
Abstract
Hepatocellular carcinoma (HCC) is one of the most lethal and prevalent human malignancies, leading to poor prognosis due to its high recurrence and metastasis rates. In recent years it has become increasingly evident that the tumor microenvironment (TME) plays an important role in tumor progression and metastasis. Tumor microenvironment (TME) refers to the complex tissue environment of tumor occurrence and development. Here, we summarize the development of HCC and the role of cellular and non-cellular components of the TME in the metastasis HCC, with particular reference to tumor-infiltrating immune cells. We also discuss some of the possible therapeutic targets for the TME and the future prospectives of this evolving field. SIGNIFICANCE: This review provides a comprehensive analysis of the role of the infiltrating immune cells in TME in the metastasis of HCC and highlights the future outlook for targeted therapy of the TME in the context of recent experiments revealing a number of therapeutic targets targeting the TME.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignancies in the world, mainly occurring in developing countries, and the fourth leading cause of cancer-related death [1]. Common risk factors for HCC is liver damage caused by chronic hepatitis B and C infections, alcohol, and diabetes, followed by liver inflammation, necrosis, and hepatocyte proliferation. This continuous cycle of destruction-regeneration process leads to cirrhosis characterized by dysplastic nodules [2], which eventually leads to HCC.
Despite advances in surgical excision and transplantation, the prognosis of HCC patients remains unsatisfactory, which is associated with a high rate of metastasis of HCC [3]. The process of HCC metastasis can be simplified as: (a) continuous expression of adhesion molecules and an intact substrate, confining cancer cells to the tumor site; (b) epithelial-mesenchymal transition (EMT) occurred in some cells, promoting local infiltration and invasion; (c) Tumor cells enter the circulation, reach the site of secondary tumor, colonize and grow, and achieve metastasis. According to current studies, tumor cells, immune cells, stromal cells, endothelial cells and cancer-related fibrocytes all exist in the tumor microenvironment (TME) [4] and regulate HCC progression differently. In this review, we mainly provide an overview of the role of immune cells in the tumor microenvironment and highlight their possible regulatory mechanisms for HCC metastasis.
2 Development of HCC
As the main metabolic organ of the body, liver is constantly targeted by intestinal pathogens, microbiome associated molecules, toll-like receptor agonists and various metabolites. Thus, a healthy liver has immunosuppressive polarity that weakens T-cell-mediated antigenic responses and is maintained by other liver-resident cells, including Kupffer cells (KCs), dendritic cells (DCs), regulatory T (Treg) cells, and liver sinusoidal endothelial cells (LSECs) [5]. However, this immunosuppressive microenvironment, maintained by the cooperation between liver resident cells and peripheral white blood cells or bone marrow cells, is destroyed in the development of HCC and further exhibits a malignant effect of tumor promotion. The persistence of chronic inflammation is believed to be the beginning of this event.
Current studies believe that chronic inflammation is the leading risk factor inducing the transformation of HCC microenvironment [6]. In inflammatory liver disease, inflammatory abnormalities may promote the activation of innate immune responses including recruitment of monocytes, neutrophils, and dendritic cells (DC), as well as specific immune responses at sites of stress. Furthermore, inflammation can also alter the homeostasis of liver-resident cells [7], leading to liver fibrosis and further transformation to cirrhosis.
Tumor initiation is thought to be the process by which normal cells gain a survival advantage and gradually accumulate cancer-causing mutations [8]. Chronic liver injury and inflammation induce compensatory regeneration of the liver to restore organ structure and maintain its function. This unique liver response is characterized by hyper- proliferation of hepatic A6 + KRT19 + progenitor- like cells around bile ducts near to the portal vein, known as the ductal response. Ductal response is considered to be a key step in carcinogenic transformation. Cells proliferate rapidly in the microenvironment induced by carcinogenic pathway, and gene mutations accumulate rapidly and are endowed with malignant potential [9, 10].
Despite tumor metastasis is mainly relevant to the malignant degree and type of the primary tumor, the so-called “invasion-metastasis cascade” is one of the common features that all metastases need to go through. After tumor cells gain invasiveness and lose adhesion to the surrounding matrix, the metastasis process begins and then tumor cells shed from the primary tumor nest. The disseminated tumor cells will penetrate blood vessels or lymph vessels into the circulation and make responses to diverse resistance conditions such as immune surveillance. Only a small amount of tumor cells can enter the pre-metastatic niche and seed in the niche [11]. After seeding, tumor cells either enter a long-term dormant state or micro metastasis, and finally develop into clinical metastases.
3 Non-immune cells in TME
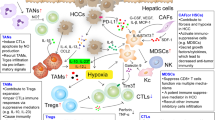
Hepatic stellate cells (HSCs), tumor-associated endothelial cells (TAECs) and cancer-associated fibroblasts (CAFs) are important non-immune cells in TME, which play important structural functions in normal liver tissues and provide assistance in the process of HCC metastasis. TAECs participate in the formation of tumor neovascularization, and are irregularly arranged, loosely connected, and have large gaps in HCC tissues, providing the structural basis for HCC metastasis [12]. Further enhancement of HSC activation and significantly increased α-SMA expression were observed in the acidic tumor microenvironment of HCC, which was proved to be due to the activation of the Erk1/2 signaling pathway. Activated HSC promotes HCC metastasis through OPN, a key executive molecule [13] (Fig. 1).

