
Abstract
The tumor microenvironment is a complex and heterogeneous milieu in which multiple interactions occur between tumor and host cells. Immunosuppressive cells which are present in this microenvironment, such as regulatory T (Treg) cells and myeloid-derived suppressor cells (MDSCs), play an important role in tumor progression, via down-regulation of antitumor responses. MDSCs represent a heterogeneous group of cells originated from the myeloid lineage that are in the immature state. These cells markedly accumulate under pathologic conditions, such as cancer, infection, and inflammation, and use various mechanisms to inhibit both adaptive and innate immune responses. These immunosuppressive mechanisms include deprivation of T cells from essential amino acids, induction of oxidative stress, interference with viability and trafficking of T cells, induction of immunosuppressive cells, and finally polarizing immunity toward a tumor-promoting type 2 phenotype. In addition to suppression of antitumor immune responses, MDSCs can also enhance the tumor metastasis and angiogenesis. Previous studies have shown that increased frequency of MDSCs is related to the tumor progression. Moreover, various drugs that directly target these cells or reverse their suppressive activity can improve antitumor immune responses as well as increase the efficacy of immunotherapeutic intervention. In this review, we will first discuss on the immunobiology of MDSCs in an attempt to find the role of these cells in tumor progression and then discuss about therapeutic approaches to target these cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Suppression of antitumor immune responses is one of the main mechanisms by which tumor cells escape from destruction by the immune system [1, 2]. While the immune system restrains the tumor growth, immunosuppressive cells in the tumor microenvironment, such as myeloid-derived suppressor cells (MDSCs), type 2 natural killer (NK) T cells, regulatory T (Treg) cells, tumor-associated macrophages (TAMs), etc., can suppress the immune system and accelerate tumor growth [3–6]. Moreover, these cells reduce the efficacy of immunotherapeutic interventions and until the imposed suppression is not resolved, the immunotherapy of cancer will have little effect [7–9]. So, MDSC targeting may be a beneficial strategy for improvement of efficiency of immunotherapeutic interventions [10].
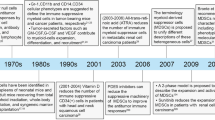
MDSCs were the first identified in the late 1970s in tumor-bearing mice and introduced as null cells, veto cells, or as natural suppressor (NS) cells which were able to suppress T cell responses [11–14]. These cells did not express markers which are conventionally expressed on the surface of mature B cells, T cells, macrophages, dendritic cells (DCs), monocytes, and natural killer (NK) cells; they were, therefore, originally called null cell [13, 15–17]. Then, these cells were called with different names, such as myeloid suppressor cells (MSCs), immature myeloid cells (IMCs), or natural suppressors cells [18]. Indeed, no unique specific term was assigned for these cells by 2007, until Gabrilovich et al. in a Letter to the Editor entitled “The Terminology Issue for Myeloid-Derived Suppressor Cells,” suggested “MDSC” as a name for these cells and partly resolved the controversies about their nomenclature [19, 20].
MDSCs are one of the most important immune regulatory cells which expand under different pathological conditions such as cancer, inflammation, infection, and other damages [21]. These cells use different mechanisms to suppress both adaptive and innate immune responses [22, 23]. MDSCs are in the immature state and have remarkable potential to suppress the immune system [24]. Moreover, these cells have also non-immunological functions, like promoting tumor angiogenesis and increasing metastatic potential of cancer cells [9, 25]. Studies have shown that the accumulation of MDSCs in the blood and lymphoid tissues is increased as the tumor develops [26]. Furthermore, the number of MDSCs is negatively correlated with the survival rate of tumor-bearing hosts [24, 27]. Additionally, the association of MDSCs with non-malignant settings such as various infections, graft versus host disease, different autoimmune diseases, and traumatic stress has also been described [28–30]. Moreover, various protocols have been introduced for the generation of MDSCs in vitro, and several studies have been performed to evaluate the possibility of application of in vitro-generated MDSCs for regulation of immune responses in autoimmune settings and transplantation [17, 30, 31]. In this review, we will just keep to the role of MDSCs in tumors, and the association of MDSCs with non-malignant settings will not be discussed.
