nature, 2023, 623(7986): 415– 22. https://doi.org/10.1038/s41586-023-06685-2
D.S. Foster, M. Januszyk, D. Delitto et al., 1392– 406.e7. Cancer Cell. 40(11) (2022). https://doi.org/10.1016/j.ccell.2022.09.015. Multiomic analysis reveals conservation of cancer-associated fibroblast phenotypes across species and tissue of origin
W. Lin, P. Noel, E.H. Borazanci et al., Single-cell transcriptome analysis of tumor and stromal compartments of pancreatic ductal adenocarcinoma primary tumors and metastatic lesions. Genome Med. 12(1), 80 (2020). https://doi.org/10.1186/s13073-020-00776-9
K. Oh, Y.J. Yoo, L.A. Torre-Healy et al., Coordinated single-cell tumor microenvironment dynamics reinforce pancreatic cancer subtype. Nat. Commun. 14(1), 5226 (2023). https://doi.org/10.1038/s41467-023-40895-6
C. Feig, J.O. Jones, M. Kraman et al., Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. U S A 110(50), 20212–20217 (2013). https://doi.org/10.1073/pnas.1320318110
E. Sahai, I. Astsaturov, E. Cukierman et al., A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 20(3): 174– 86 (2020). https://doi.org/10.1038/s41568-019-0238-1
G. Biffi, D.A. Tuveson, Diversity and Biology of Cancer-Associated Fibroblasts. Physiol Rev, 2021, 101(1): 147– 76. https://doi.org/10.1152/physrev.00048.2019
A.N. Hosein, R.A. Brekken, A. Maitra, Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 17(8), 487–505 (2020). https://doi.org/10.1038/s41575-020-0300-1
D. Öhlund, A. Handly-Santana, G. Biffi et al., Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 214(3), 579–596 (2017). https://doi.org/10.1084/jem.20162024
T. Zhang, Y. Ren, P. Yang et al., Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Cell. Death Dis. 13(10), 897 (2022). https://doi.org/10.1038/s41419-022-05351-1
K. Yoshikawa, M. Ishida, H. Yanai et al., Prognostic significance of PD-L1-positive cancer-associated fibroblasts in patients with triple-negative breast cancer. BMC Cancer. 21(1), 239 (2021). https://doi.org/10.1186/s12885-021-07970-x
C. Iwamoto, K. Ohuchida, T. Shinkawa et al., Bone marrow-derived macrophages converted into cancer-associated fibroblast-like cells promote pancreatic cancer progression. Cancer Lett. 512, 15–27 (2021). https://doi.org/10.1016/j.canlet.2021.04.013
H. Liu, Y. Shi, F. Qian, Opportunities and delusions regarding drug delivery targeting pancreatic cancer-associated fibroblasts. Adv. Drug Deliv Rev. 172, 37–51 (2021). https://doi.org/10.1016/j.addr.2021.02.012
I. Stouten, Van N. Montfoort, L. Hawinkels, The Tango between Cancer-Associated fibroblasts (CAFs) and Immune cells in affecting Immunotherapy Efficacy in Pancreatic Cancer. Int. J. Mol. Sci. 24(10) (2023). https://doi.org/10.3390/ijms24108707
C. Tian, K.R. Clauser, D. Öhlund et al., Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. U S A 116(39), 19609–19618 (2019). https://doi.org/10.1073/pnas.1908626116
Y. Chen, J. Kim, S. Yang et al., Type I collagen deletion in αSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell, 2021, 39(4): 548– 65.e6. https://doi.org/10.1016/j.ccell.2021.02.007
F. Kai, A.P. Drain, V.M. Weaver, The Extracellular Matrix modulates the metastatic journey. Dev. Cell. 49(3), 332–346 (2019). https://doi.org/10.1016/j.devcel.2019.03.026
C.S. Moreno, SOX4: the unappreciated oncogene. Semin Cancer Biol. 67(Pt 1), 57–64 (2020). https://doi.org/10.1016/j.semcancer.2019.08.027
A. Bagati, S. Kumar, P. Jiang et al., Integrin αvβ6-TGFβ-SOX4 pathway drives Immune Evasion in Triple-negative breast Cancer. Cancer Cell. 39(1), 54–67e9 (2021). https://doi.org/10.1016/j.ccell.2020.12.001