A general overview of mechanisms and interactions of the HCC microenvironment. Effect of cellular components in TME on metastasis of hepatocellular carcinoma. TAECs indicates tumor-associated endothelial cells, HSCs indicates hepatic stellate cells, CAFs indicates tumor-associated fibroblasts, CTL indicates cytotoxic T lymphocytes, Treg indicates regulatory T cells, MDSCs indicates myeloid-derived suppressor cells, TAM indicates tumor-Associated Macrophages, TAN indicates tumor-Associated Neutrophils
A large number of cancer-associated fibroblasts is a prominent feature of HCC-TME, which significantly increases the secretion of matrix metalloproteinases (MMP) and acts on the extracelluar matrix (ECM) barrier to promote tumor invasion. Meanwhile, CAFs can secrete a variety of cytokines and chemokines, such as SDF-1, which recruits endothelial cells to the tumor site and promotes angiogenesis; IL-6 can promote bone marrow-derived inhibition of cell proliferation, and improve the dryness of liver cancer cells by enhancing STAT3/Notch signaling [14]. It should be emphasized that, different from the apoptosis process of normal cells, CAFs can remain active even after the original stimulus is removed during HCC progression, which in a sense demonstrates the “uncontrolled” characteristics of tumors [15].
4 The immune cells in TME
4.1 Cytotoxic T lymphocyte
Studies have shown that CD8 cytotoxic T cells (CTLs) are the immune cells that primarily kill tumor cells and are the preferred immune cells targeting cancer. CD8+ T lymphocytes, after activation and modification to CTLs, utilize two main pathways for killing their target cells: Granule exocytosis and Fas ligand (FasL)-mediated apoptosis. In the granular exocytosis pathway, when CTLs encountered cancer cells, perforin will be produced to make cell membrane pores acting as a channel to release granzyme A and B into the TME. The released granzyme then enter the target cells and dissociation them. The FasL pathway activates FasL through CTL to release cytochrome C and activate caspase-mediated apoptosis. In addition, apart from increasing proinflammatory cytokine production and antigen presentation, CTLs make it to directly killed tumor cells by producing interferon γ (IFN-γ), a key factor of anti-tumor immunity. Clinical studies had found that tumor cells exposed to IFN-γ secreted by CTL occurred genetic instability, including DNA damage response associated with copy number variation [16].
It’s worth noting that the specific antitumor effect of CTL in HCC metastasis has not yet been found, and CTL appears to kill tumor cells in a broad-spectrum form.
4.2 NK cells
Apart from exhibiting an inherent capacity to kill tumor cells without prior sensitization, NK cells also affect the activity of other immune cells via the production of cytokines, such as interferon-gamma (IFN-γ). NK cells are mainly subdivided into two subsets, CD56brightCD16dim and CD56dimCD16bright NK cells [17]. Over the total NK cells, CD56dimCD16bright cells account for approximately 90% and show mature phenotype, mediating cytolytic reactions while the immature CD56brightCD16dim ones mainly produce cytokines [18]. Approximately 90% of peripheral blood and spleen NK cells are CD56dimCD16bright ones that exhibit strong cytotoxic effects [19]. CD56brightCD16dim cells are the main NK cell subsets in liver, which exhibits a strong ability of producing cytokine [20].
In most healthy nucleated cells [20], highly expressed MHC class I can connect with inhibitory receptors on the surface of NK cells, removing the activation signals of activating receptors [21]. While abnormal cells with downregulated expression of fail to activate inhibitory receptors, in which case the killing of target cells is triggered by the uninhibited activation signal produced by the activated receptor [22]. Activated NK cells first make contact with target cells via integrins such as leukocyte functional antigen-1, and then induce apoptosis with the exocytosis of perforins and granzymes [21]. NK cells can also secrete proinflammatory cytokines and chemokines exerting immunomodulatory effects on the function of Treg cell and dendritic cells (DCs) [23].
The effect of tumor-induced microenvironment on tumor escape from NK cell-mediated cytotoxicity is mainly through the following two mechanisms: the imbalance between NK cell-killing active and killer-inhibiting receptors inhibits the effect of NK cell function; Tumor cells self-modify by selecting/editing low immunogenicity antigens to avoid detection or destruction of NK cells [24]. For example, DC cells can promote the survival, differentiation, cytotoxicity and secretion of interferon-γ of NK cells by secreting cytokines [25].
On account of the median oxygen level of 0.8%, Hepatic cellular carcinoma is always associated with hypoxia [26]. Under conditions of prolonged hypoxia, NK cells themselves upregulate HIF-1α to downregulates the expression of activators of NK cells, such as NKp30, NKp44, NKp46 and the natural killer group 2D (NKG2D) receptor [27]. In addition, HIF-1α as well alters the expression of glycolytic enzymes, metabolite transporters and enzymes involved in biosynthesis (e.g., PMK2, GLUT1 and FAS) [28, 29], and further affect the metabolism of NK cells.
4.3 Regulatory T cells
Regulatory T cells (Treg), an immune subset of CD4 + T cells characterized by expression of Foxp3 and CD25.While Tregs have an impact on maintaining self-tolerance of the immune system, they can also suppress antitumor immunity within the TME and associated with a range of disease progression, including cancer development and progression. Infiltration of a large number of Tregs within a tumor are associated with worse prognosis across many cancers (such as breast, ovarian, and hepatocellular cancers). But in settings of chronic inflammation such as colorectal cancer, the presence of Tregs suppressing chronic inflammation is beneficial, which associated with a good prognosis [30].