The origin of MDSCs
MDSCs originate from myeloid progenitor cells in the bone marrow that do not pass the final stages of differentiation. These cells are identified by the expression of specific cell surface markers and their ability to suppress the immune system [8, 32]. Bone marrow hematopoietic stem cells differentiate into the common myeloid progenitor cells, and then these cells differentiate into IMCs, which did not show any inhibitory activity [17, 24]. Because they immediately differentiate into macrophages, granulocytes, or dendritic cells, IMCs are present only in the bone marrow and are not found in the secondary lymphoid organs in healthy people and under physiological conditions. But a series of mediators are produced in pathological conditions such as cancer, inflammation, infection, sepsis, and some autoimmune diseases that disrupt differentiation of IMCs and lead to activation of these cells in the immature state. These activated immature cells represent inhibitory functions and called MDSCs [24, 29] (Fig. 1).

MDSCs originate from myeloid progenitor cells in the bone marrow and divided into two subsets. MDSC myeloid-derived suppressor cell, STAT signal transducer and activator of transcription, ROS reactive oxygen species, iNOS inducible nitric oxide synthase, NO nitric oxide
MDSC surface markers and subsets
Different types of mouse MDSCs
Mouse MDSCs are identified by co-expression of CD11b and GR1 [33]. GR1 molecules, which are expressed on the surface of these cells, have two isoforms, LY6G and LY6C, each are coded by separate genes and identified by specific antibodies. Based on these isoforms, mice-derived MDSCs are divided into two groups including monocytic MDSCs (M-MDSCs) which have a CD11b+LY6Glow/−LY6Chigh phenotype and granulocytic MDSCs (G-MDSCs) with CD11b+LY6G+LY6Clow/− phenotype [24, 34, 35]. In addition to cell surface markers, nuclear morphology as well inhibitory mechanisms of these subsets are different [24, 34]. Nuclear morphology is mononuclear and multiplied in monocytic and granulocytic lineages, respectively [32]. Suppressive activity of granulocytic MDSCs is mostly through reactive oxygen species (ROS) which are produced following the activation of signal transducer and activator of transcription 3 (STAT3) and NADPH [36]. In monocytic MDSCs, the activation of STAT1 pathway is resulted in enhancement of inducible nitric oxide synthase (iNOS) expression and production of nitric oxide (NO) [8, 34, 37] (Fig. 1). Heterogeneity of MDSCs is not limited to these two subtypes, and several other cell surface markers are introduced such as F4/80، CD124 (IL-4Rα) ،CD115 (M-CSF-1R) and CD80 (B7.1), which are used for identification of MDSC subsets [24, 32, 38]. With the identification of these markers, probably more subsets will be identified in the future.
Different types of human MDSCs
In human, CD33+ CD11b+ HLA-DRlow/neg cells, which lack markers of mature lymphoid and myeloid cells, are known as MDSCs and according to the expression of CD14 and CD15 are divided into monocytic and granulocytic subpopulations. Granulocytic MDSCs are CD15+ and monocytic MDSCs express CD14 [30, 39–41]. It has been observed that diversity of cell surface markers in tumor-derived MDSCs is very high, which is in part due to the differences in the factors that are involved in the development and activation of MDSCs [8] (Table 1). In spite of identification of various markers for these cells, the best characteristic to identify these cells is still their inhibitory function [24].
Factors involved in the differentiation, expansion, and activation of MDSCs
Factors involved in the differentiation, expansion, and activation of MDSCs are divided into two groups. The first group includes factors that stimulate myelopoiesis and inhibit differentiation of IMCs into mature myeloid cells, as a result, enhance the accumulation of IMCs in lymphoid organs and tumor microenvironment. These factors are mainly secreted by tumor cells and activate STAT3 signaling pathway [24, 60, 61]. The second group factors are involved in activation of IMCs. These activated IMCs which have inhibitory functions are named MDSCs [24]. These factors are mainly secreted by activated T cells and tumor stromal cells and activate STAT1, STAT6, and NFκB signaling pathways [10, 60, 62–64] (Fig. 2).