M. Perez-Penco, S.E. Weis-Banke, A. SCHINA et al., TGFβ-derived immune modulatory vaccine: targeting the immunosuppressive and fibrotic tumor microenvironment in a murine model of pancreatic cancer. J. Immunother Cancer. 10(12) (2022). https://doi.org/10.1136/jitc-2022-005491
J. Chen, Z.Y. Ding, S. LI et al., Targeting transforming growth factor-β signaling for enhanced cancer chemotherapy. Theranostics. 11(3), 1345–1363 (2021). https://doi.org/10.7150/thno.51383
A. Naik, A. Leask, Tumor-associated fibrosis impairs the response to immunotherapy. Matrix Biol. 119, 125–140 (2023). https://doi.org/10.1016/j.matbio.2023.04.002
C.J. Whatcott, C.H. Diep, P. Jiang et al., Desmoplasia in primary tumors and metastatic lesions of pancreatic Cancer. Clin. Cancer Res. 21(15), 3561–3568 (2015). https://doi.org/10.1158/1078-0432.Ccr-14-1051
N.M. Aiello, D.L. Bajor, R.J. Norgard et al., Metastatic progression is associated with dynamic changes in the local microenvironment. Nat. Commun. 7, 12819 (2016). https://doi.org/10.1038/ncomms12819
V.R. Placencio-Hickok, M. Lauzon, N. Moshayedi et al., Hyaluronan heterogeneity in pancreatic ductal adenocarcinoma: Primary tumors compared to sites of metastasis. Pancreatology. 22(1): 92– 7 (2022). https://doi.org/10.1016/j.pan.2021.09.015
Van E. Cutsem, M.A. Tempero, D. Sigal et al., Randomized Phase III Trial of Pegvorhyaluronidase Alfa with Nab-Paclitaxel Plus Gemcitabine for patients with Hyaluronan-High Metastatic pancreatic adenocarcinoma. J. Clin. Oncol. 38(27), 3185–3194 (2020). https://doi.org/10.1200/jco.20.00590
J.D. Martin, G. Seano, R.K. Jain, Normalizing function of Tumor vessels: Progress, opportunities, and challenges. Annu. Rev. Physiol. 81, 505–534 (2019). https://doi.org/10.1146/annurev-physiol-020518-114700
Van Der J.A. Zee, Van C.H. Eijck, W.C. Hop et al., Angiogenesis: a prognostic determinant in pancreatic cancer?. Eur. J. Cancer. 47(17), 2576–2584 (2011). https://doi.org/10.1016/j.ejca.2011.08.016
J. Tao, G. Yang, W. Zhou et al., Targeting hypoxic tumor microenvironment in pancreatic cancer. J. Hematol. Oncol. 14(1), 14 (2021). https://doi.org/10.1186/s13045-020-01030-w
E. Katsuta, Q. Qi, X. Peng et al., Pancreatic adenocarcinomas with mature blood vessels have better overall survival. Sci. Rep. 9(1), 1310 (2019). https://doi.org/10.1038/s41598-018-37909-5
A. Ene-Obong, A.J. Clear, J. Watt et al., Activated pancreatic stellate cells sequester CD8 + T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 145(5), 1121–1132 (2013). https://doi.org/10.1053/j.gastro.2013.07.025
B. Keith, R.S. Johnson, M.C. Simon, HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer. 12(1), 9–22 (2011). https://doi.org/10.1038/nrc3183
C.S. Mundry, K.C. Eberle, P.K. Singh et al., Local and systemic immunosuppression in pancreatic cancer: targeting the stalwarts in tumor’s arsenal. Biochim. Biophys. Acta Rev. Cancer. 1874(1), 188387 (2020). https://doi.org/10.1016/j.bbcan.2020.188387
A.D. Rhim, P.E. Oberstein, D.H. Thomas et al., Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 25(6), 735–747 (2014). https://doi.org/10.1016/j.ccr.2014.04.021
E.B. Rankin, A.J. Giaccia, The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 15(4): 678– 85 (2008). https://doi.org/10.1038/cdd.2008.21
S. Schwörer, F.V. Cimino, M. Ros et al., Hypoxia potentiates the inflammatory fibroblast phenotype promoted by pancreatic Cancer cell-derived cytokines. Cancer Res. 83(10), 1596–1610 (2023). https://doi.org/10.1158/0008-5472.Can-22-2316
C.J. Garcia Garcia, Y. Huang, N.R. Fuentes et al., Stromal HIF2 regulates Immune suppression in the pancreatic Cancer microenvironment. Gastroenterology. 162(7), 2018–2031 (2022). https://doi.org/10.1053/j.gastro.2022.02.024
L. Ye, K. Jin, Z. Liao et al., Hypoxia-reprogrammed regulatory group 2 innate lymphoid cells promote immunosuppression in pancreatic cancer. EBioMedicine. 79, 104016 (2022). https://doi.org/10.1016/j.ebiom.2022.104016