During liver cancer metastasis, tumor cells need to evade surveillance and elimination of abnormal tissue cells by the immune system, and Treg cells can play this role. Tregs can promote tumor metastasis by suppressing tumor immune responses and/or directly promoting tumor invasion and migration. This includes competing for and consumption of IL-2, producing immunosuppressive factors such as IL-10, IL-35 [31] and TGF-β, secreting granzyme and/or perforin [32] to destroy effector cells, and exerting their immunosuppressive effects through the interaction of target cell surface receptors (e.g., CTLA-4) with target cells (e.g., CTL) in various ways. Proliferating cancer cells often cause a hypoxic microenvironment, and HIF1α produced by hepatocellular carcinoma cells during hypoxia can upregulate the expression of CCL28 [33], which can effectively recruit CCR10 + Treg cells to tumor sites, thus promoting tumor metastasis. Meanwhile, the abundant liver-resident cells like Kupffer cells, LSECs and HSCs [34], also play an important role in Treg generation. Clinically, elevated Treg levels in peripheral blood have also been observed in patients with liver metastases.
4.4 Myeloid-derived suppressor cells
Myeloid-derived suppressor cells (MDSCs) are immature myeloid cells classified as granulocytic-MDSCs (G-MDSCs) and monocytic-MDSCs (M-MDSCs) based on their phenotypic and morphological features [35]. MDSCs are the primary immunosuppressive cells present in TME that sustain cancer progression, involved in immune escape and immune tolerance. During the progression of the tumor, tumor cells can secrete factors such as stem cell factor (SCF) to stimulate MDSC production by activating JAK and STAT pathways.
MDSC can have a dual effect on immune cells through different pathways. MDSC can inhibit T cell activity by secreting immunosuppressive cytokines, reducing T cell I-selectin expression, oxidative stress induction (inducing production of nitric oxide synthase), depriving T cells of essential amino acids (inducing production of arginase), on the one hand. On the other hand, it stimulates the production of Th17 cells, Treg and M2-type TAM [36], inhibits the proliferation and activation of CTL, and abrogates hepatic NK-cell activity via membrane-bound TGF-β [37]. Among them, M-MDSC and G-MDSC can suppress the immune response using different mechanisms. M-MDSC expresses high levels of arginase 1 and inducible nitric oxide synthase, which inhibit T cell responses. In contrast, G-MDSC acts by producing high levels of activated oxygen ROS [38]. These immunosuppressive mechanisms can lead to failure of antitumor immune responses and promote cancer progression, metastasis, and chemoresistance.
MDSCs function not only to suppress immune surveillance, but also to directly promote tumor metastasis through non-immune functions. The mechanisms include influencing TME remodeling and tumor angiogenesis via production of VEGF, bFGF, Bv8, and matrix metalloproteinase MMP-9 [39]. MDSC also increases CXCL16 expression, and the importance of the CXCL16-CXCR6 axis in cancer development, metastasis is well established, including its promotion of Treg growth [40], promotion of MDSC survival, and induction of monocyte differentiation into TAM [41]. However, its importance in tumor resistance to anticancer drugs has not been revealed [42].
4.5 Tumor-Associated Macrophages
Macrophages are leukocytes with antigen-presenting capacity and play an important role in the modification of tissue structure and the removal of self and foreign components [43]. Macrophages present in tumor tissue are called tumor-associated macrophages (TAMs). In some studies, TAMs have been reported to be actively involved in tumor development, promoting cell proliferation, angiogenesis, metastasis, and invasion [44]. Depending on the embedded microenvironment, macrophages can show different phenotypic states upon activation. This change is called macrophage polarization. Macrophages are subdivided into antitumorigenic M1 type (classically activated macrophages) and pro-tumorigenic M2 type (alternatively activated macrophages) (Fig. 2). M2 macrophages can be further classified into M2a,M2b,M2c and M2d subtypes [45].

A schematic overview of mechanisms and interactions of the M1/M2 model of macrophage. M1 cells exert an inflammatory phenotype and are involved in killing tumor cells, while M2 are involved in inhibiting anti-tumor immunity, impair cytotoxic function
Induced by Th1 cytokine INF-γ or lipopolysaccharide (LPS), M1 is a typical macrophage that expresses lots of IL-12, triggers inflammation by secreting pro-inflammatory cytokines (e.g., IL-6, IL-12), and releases reactive oxygen species (ROS) or toxic intermediates to exert cytotoxic effects [46]. Luo et al. also found that lnc-Ma301 expressed significantly in M1 was lower in hepatocellular carcinoma tissues than in normal tissues, suggesting that it was associated with lower overall survival in HCC patients, and found that lnc-Ma301 inhibited the proliferation, migration, and epithelial–mesenchymal transition (EMT) of HCC cell by targeting cytoplasmic activation/proliferation-associated protein-1 (caprin-1) in a subsequent lung metastasis models [47]. IL-4 or IL-13, TGF-β or glucocorticoids can polarize macrophages into M2 type with low antigen presentation capacity, which secret anti-inflammatory cytokines, express arginase-1, mannose receptors and scavenger receptors in high levels, and prevent infiltration and activation of DC cells and CTLs, thereby inhibiting anti-tumor immunity and promoting tissue repair. In addition, the M2 type produces high levels of PD-L1, which interacts with PD1 on CTL and impair cytotoxic function [48]. M2 type can also promote vascularization through stimulating the formation of new tumor vessels and the remodeling of the established vascular system into a more tortuous and leaky form to promote tumor metastasis [49].