Signaling pathways and transcription factors involved in the differentiation, expansion, and activation of MDSCs. VEGF vascular endothelial growth factor, MYD88 myeloid differentiation primary-response gene 88, M-CSF macrophage colony-stimulating factor, JAK Janus kinase, IL interleukin, IFN-γ interferon-γ, GM-CSF granulocyte-macrophage colony-stimulating factor, G-CSF granulocyte colony-stimulating factor, STAT signal transducer and activator of transcription, PGE2 prostaglandin E2, iNOS inducible nitric oxide synthase, BCL-XL B cell lymphoma XL
Tumor cell-derived granulocyte-macrophage colony-stimulating factor (GM-CSF) enhances the expansion and accumulation of MDSCs in tumor site and peripheral lymph nodes. In addition, treatment of mice with recombinant GM-CSF has the similar results [17, 65–68]. Interestingly, GM-CSF is used as a vaccine adjuvant in human for immune system stimulation [17, 69, 70]. This controversy can be explained by the dual role of GM-CSF in the immune system, stimulatory and suppressive, according to the dose, duration of exposure, and its concentration [17].
Studies showed that stem cell factor (SCF) is one of the most important factors involved in MDSC accumulation. Tumor cell-secreted SCF contributes to the expansion of MDSC population by accelerating myelopoiesis and attenuating myeloid cells differentiation. Accumulation of MDSCs is significantly reduced through inhibition of SCF expression in tumor cells or blocking c-kit-SCF interaction by using anti-c-kit [71, 72].
The S100A8 and S100A9, calcium-binding proteins, are involved in the regulation of MDSC suppressive activity and accumulation [32, 73, 74]. These proteins enhance the suppressive functions of MDSCs and contribute to MDSC recruitment to tumor sites [24].
C5a is a component of the complement system which is also known as anaphylatoxin. C5a binds to its receptor on the surface of MDSCs and regulates their functions. Studies have revealed that C5a receptor is expressed on both MDSC subpopulations, G-MDSCs and M-MDSCs, in tumor-bearing mice. However, C5a receptor signaling has different effects on the M-MDSCs versus G-MDSCs. While the C5a increases the production of reactive nitrogen species (RNS) and ROS in M-MDSCs, it enhances the migration of G-MDSCs toward the tumor sites and peripheral lymph nodes [32, 75].
Interferon-γ (IFN-γ) can also affect the expansion and activation of MDSCs. It has been shown that IFN-γ signaling increases the expression of iNOS and arginase 1 (ARG1) in MDSCs [76, 77]. Consistently, MDSCs are not able to increase the expression of ARG1 and iNOS in the STAT1-knockout mice, thereby cannot inhibit T cells [10, 24, 78]. In contrast, Sinha et al. demonstrated that MDSCs derived from the IFN-γ receptor-deficient mice have intact inhibitory function. This controversy may be in part related to the different responses of MDSC subsets to IFN-γ [79].
Hypoxia-inducible factor (HIF)-1α in the tumor microenvironment upregulates the expression of NOS and ARG1 in MDSCs and enhances their functions. It also regulates the differentiation of MDSCs into TAMs [80, 81].
Endoplasmic reticulum disulfide oxidase ERO 1-α, which is overexpressed in a variety of tumor cells, is a predictor of poor prognosis in breast cancer [82]. Recently, Tanaka et al. demonstrated that overexpression of ERO1-α in tumor cells resulted in the induction and recruitment of G-MDSCs [83].
In addition to factors described above, other mediators such as macrophage colony-stimulating factor (M-CSF), granulocyte colony-stimulating factor (G-CSF), matrix metalloproteinases-9 (MMP-9), transforming growth factor-β (TGF-β), IL-1β, IL-4, IL-6, IL-10, IL-12, IL-13, vascular endothelial growth factor (VEGF), gangliosides, prostaglandins, CCL2, CXCL5, ligands for Toll-like receptors, and CXCL12 are also involved in the expansion and activation of MDSCs [17, 24, 84–87].
Transcription factors and molecular mechanisms involved in the differentiation, expansion, and activation of MDSCs
Differentiation, expansion, and activation of MDSCs are regulated via a series of transcription factors including STAT1, STAT3, STAT5, STAT6, and nuclear factor-κB (NF-κB) [88] (Fig. 2).