V.P. Balachandran, G.L. Beatty, S.K. Dougan, Broadening the impact of Immunotherapy to Pancreatic Cancer: challenges and opportunities. Gastroenterology. 156(7), 2056–2072 (2019). https://doi.org/10.1053/j.gastro.2018.12.038
A.S. Bear, R.H. Vonderheide, M.H. O’hara, Challenges and opportunities for Pancreatic Cancer immunotherapy. Cancer Cell. 38(6), 788–802 (2020). https://doi.org/10.1016/j.ccell.2020.08.004
L.D. Wood, M.I. Canto, E.M. Jaffee et al., Pancreatic Cancer: Pathogenesis, screening, diagnosis, and treatment. Gastroenterology. 163(2), 386–402e1 (2022). https://doi.org/10.1053/j.gastro.2022.03.056
H.W. Jackson, J.R. Fischer, V.R.T. Zanotelli et al., The single-cell pathology landscape of breast cancer. Nature, 2020, 578(7796): 615– 20. https://doi.org/10.1038/s41586-019-1876-x
C. Hutton, F. Heider, A. Blanco-Gomez et al., Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell. 39(9), 1227–44e20 (2021). https://doi.org/10.1016/j.ccell.2021.06.017
R. Moncada, D. Barkley, F. Wagner et al., Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 38(3), 333–342 (2020). https://doi.org/10.1038/s41587-019-0392-8
J. Leinwand, G. Miller, Regulation and modulation of antitumor immunity in pancreatic cancer. Nat. Immunol. 21(10), 1152–1159 (2020). https://doi.org/10.1038/s41590-020-0761-y
Y.D. Seo, X. Jiang, K.M. Sullivan et al., Mobilization of CD8(+) T cells via CXCR4 blockade facilitates PD-1 checkpoint therapy in human pancreatic Cancer. Clin. Cancer Res. 25(13), 3934–3945 (2019). https://doi.org/10.1158/1078-0432.Ccr-19-0081
C.M. Schürch, S.S. Bhate, G.L. Barlow et al., Coordinated Cellular neighborhoods Orchestrate Antitumoral immunity at the Colorectal Cancer Invasive Front. Cell. 182(5), 1341–59e19 (2020). https://doi.org/10.1016/j.cell.2020.07.005
M.C. Dieu-Nosjean, N.A. Giraldo, H. Kaplon et al., Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol. Rev. 271(1), 260–275 (2016). https://doi.org/10.1111/imr.12405
C. Sautès-Fridman, M. Lawand, N.A. Giraldo et al., Tertiary lymphoid structures in cancers: Prognostic Value, Regulation, and manipulation for therapeutic intervention. Front. Immunol. 7, 407 (2016). https://doi.org/10.3389/fimmu.2016.00407
A.B. Rodriguez, V.H. Engelhard, Insights into Tumor-Associated Tertiary lymphoid structures: novel targets for Antitumor Immunity and Cancer Immunotherapy. Cancer Immunol. Res. 8(11), 1338–1345 (2020). https://doi.org/10.1158/2326-6066.Cir-20-0432
C. Sautès-Fridman, F. Petitprez, J. Calderaro et al., Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer. 19(6), 307–325 (2019). https://doi.org/10.1038/s41568-019-0144-6
C. Gu-Trantien, S. Loi, S. Garaud et al., CD4⁺ follicular helper T cell infiltration predicts breast cancer survival. J. Clin. Invest. 123(7), 2873–2892 (2013). https://doi.org/10.1172/jci67428
D.R. Kroeger, K. Milne, B.H. Nelson, Tumor-infiltrating plasma cells are Associated with Tertiary lymphoid structures, cytolytic T-Cell responses, and Superior Prognosis in Ovarian Cancer. Clin. Cancer Res. 22(12), 3005–3015 (2016). https://doi.org/10.1158/1078-0432.Ccr-15-2762
A. Hennequin, V. Derangère, R. Boidot et al., Tumor infiltration by tbet + effector T cells and CD20 + B cells is associated with survival in gastric cancer patients. Oncoimmunology. 5(2), e1054598 (2016). https://doi.org/10.1080/2162402x.2015.1054598
J. Goc, C. Germain, T.K. Vo-Bourgais et al., Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8 + T cells. Cancer Res. 74(3), 705–715 (2014). https://doi.org/10.1158/0008-5472.Can-13-1342
F. Bergomas, F. Grizzi, A. Doni et al., Tertiary intratumor lymphoid tissue in colo-rectal cancer. Cancers (Basel). 4(1), 1–10 (2011). https://doi.org/10.3390/cancers4010001