Clinical results show that M1 type macrophages participate in the immune elimination of tumor cells during the initial stages of tumorigenesis, but in the late stage, M1 type is replaced by M2 type, involving in inhibiting the adaptive immune system and promoting tumor cell proliferation, angiogenesis and extracellular matrix (ECM) structure remodeling, so that tumor cells avoid the response of the immune system and metastasize. The conversion between M1 (anti-tumorigenesis) and M2 (pro-tumorigenesis) is a polarization process in response to microenvironment signals, and the polarization process of macrophages mediated by TH2 transforms the anti-tumor environment into a pro-tumor immunosuppressive niche [50].
The recruitment of macrophages in tissues is closely related to chemokines. Studies have found that monocytes can produce more TH2 cytokines and less TH1 cytokines when induced by colony-stimulating factor CSF-1 [51], and increase the expression of allogeneic graft inflammatory factor-1 (AIF-1) through the CSF1R-MEK1/2-Erk1/2-c-Jun axis, which promotes the increase of CXCL16 in macrophages and HCC cell migration [52]. Clinical HCC samples showed a positive correlation between CSF-1 levels and circASAP1 levels, which contributed to cell migration [53]. In addition, peritumoral tissue in HCC is rich in CSF-1 and macrophages, which provides a good pre-metastatic niche for tumor cell metastasis, and may be related to the development of hepatocellular carcinoma, more metastasis in the liver and poor survival after hepatocellular carcinoma resection [54], highlighting the important role of peritumoral tissue in HCC metastasis and recurrence.
Recent studies found a new potential target, carbonic anhydrase X.II. (ca12) in TAMs, which can stimulate CCL8 secretion in a p38-dependent manner, inducing epithelial mesenchymal transformation (EMT) in tumor cells and favoring tumor growth and metastasis [55]. And the studies also showed inhibiting the expression of carbonic anhydrase X.II can reduce tumor growth and lung metastasis in HCC mouse models. In addition, OIT3, as a liver-specific zona pellucida domain protein (LZP), has been validated as a novel marker for replacing activated macrophages. Its overexpression enhances the cell migration and invasion of HCC cells and promotes cancer metastasis [56].
4.6 Tumor-Associated Neutrophils
Neutrophils play a key role in the innate response, exerting antimicrobial and inflammatory functions through different mechanisms such as phagocytosis, degranulation and release of neutrophil extracellular traps (NETs). In response to inflammatory stimuli, a large number of neutrophils are recruited in the TME and transformed into tumor-associated neutrophils (TANs). Related studies showed that, similar to TAMs, TANs are not terminally differentiated immune effector cells, but polarized to N1 (antitumorigenic phenotype) and N2 (pro-tumorigenic phenotype) under the complex regulation of tumor microenvironment [57].
N1 neutrophils inhibit tumor growth by producing ROS, TNF-α and reducing arginase expression, while producing a variety of chemokines to recruit and activate immune cells. N2 neutrophils promote tumor cell proliferation, migration, and angiogenesis through MMPs, VEGF, and arginase-expressing enzymes [58]. Mouse models of tumor demonstrated that type I interferon or TGF-β can induce the conversion between N1 and N2, indicating the presence of an antagonistic signaling pathway [59]. In addition, increased oxygen supply was shown to induce the transformation of N2 to N1 phenotype, suggesting the facilitative effect of hypoxia on tumor metastasis [60].
Hepatocyte growth factor (HGF) can stimulate the proliferation of hepatocyte cells and is involved in embryogenesis, wound healing, angiogenesis, tissue and organ regeneration, morphogenesis and carcinogenesis. Studies showed that TAN-derived HGF can directly enhance the transfer ability of HCC cells by activating the HGF receptor (MET) [61]. The granules of TANs are involved in cancer metastasis. For example, MMPs can promote epithelial-mesenchymal transformation in HCC-TME and disrupt the degradation balance of the stroma, thereby promoting tumor cells to break through the basement membrane and extracellular matrix, invade and metastasize to surrounding tissues. Interestingly, the antitumor effect of N1 type is mediated by expressing enhanced NADPH oxidase activity, which increases the production of ROS to exerts cytotoxic effects. However, ROS may damage DNA bases and lead to gene mutations, which promote tumor development conversely [62]. Feng et al. also demonstrated that ROS can produce activated p38MARK to promote the expression of β3 integrins on the surface of HCC cells, allowing HCC cells to be more invasive and metastatic [63].
Neutrophil extracellular traps (NETs) are web-like structure composed of DNA fibers, histone and antimicrobial proteins extruded by activated neutrophils. NETs can block the cytotoxic effect of immune cells by covering tumor cells, and involve in lung metastasis from hepatocellular tumor [64]. Studies showed that TANs in HCC patients exhibits greater NETs secretion and more NETs are found in metastatic cases. And in subsequent model studies, inhibition of NETs by DNase reduces liver metastasis as direct evidence of the role of NETs in HCC metastasis [65].
Following HCC cells capture, NETs induce resistance to death and enhance invasion capacity of HCC cells, and NETs are internalized into captured HCC cells, activating the TLR4/9-COX2 signaling pathway to trigger their metastatic potential [66]. Activation of the TLR4/9-COX2 pathway is an important molecular event in which NETs enhance the metastasis ability of HCC cells. In addition to inducing COX2 upregulation and triggering an aggressive inflammatory response, the upstream molecule TLR4/9 itself is an important sensor of multiple damage-related molecular patterns, mediating cellular contact between host cells, and the activation of TLR4/9 also indicates a highly metastatic phenotype of HCC cells [67]. Besides, in normal tissues, NETs can exert potentially cytotoxic effect against captured pathogens, and this mechanism can also act on the captured HCC cells to trigger potentially cytotoxic resistance and enhance the viability of tumor cells. The adhesion of HCC cell in the liver and lung was significantly increased in LPS-induced NETs models, and this increase was abolished by DNase1, suggesting the importance of NETs on HCC metastasis [65].