STAT3 serves as main signal transducer protein involved in MDSC expansion [89]. STAT3 signaling in myeloid cells inhibits their differentiation into mature cells, promotes their proliferation, and increases the expression of cyclin D1, B cell lymphoma XL (BCL-XL), survivin, and MYC in these cells, therefore, induces an anti-apoptotic state in these cells [22, 24, 90]. The calcium-binding proteins S100A8 and S100A9 are involved in the differentiation of myeloid cells, and their expression is regulated by STAT3 signaling [71, 91]. STAT3 signaling leads to increment of the expression of these proteins in myeloid progenitors. These proteins not only inhibit DC differentiation but are also involved in MDSC recruitment and chemotaxis, through binding to their receptor, receptor for advanced glycation end-products (RAGE), which are expressed on the surface of MDSCs [22, 92]. Additionally, the expression of S100A8 and S100A9 is also increased in MDSCs via STAT3 signaling, and these proteins contribute to MDSC suppressive functions by binding to NADPH complex and enhancing ROS production [71].
STAT3 signaling has different effects on the MDSC functions and expansion. For example, it increases the production of ROS via up-regulation of component of NADPH oxidase complex such as p47phox (NCF1) and gp91phox (CYBB), and thus enhances the inhibitory function of MDSCs [47]. STAT3 can also disrupt the maturation of DCs by down-regulation of PKCβII (protein kinase Cβ isoform II), thereby leads to the expansion of MDSCs [93]. C/EBPβ (CCAAT/enhancer-binding protein-β) is a transcription factor which is involved in the regulation of myeloid progenitor’s differentiation into functional MDSCs. STAT3 upregulates C/EBPβ which leads to the MDSC expansion [94, 95]. Moreover, inhibition of STAT3 signaling leads to the elimination of inhibitory function of MDSCs, in vitro. It has been also shown that the application of sunitinib in tumor-bearing mice leads to down-regulation of MDSCs by inhibition of STAT3 signaling in myeloid cells. All these findings emphasize the importance of STAT3 signaling in the MDSCs [96].
Another transcription factor involved in the expansion and activation of MDSCs includes STAT1, which is activated by IFN-γ and IL-1β and involved in the up-regulation of ARG1 and iNOS in MDSCs [10, 24, 78]. It has been shown that the STAT1-deficient MDSCs are not able to up-regulate iNOS and ARG1, and thus cannot inhibit T lymphocytes activation [22]. STAT5, which is activated by GM-CSF signaling, plays a pivotal role in the regulation of MDSC survival by induction of MYC, survivin, BCL-XL, and cyclins [63, 71, 78, 97]. STAT6, which is activated subsequent to the binding of IL-4 and IL-13 to CD124 receptor, is implicated in the up-regulation of ARG1 and iNOS in MDSCs [63, 71, 78, 97]. Finally, NF-κB is the last transcription factor involved in the regulation of MDSC activity. NF-κB, which is activated by TLR family through myeloid differentiation primarily-response protein 88 (MYD88) signaling pathway, has a role in the enhancement of MDSC suppressive potential and their accumulation via targeting cyclooxygenase 2 (COX2), prostaglandin E2 (PGE2), iNOS, and ARG1 [71, 98, 99].
Factors involved in the recruitment of MDSCs
In addition to factors involved in the expansion and activation of MDSCs, there are a series of factors that contribute to the recruitment of MDSCs toward the tumor microenvironment and peripheral lymph nodes. Elevated levels of BV8 (PROK2) associates with increased frequency of circulating MDSCs in tumor-bearing mice. This cytokine is also involved in the recruitment of MDSCs to tumor sites by ligation with EG-VEGFR1 and EG-VEGFR2 on the surface of these cells [100, 101]. Moreover, recruitment of MDSCs to tumor sites is mediated by other chemoattractants released from tumor, such as CXCL5 (ENA-78), CCL2, CXCL12, and SCF (Kit ligand), which bind to CXCR2 (IL8RB), CCR2, CXCR4, and CD117 (KIT) on the surface of MDSCs, respectively [85, 87, 100–102].