Di G. Caro, F. Bergomas, F. Grizzi et al., Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin. Cancer Res. 20(8), 2147–2158 (2014). https://doi.org/10.1158/1078-0432.Ccr-13-2590
T.P. Mcmullen, R. Lai, L. Dabbagh et al., Survival in rectal cancer is predicted by T cell infiltration of tumour-associated lymphoid nodules. Clin Exp Immunol. 161(1): 81– 8 (2010). https://doi.org/10.1111/j.1365-2249.2010.04147.x
M. Baratin, L. Simon, A. Jorquera et al., T Cell Zone Resident Macrophages Silently Dispose of Apoptotic Cells in the Lymph Node. Immunity. 47(2): 349– 62.e5 (2017). https://doi.org/10.1016/j.immuni.2017.07.019
F. BARONE, D.H. Gardner, S. Nayar et al., Stromal fibroblasts in Tertiary lymphoid structures: a Novel Target in Chronic inflammation. Front. Immunol. 7, 477 (2016). https://doi.org/10.3389/fimmu.2016.00477
A.J. Aguirre, J.A. Nowak, N.D. Camarda et al., Real-time genomic characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov. 8(9), 1096–1111 (2018). https://doi.org/10.1158/2159-8290.Cd-18-0275
A.J. Mcguigan, H.G. Coleman, R.S. Mccain et al., Immune cell infiltrates as prognostic biomarkers in pancreatic ductal adenocarcinoma: a systematic review and meta-analysis. J. Pathol. Clin. Res. 7(2), 99–112 (2021). https://doi.org/10.1002/cjp2.192
V.P. Balachandran, M. Łuksza, J.N. Zhao et al., Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 551(7681), 512–516 (2017). https://doi.org/10.1038/nature24462
K.T. Byrne, C.B. Betts, R. Mick et al., Neoadjuvant Selicrelumab, an agonist CD40 antibody, induces changes in the Tumor Microenvironment in patients with resectable pancreatic Cancer. Clin. Cancer Res. 27(16), 4574–4586 (2021). https://doi.org/10.1158/1078-0432.Ccr-21-1047
K.E. Blise, S. Sivagnanam, C.B. Betts et al., Machine learning links T-cell function and spatial localization to Neoadjuvant Immunotherapy and Clinical Outcome in Pancreatic Cancer. Cancer Immunol. Res. 12(5), 544–558 (2024). https://doi.org/10.1158/2326-6066.Cir-23-0873
R.D. Gartrell, T. Enzler, P.S. Kim et al., Neoadjuvant chemoradiation alters the immune microenvironment in pancreatic ductal adenocarcinoma. Oncoimmunology. 11(1), 2066767 (2022). https://doi.org/10.1080/2162402x.2022.2066767
E. Karamitopoulou, A.S. Wenning, A. Acharjee et al., Spatially restricted tumour-associated and host-associated immune drivers correlate with the recurrence sites of pancreatic cancer. Gut. 72(8), 1523–1533 (2023). https://doi.org/10.1136/gutjnl-2022-329371
Acknowledgements
Not applicable.
Funding
This work was supported “The Clinical Research Incubation Project, West China Hospital, Sichuan University (2020HXFH027)” and “1·3·5 project for disciplines of excellence–Clinical Research Incubation Project, West China Hospital, Sichuan University”.
Author information
Authors and Affiliations
Contributions
Xiaoying Li and Wanting Hou drafted the original article; Chaoxin Xiao prepared the figures, and Heqi Yang critically revised the article for intellectual content, Chengjian Zhao and Dan Cao approved the final version to be submitted.
Corresponding author
Ethics declarations
Ethical approval of the research protocol by an institutional review board
Not applicable.
Informed consent
Not applicable.
Competing interests
The authors declare no competing interests.
Registry and registration no. of the study/trial
Not applicable.
Animal studies
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, X., Hou, W., Xiao, C. et al. Panoramic tumor microenvironment in pancreatic ductal adenocarcinoma. Cell Oncol. (2024). https://doi.org/10.1007/s13402-024-00970-6
Accepted:
Published:
DOI: https://doi.org/10.1007/s13402-024-00970-6