New studies found that NETs also promote epithelial-mesenchymal-transformation (EMT) in tumor cells and downregulate the expression of the adhesion molecule VE-cad on endothelial cells, thereby disrupting the integrity of vascular system in tumor tissues and enhancing the metastasis of HCCs [68]. The contents of NETs were also found to be involved in HCC metastasis. The highly oxidized mtDNA in HCC-NETs can cause significant inflammation [69], and NETs-associated cathepsin G (cG) component can decrease E-cadherin expression in vitro and promote HCC cells invasion [70], which provides a potential target for HCC treatment.
4.7 B cells
According to current studies, the role of B cells in HCC metastasis can’t be clearly defined as inhibition or promotion. On the one hand, B cells are effective antigen-presenting cells (APCs), which can activate CTL activity and secrete cytokines that contribute to liver cancer metastasis, and can also directly kill tumor cells by secreting cell granzyme B [71, 72]. On the other hand, cytokines produced by B cells can also recruit MDSC and promote angiogenesis, and its subgroup B-REG can also convert resting CD4 + T into Treg to promote tumor metastasis. Studies have shown that the loss of specific TGF-β on the surface of B cells can inhibit the development of HCC [73]. Liu et al. also found that CXCR3 + B cells can interact with the HCC microenvironment to promote the polarization of M2b macrophages at the edge of invasive tumors [74] and stimulate the potential of metastasis.
5 The non-cellular component of TME
Non-cellular components of TME cover a wide range, including inflammatory cytokines (such as interleukin-IL-6), proteolytic enzymes (such as matrix metalloproteinase MMPs), proteins (such as histidine rich glycoprotein HRG) and exosome special structures. In addition, hypoxia environment also counts a lot in the metastasis of liver cancer.
Due to the specific physiological structure and functional characteristics of liver, hypoxia is considered to be a characteristic indicator of HCC microenvironment [75], which can promote HCC metastasis through interaction with cytokines (such as TGF-β, HIF-α, etc.). Histidine rich glycoprotein (HRG) is a plasma protein synthesized by the liver and plays a role in inhibiting insoluble immune complexes and promoting the clearance of apoptotic necrotic cells in healthy liver. In HCC-TME, HRG can mediate phenotypic transformation of TAMs, down-regulate M2 markers such as MRC1, Arg1, IL10 and CCL-22, and up-regulate M1 markers such as IL6 and CXCL-9, thus promoting the occurrence of anti-tumor polarization of TAMs. Recent studies have found that increased oxygenation caused by vascular normalization in HRG+ tumors provide stimulation for TAMs to move away from M2-type polarization to some extent [76].
Inflammation is a non-specific manifestation of tumor TME, which is manifested by significant TH1 and TH2 cytokine conversion in HCC, and this anti-inflammatory state can promote HCC metastasis [77]. Matrix metalloproteinase MMPs, a typical proteolytic enzyme, promotes EMT through hydrolysis, activates the metastasis potential of tumor cells, and participates in the regulation of signaling pathways to enhance the metastasis activity of HCC [78]. Recent studies have also pointed to exosomes as a target, but it should be noted that the action properties of exosomes are influenced by their donor cells and inclusions. When tumor cells impact neighboring cells through exosomes to establish tumorigenic microenvironment, stromal cells (such as stellate cells and mesenchymal stem cells) and immune cells also interfere with tumor cells through exosomes to promote or prevent tumorigenesis [79].
6 Conclusion and discussion
The immune microenvironment of HCC is a complex mixture of hepatocellular carcinoma cells, stromal cells, and various cytokines and proteins, which together lead to epigenetic changes in HCC and a high incidence of metastasis. Tumor cells do not exhibit disease alone but form a complex signal interaction system with the matrix. In this way, all cellular and non-cellular components of TME prepare a tumor niche, providing rapid proliferation of HCC cells while also escaping the body’s defenses. TME components also provide an opportunity for cancer cells to pass through the basement membrane and invade the circulation, forming a post-metastatic microenvironment suitable for colonization and promoting metastasis.
The multiple immune cells infiltrating in the TME not only provides suitable soil for the growth of tumor cells and mediates immune tolerance through the formation of an immunosuppressive microenvironment, but also creating conditions for immune escape, mediating the metastatic process of cancer cells. In general, cytotoxic T cells and NK cells in the TME were intended to construct immune defense nonspecifically, while Regulatory T cells and Myeloid-derived suppressor cells play an immunosuppressive function by inhibiting effector cells. When it comes to myeloid cells, their functions in the TME are usually controversial. As for macrophages, the M1 macrophages with pro-inflammatory properties are usually considered as anti-tumor and anti-metastasis cells, while the M2 macrophages with anti-inflammatory functions are equipped with tumor-promoting capacities. Neutrophils have also been identified with varied functions and been classified using different terms, including N1/N2 neutrophils, tumor-associated neutrophils, and polymorphonuclear neutrophil myeloid-derived suppressor cells.