MDSCs as negative regulators of immune responses
MDSCs are able to suppress both adaptive and innate immune responses via a series of inhibitory mechanisms, which lead to tumor growth. Data on the interaction of MDSCs and B cells are spare. However, Crook et al. recently reported that M-MDSCs isolated from collagen-induced arthritis (CIA) mice are also able to inhibit autologous B cell proliferation and antibody production [103]. On the other hand, extensive studies have been done in the case of T cells and it is known that MDSCs can inhibit T cell responses via multiple mechanisms such as deprivation of T cells from essential amino acids, production of oxidative stress, and interfering with the viability and trafficking of T lymphocytes. In addition, the interaction of MDSCs with other immune cells has also been reported (Figs. 3 and 4).

Amino-acids metabolism and deprivation of T cells from essential amino acids. a Cysteine metabolism. b l-arginine metabolism. c Tryptophan metabolism. DC dendritic cells, MQ macrophage, Xc − cystine/glutamate antiporter, ASC asc-type amino acid transporter, MDSC myeloid-derived suppressor cell, COX-2 cyclooxygenase 2, PGE2 prostaglandin E2, EP E-prostanoid receptor, CAT-2B cationic amino acid transporter 2 isoform 1, IDO indoleamine 2, 3 dioxygenase

MDSCs as negative regulators of immune responses. a Suppression of innate immunity. b Generation of oxidative stress. c Interference with viability and trafficking of T cells. d Induction and expansion of Treg cells and interaction with T helper 17 (Th17) cells. DC dendritic cells, MQ macrophage, MDSC myeloid-derived suppressor cell, NK cells natural killer cells, FOXP3 forkhead box P3, CCL2 CC chemokine ligand 2, CCR2 CC chemokine receptor 2, iNOS inducible nitric oxide synthase, mTOR mammalian target of rapamycin, MYD88 myeloid differentiation primary-response protein 88, STAT signal transducer and activator of transcription, CAF cancer-associated fibroblasts, Th17 cells T helper 17 cells, Treg cells regulatory T cells, TIM-3T cell immunoglobulin and mucin domain-containing protein 3, ADAM17 a disintegrin and metalloproteinase domain 17
Amino-acids metabolism and deprivation of T cells from essential amino acids
l-arginine metabolism
l-arginine metabolism is one of the inhibitory mechanisms of MDSCs that is mediated by arginase 1 activity [104]. l-arginine is required for protein synthesis in T cells and MDSCs, like other cells. PGE2 and COX2, which are involved in the conversion of arachidonic acid to PGE2, are produced by tumor cells [105]. PGE2 induces the up-regulation of arginase 1 and cationic amino acid transporter 2B (CAT-2B) expression in MDSCs via E-prostanoid receptor (EP4) [99]. This up-regulation leads to the increased consumption of arginine in MDSCs and, therefore, up-taking of l-arginine by these cells is increased, resulting in decreased extracellular arginine. l-arginine starvation induces the accumulation of empty aminoacyl-tRNAs in T cells and leads to activation of an enzyme called GCN2 kinase, which phosphorylates the translation initiation factor eIF2α. Phosphorylated eIF2α binds very tightly to eIF2β and inhibits its function through exchanging guanosine triphosphate (GTP) for guanosine diphosphate (GDP). Ultimately, the initiation of the translation process is blocked by inhibition of binding of eIF2 complex to methionine aminoacyl-tRNA. As a result, l-arginine starvation in T cells leads to inhibition of expression of CD3 ζ chain, cyclin D3, and cdk4 in T cells. Moreover, the PI3K/mTOR pathway is inhibited in T cells and their cell cycle is arrested in G0/G1 phase [30, 105–109] (Fig. 3). Recently, Gey et al. reported that G-MDSCs are increased in the critically ill patient, which this increment was negatively associated with the plasma level of arginine and patients survival [110].
Cysteine metabolism
Cysteine is required by T cells and other cells for protein synthesis, and this requirement is increased during proliferation, differentiation, and activation by antigen. Usually, cells synthesize their required cysteine from intracellular methionine via the action of cystathionase [111, 112]. In addition, cells import oxidized form of cysteine, cystine, from extracellular milieu through Xc− cystine/glutamate antiporter, which is present in the plasma membrane [113]. In the cytoplasm, the imported cystine is reduced and converted into cysteine [114]. As T lymphocytes do not express xCT chain of the Xc− cystine/glutamate antiporter and cystathionase enzyme, they cannot generate cysteine [115]. So, they should provide their required cysteine from exogenous sources, which is imported through asc-type amino acid transporter (ASC) [8].