For most patients with advanced-stage cancer, surgery and radiotherapy are difficult to achieve satisfactory therapeutic effects. While in the process of tumor metastasis from initiation to colonization, the occurrence of each step is the result of the joint action of some specific genes and signaling pathways which hints that blocking one of these steps may block the formation of metastases. Those discoveries cells have brought new insight into our understanding of infiltrating immune cells in HCC-TME. In-depth study of these mechanisms will help us to find possible immunotherapy for more effective treatment of HCC patients by weakening the pro-tumor or enhancing the anti-tumor effect. Recently, there is increasing evidence that TAMs and Tregs in the microenvironment promote tumor development and metastasis. Despite the lack of research on HCC, inhibitors aiming at CC chemokine receptors have been proved to inhibit lung metastasis in some tumors [80], which may be an effective target for HCC metastasis. Besides, NETs released by neutrophils also suggest strong inhibition of tumor metastasis.
Although the complex and diverse functions performed by immune cells in TME make therapeutic strategies targeting specific elements often ineffective, all these characteristics make the immune microenvironment a powerful target for cancer therapy. Finding suitable targets in TME is only the first step on the long road to clinical application, and we need to explore further roads.
Data availability
Not applicable.
References
J. Ferlay, I. Soerjomataram, R. Dikshit et al., Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136(5), E359–E386 (2015)
M.T. Birgani, V Carloni, Tumor microenvironment, a paradigm in hepatocellular carcinoma progression and therapy [J]. Int. J. Mol. Sci 18(2), 19 (2017)
K.O. Asafo-Agyei, H. Samant, Hepatocellular carcinoma. 2023 Mar 27. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing (2023)
D. Hanahan, R.A. Weinberg, Hallmarks of cancer: the next generation. Cell 144(5), 646–674 (2011)
U. Protzer, M.K. Maini, P.A. Knolle, Living in the liver: hepatic infections. Nat. Rev. Immunol 12(3), 201–213 (2012)
B.P. Keenan, L. Fong, R.K. Kelley, Immunotherapy in hepatocellular carcinoma: the complex interface between inflammation, fibrosis, and the immune response. J. Immunother. Cancer 7(1), 267 (2019)
X. li, P. Ramadori, D. Pfister, The immunological and metabolic landscape in primary and metastatic liver cancer. Nat. Rev. Cancer 21(9), 541–557 (2021)
S.I. Grivennikov, F.R. Greten, M. Karin, Immunity, inflammation, and cancer. Cell 140(6), 883–899 (2010)
T. Flecken, N. Schmidt, S. Hild et al., Immunodominance and functional alterations of tumor-associated antigen-specific CD8 + T-cell responses in hepatocellular carcinoma. Hepatology 59(4), 1415–1426 (2014)
R.A. MacDonald, “Lifespan” of liver cells. Autoradio-graphic study using tritiated thymidine in normal, cirrhotic, and partially hepatectomized rats. Arch. Intern. Med 107, 335–343 (1961)
Y. Liu, X.T. Cao, Characteristics and significance of the pre-metastatic niche. Cancer Cell 30(5), 668–681 (2016)
Y.S. Chang, E. Di Tomaso, D.M. McDonald et al., Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood. Proc. Natl. Acad. Sci. U S A 97(26), 14608–14613 (2000)
S. Xiong, R. Wang, Q. Chen et al., Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Am. J. Cancer Res 8(2), 302–316 (2018)
J. Song, Z.H. Ge, X.R. Yang et al., Hepatic stellate cells activated by acidic tumor microenvironment promote the metastasis of hepatocellular carcinoma via osteopontin [J]. Cancer. Lett 356(2), 713–20 (2015)
P. Heneberg, Paracrine tumor signaling induces transdifferentiation of surrounding fibroblasts. Crit. Rev. Oncol. Hematol 97, 303–311 (2016)
M. Shimoda, Y. Tomimaru, K.P. Charpentier et al., Tumor progression-related transmembrane protein aspartate-β-hydroxylase is a target for immunotherapy of hepatocellular carcinoma. J. Hepatol 56(5), 1129–1135 (2012)
A. Poli, T. Michel, M. Thérésine et al., CD56bright natural killer (NK) cells: an important NK cell subset. Immunology 126(4), 458–465 (2009)
E. Alari-Pahissa, C. Grandclément, B. Jeevan-Raj et al., Inhibitory receptor-mediated regulation of natural killer cells. Crit. Rev. Immunol 34(6), 455–465 (2014)
M. Amand, G. Iserentant, A. Poli et al., Human CD56(dim)CD16(dim) cells as an Individualized Natural Killer Cell subset. Front. Immunol 8, 699 (2017)
F.D. Shi, H.G. Ljunggren, A. La Cava et al., Organ-specific features of natural killer cells. Nat. Rev. Immunol 11(10), 658–671 (2011)
M.G. Morvan, L.L. Lanier, NK cells and cancer: you can teach innate cells new tricks. Nat. Rev. Cancer 16(1), 7–19 (2016)
E.O. Long, H.S. Kim, D. Liu et al., Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu. Rev. Immunol 31, 227–258 (2013)
C. Guillerey, N.D. Huntington, M.J. Smyth, Targeting natural killer cells in cancer immunotherapy. Nat. Immunol 17(9), 1025–1036 (2016)