Antigen-presenting cells (APCs) such as macrophages and dendritic cells uptake cystine via their Xc− cystine/glutamate antiporter and then reduce it to cysteine in the intracellular environment. During antigen processing and presentation, these APCs release cysteine to the extracellular through their ASC, thereby provide cysteine which is required for T lymphocytes [8, 116]. Furthermore, these APCs release thioredoxin, an enzyme that reduces extracellular cystine to cysteine. T cells can uptake cysteine via their ASC transporter [117, 118]. It should be noted that the extracellular cysteine should be immediately taken up by T lymphocytes which, otherwise, oxidized back to cystine that is not useable for T lymphocytes. So, during antigen presentation, since APCs and T cells are next to each other, there is a chance that exported cysteine from APCs is quickly taken up by T cells [8]. In contrast, since MDSCs do not express cystathionase and ASC transporter, they should import cystine through their X− c transporter and reduce it to cysteine for intracellular utilization. Therefore, MDSCs deplete cystine from the extracellular environment and do not export cysteine into the extracellular environment because they lack the ASC transporter. In conclusion, MDSCs prevent the proliferation and activation of T lymphocytes through depletion of cystine and deprivation of T lymphocytes from cysteine [1, 8]. In addition, MDSCs sequester cystine and deprive macrophages and dendritic cells from obtaining cysteine. Therefore, these cells can only provide their required cysteine through cystathionase enzyme [8]. MDSCs also disrupt the function of thioredoxin and, thereby, reduce extracellular cysteine [1] (Fig. 3).
In addition to protein synthesis, cysteine is essential for the production of glutathione and protection of cells against oxidative stress. Considering the increased oxidative stress in the tumor microenvironment, cysteine-deprived T cells even if activated are not able to survive because of inadequate glutathione [8, 119]. Regarding the importance of cysteine in cancer patients, Zhang et al. also reported that the total plasma cysteine is correlated with risk of breast cancer and high level of serum cysteine reduces the risk of breast cancer [120].
Tryptophan metabolism
Studies show that the expression of indoleamine 2, 3 dioxygenase (IDO) is correlated with tumor-induced immunosuppression [121–123]. The expression of IDO can inhibit the proliferation of T cells and induce apoptosis in these cells through the depletion of local tryptophan and production of cytotoxic metabolites [109]. Recently, Yu et al. reported that MDSCs express IDO and suppress the immune system through this enzyme [124]. The inhibitory mechanism of IDO on T cells is resembled to that of ARG1, but currently, there is not enough information about the role of IDO in MDSCs and more investigation is required (Fig. 3).
Generation of oxidative stress
INOS, which is also known as NOS2, is another enzyme that is expressed in MDSCs and catalyzes the reaction between l-arginine and oxygen and produces l-citrulline and NO. NO can then inhibit T cell responses by different mechanisms. [24] NO inhibits the IL-2 receptor signaling and prevents the activation of T cells. NO performs this function by activation of cyclic guanosine-3′, 5′-monophosphate-dependent protein kinase or directly through S-nitrosylation of cysteine residues of intracellular signaling proteins [61, 125–127]. In addition, NO reduces the stability of IL-2 mRNA and its production [128] (Fig. 4). Gehad et al. showed that NO can also inhibit the expression of E-selectin in human endothelial cells [129]. In another study, it is observed that NO accelerated tumor growth via the nitration of STAT1 and blocking interferon signaling [130]. It should be noted that ARG1 depletes intracellular l-arginine in MDSCs, and consequently the activity of iNOS is switched from NO production to O2 − (superoxide anion) generation. The produced O2 − reacts with other molecules to form ROS and RNS [60, 63, 131].