N.J.W. Easom, K.A. Stegmann, L. Swadling et al., IL-15 overcomes Hepatocellular Carcinoma-Induced NK Cell dysfunction. Front. Immunol 9, 1009 (2018)
M. Yamamoto, T. Tatsumi, T. Miyagi et al., α-Fetoprotein impairs activation of natural killer cells by inhibiting the function of dendritic cells. Clin. Exp. Immunol 165(2), 211–219 (2011)
C. CHEN, T. LOU, Hypoxia inducible factors in hepatocellular carcinoma. Oncotarget 8(28), 46691–46703 (2017)
M. Balsamo, C. Manzini, G. Pietra et al., Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur. J. Immunol 43(10), 2756–2764 (2013)
V.L. Dengler, M. Galbraith, J.M. Espinosa, Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol 49(1), 1–15 (2014)
E. Furuta, S.K. Pai, R. Zhan et al., Fatty acid synthase gene is up-regulated by hypoxia via activation of akt and sterol regulatory element binding protein-1. Cancer Res 68(4), 1003–1011 (2008)
F. Shan, A. Somasundaram, T.C. Bruno et al., Therapeutic targeting of regulatory T cells in cancer. Trends Cancer 8(11), 944–961 (2022)
M.E. Turnis, D.V. Sawant, A.L. Szymczak-Workman et al., Interleukin-35 limits anti-tumor immunity. Immunity 44(2), 316–329 (2016)
W.J. Grossman, J.W. Verbsky, W. Barchet et al., Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 21(4), 589–601 (2004)
L. Ren, Y. Yu, L. Wang et al., Hypoxia-induced CCL28 promotes recruitment of regulatory T cells and tumor growth in liver cancer. Oncotarget 7(46), 75763–75773 (2016)
A.W. Thomson, P.A. Knolle, Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol 10(11), 753–766 (2010)
P. De Cicco, G. Ercolano, A. Ianaro, The New Era of Cancer Immunotherapy: targeting myeloid-derived suppressor cells to overcome Immune evasion. Front. Immunol 11, 1680 (2020)
J. Prieto, I. Melero, B. Sangro, Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol 12(12), 681–700 (2015)
H. Li, Y. Han, Q. Guo et al., Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol 182(1), 240–249 (2009)
J.I. Youn, M. Collazo, I.N. Shalova et al., Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J. Leukoc. Biol 91(1), 167–181 (2012)
M. Motallebnezhad, F. Jadidi-Niaragh, E.S. Qamsari et al., The immunobiology of myeloid-derived suppressor cells in cancer. Tumour Biol 37(2), 1387–1406 (2016)
Y.N. Xing, J.Y. Zhang, H.M. Xu, The roles of serum CXCL16 in circulating Tregs and gastrointestinal stromal tumor cells. Onco Targets Ther 9, 3939–3949 (2016)
R. Allaoui, C. Bergenfelz, S. Mohlin et al., Cancer-associated fibroblast-secreted CXCL16 attracts monocytes to promote stroma activation in triple-negative breast cancers. Nat. Commun 7, 13050 (2016)
J. Korbecki, K. Bajdak-Rusinek, P. Kupnicka et al, The role of CXCL16 in the pathogenesis of cancer and other diseases. Int. J. Mol. Sci 22(7), 3490 (2021)
P.J. Murray, T.A. Wynn, Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol 11(11), 723–737 (2011)
Y. Liu, X. Cao, The origin and function of tumor-associated macrophages. Cell. Mol. Immunol 12(1), 1–4 (2015)
S. Olin, G. Chinetti-Gbaguidi, B. Staels, Macrophage phenotypes in atherosclerosis. Immunol. Rev 262(1), 153–166 (2014)
J.Y. Ao, X.D. Zhu, Z.T. Chai et al., Colony-stimulating factor 1 receptor blockade inhibits tumor growth by altering the polarization of tumor-associated macrophages in hepatocellular Carcinoma. Mol. Cancer Ther 16(8), 1544–1554 (2017)
H.L. Luo, T. Luo, J.J. Liu et al., Macrophage polarization-associated lnc-Ma301 interacts with caprin-1 to inhibit hepatocellular carcinoma metastasis through the Akt/Erk1 pathway. Cancer Cell. Int 21(1), 422 (2021)
K. Wu, I. Kryczek, L. Chen et al., Kupffer cell suppression of CD8 + T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res 69(20), 8067–8075 (2009)
M.M. Liu, J. Yang, B.S. Xu et al., Tumor metastasis: mechanistic insights and therapeutic interventions [J]. MedComm 2(4), 587–617 (2021)
R. Noy, J.W. Pollard, Tumor-associated macrophages: from mechanisms to therapy. Immunity 41(1), 49–61 (2014)
F.A. Verreck, T. De Boer, D.M. Langenberg et al., Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc. Natl. Acad. Sci. U S A 101(13), 4560–4565 (2004)
H. Cai, X.D. Zhu, J.Y. Ao et al., Colony-stimulating factor-1-induced AIF1 expression in tumor-associated macrophages enhances the progression of hepatocellular carcinoma. Oncoimmunology 6(9), e1333213 (2017)
Z.Q. Hu, S.L. Zhou, J. Li et al., Circular RNA sequencing identifies CircASAP1 as a key regulator in hepatocellular carcinoma metastasis. Hepatology 72(3), 906–922 (2020)
X.D. Zhu, J.B. Zhang, P.Y. Zhuang et al., High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J. Clin. Oncol 26(16), 2707–2716 (2008)
N. Graham, J.W. Pollard, An acid trip activates protumoral macrophages to promote hepatocellular carcinoma malignancy. J. Clin. Invest 132(7), e158562 (2022)
S. Yang, J. Zhang, Y. Xu et al., OIT3 mediates macrophage polarization and facilitates hepatocellular carcinoma progression. Cancer Immunol. Immunother 71(11), 2677–2689 (2022)
M.A. Giese, L.E. Hind, A. Huttenlocher, Neutrophil plasticity in the tumor microenvironment. Blood 133(20), 2159–2167 (2019)
N. Mukaida, S.I. Sasaki, T. Baba, Two-faced roles of tumor-associated neutrophils in cancer development and progression. Int. J. Mol. Sci 21(10), 3457 (2020)
Z.G. Fridlender, J. Sun, S. Kim et al., Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 16(3), 183–194 (2009)
K. Mahiddine, A. Blaisdell, S. Ma et al., Relief of tumor hypoxia unleashes the tumoricidal potential of neutrophils. J. Clin. Invest 130(1), 389–403 (2020)
M. He, A. Peng, X.Z. Huang et al., Peritumoral stromal neutrophils are essential for c-Met-elicited metastasis in human hepatocellular carcinoma. Oncoimmunology 5(10), e1219828 (2016)
J.E. Klaunig, Oxidative stress and cancer. Curr. Pharm. Des 24(40), 4771–4778 (2018)
X.X. Feng, M. Liu, W. Yan et al., β3 integrin promotes TGF-β1/H2O2/HOCl-mediated induction of metastatic phenotype of hepatocellular carcinoma cells by enhancing TGF-β1 signaling. PLoS One 8(11), e79857 (2013)
L. Yang, Q. Liu, X. Zhang et al., DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 583(7814), 133–138 (2020)
L.Y. Yang, Q. Luo, L. Lu et al., Increased neutrophil extracellular traps promote metastasis potential of hepatocellular carcinoma via provoking tumorous inflammatory response. J. Hematol. Oncol 13(1), 3 (2020)
A.R. Thierry, S. El Messaoudi, P.B. Gahan et al., Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev 35(3), 347–376 (2016)
M. Dajon, K. Iribarren, I. Cremer, Toll-like receptor stimulation in cancer: a pro- and anti-tumor double-edged sword. Immunobiology 222(1), 89–100 (2017)
Z.Z. Jiang, Z.P. Peng, X.C. Liu et al., Neutrophil extracellular traps induce tumor metastasis through dual effects on cancer and endothelial cells. Oncoimmunology 11(1), 2052418 (2022)
L.Y. Yang, X.T. Shen, H.T. Sun et al., Neutrophil extracellular traps in hepatocellular carcinoma are enriched in oxidized mitochondrial DNA which is highly pro-inflammatory and pro-metastatic. J. Cancer 13(4), 1261–1271 (2022)
X. Guan, Y. Lu, H. Zhu et al., The crosstalk between Cancer cells and neutrophils enhances Hepatocellular Carcinoma Metastasis via Neutrophil Extracellular Traps-Associated cathepsin G component: a potential therapeutic target. J. Hepatocell Carcinoma 8, 451–465 (2021)
K.E. De Visser, L.V. Korets, L.M. Coussens, De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell 7(5), 411–423 (2005)
P. Tsou, H. Katayama, The emerging role of B cells in tumor immunity. Cancer Res 76(19), 5597–5601 (2016)
S. Shalapour, X.J. Lin, I.N. Bastian et al., Inflammation-induced IgA + cells dismantle anti-liver cancer immunity. Nature 551(7680), 340–345 (2017)
R.X. Liu, Y. Wei, Q.H. Zeng et al., Chemokine (C-X-C motif) receptor 3-positive B cells link interleukin-17 inflammation to protumorigenic macrophage polarization in human hepatocellular carcinoma. Hepatology 62(6), 1779–1790 (2015)
T. Kietzmann, E.Y. Dimova, D. Flügel et al., Oxygen: modulator of physiological and pathophysiological processes in the liver [J]. Z. Gastroenterol 44(1), 67–76 (2006)
N. Balasubramaniyan, M. Ananthanarayanan, F.J. Suchy, Nuclear factor-κB regulates the expression of multiple genes encoding liver transport proteins. Am. J. Physiol. Gastrointest. Liver Physiol 310(8), G618–G628 (2016)
A. Budhu, M. Forgues, Q.H. Ye et al., Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 10(2), 99–111 (2006)
F. Mannello, Tissue inhibitors of metalloproteinases and programmed cell death: conundrums, controversies and potential implications. Apoptosis 6(6), 479–482 (2001)
S. Zhou, M. Abdouh, V. Arena et al., Reprogramming malignant Cancer cells toward a Benign phenotype following exposure to human embryonic stem cell microenvironment. PLoS One 12(1), e0169899 (2017)
M. Miao, E. De Clercq, G.D. Li, Clinical significance of chemokine receptor antagonists. Expert Opin. Drug Metab. Toxicol 16(1), 11–30 (2020)
Funding
This study was supported by the Project of the Shanghai Municipal Health Commission (20204Y0012), Seed Fund of Renji Hospital (RJZZ18-010), Shenkang three-year action plan (SHDC2020CR2003A, SHDC2020CR5012), Innovative Research Team of High-Level Local Universities in Shanghai (SSMU-ZDCX20180802), National Natural Science Foundation of China (81972205), and the Project of Shanghai key clinical specialties (shslczdzk05801).
Author information
Authors and Affiliations
Contributions
Yiwen Chen, Yuhang Zhou, Ziyang Yan, and Peilin Tong searched and analyzed the data, and wrote the manuscript.Kang He and Qiang Xia designed the idea and revised the manuscript.
Corresponding authors
Ethics declarations
Ethical approval
This manuscript is a review article and does not involve a research protocol requiring approval by the relevant institutional review board or ethics committee.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, Y., Zhou, Y., Yan, Z. et al. Effect of infiltrating immune cells in tumor microenvironment on metastasis of hepatocellular carcinoma. Cell Oncol. 46, 1595–1604 (2023). https://doi.org/10.1007/s13402-023-00841-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-023-00841-6