RNS, like the peroxynitrite (ONOO−), acts as intercellular messenger, which can diffuse through the cell membrane and alter the functions of proteins via nitrating their amino acid residues [61, 132]. Peroxynitrite is a potent oxidizing agent, which is produced as a result of the reaction between O2 − and NO [133]. Peroxynitrite by nitration of tyrosine residues in TCR-CD8 complex affects its interaction with MHCI-peptide and inhibits antigen-specific cytotoxic T lymphocyte responses [36, 80]. Additionally, the peroxynitrite can also induce apoptosis in T cells by inhibition of phosphorylation events in signal transduction pathway of activated T lymphocytes [134, 135] or by nitration and damage to mitochondria [135, 136] (Fig. 4).
NADPH oxidase, also known as NOX2, is involved in the production of ROS such as O2 − [24]. Hydrogen peroxide (H2O2), which is produced by the reaction of H+ from H2O and O2 −, can reduce the expression of CD3ζ chain and, therefore, impair TCR signaling [33, 137]. In addition, H2O2 can also induce apoptosis in activated T cells through NF-κB signaling pathway and, then, increasing the expression of CD95 ligand as well by down-regulation of BCL-2 molecule [138]. The produced ROS from MDSCs also disrupts the maturation of DC and leads to the accumulation of MDSCs [61] (Fig. 4).
Interference with viability and trafficking of T cells
l-selectin (CD62L) is a molecule that is expressed on the surface of naive T cells and plays a key role in T cell homing to lymph nodes. MDSCs can also cleavage the l-selectin on the surface of naive T cells by expression of a disintegrin and metalloproteinase domain 17 (ADAM17), a transmembrane zinc-dependent metalloprotease. These events cause the prevention of T cell homing to lymph nodes and perturbation of their activation [8, 32, 139, 140]. Studies show that l-selectin level on circulating T cells is inversely associated with MDSC levels in tumor-bearing mice and the cancer patients [141].
MDSCs also disrupt the binding of CCL2 to CCR2 and thereby interfere with the migration of CD8+ T cells to tumor sites by the production of peroxynitrite and nitration of CCL2 [142]. CCL2 is involved in the recruitment of MDSCs to tumor sites, but unlike T cells, MDSCs can be recruited by nitrated CCL2, which is probably due to the high affinity of these cells for CCL2 [71].
MDSCs express galectin 9, a β-galactoside binding lectin, which binds to the T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3) on the surface of T cells and induces apoptosis in these cells. Moreover, this interaction leads to expansion of MDSCs, which in turn suppresses the immune system and contributes to the tumor growth [143, 144] (Fig. 4).
An important issue in this regard is the specificity of MDSC function, whether the inhibitory effect of MDSCs on T cells is antigen non-specific or antigen-specific. Based on the type and location of MDSCs, the inhibitory function of MDSCs can be antigen-specific or non-antigen-specific [145]. In cancer, the frequency of G-MDSCs in the peripheral lymph nodes is higher than M-MDSCs [33, 34, 71, 88, 146]. However, it has been suggested that the inhibitory function of M-MDSCs is outstandingly higher than G-MDSCs [37, 70, 147, 148]. In contrast, the ratio of G-MDSCs to M-MDSCs in tumor sites is very low in comparison to peripheral lymph nodes. Probably, that is why the immunosuppressive mechanism of MDSCs in tumor sites is antigen non-specific [88]. Because the specificity of MDSC functions is also related to the type of MDSCs, the inhibitory function of G-MDSCs is mainly through ROS. Since the half-life of ROS is very short, G-MDSCs can inhibit T lymphocytes only during direct cell-to-cell contact. MDSCs in the tumor-draining lymph nodes are able to uptake antigen, process and present them to T lymphocyte, so, it is thought that they are capable to suppress antigen-specific T cell responses [36, 61, 62, 145, 149]. In contrast, MDSCs in tumor sites are mainly M-MDSCs which use NO and arginase mechanisms, do not need cell-to-cell contact, and act non-specifically. However, the exact mechanism that causes preferential accumulation of M-MDSCs in tumor sites is not fully understood. Perhaps, it is mediated by chemokines, which are produced by tumor cells or that the microenvironment of tumor is not suitable for G-MDSC survival [88].
Interaction of MDSCs with immunosuppressive cells
Treg cells are present in the tumor microenvironment and one of the key players involved in the suppression of antitumor responses [80]. MDSCs are able to induce Treg cells by consumption and depletion of arginine, interaction between CD40 and CD40L, and production of IL-10 and TGF-β [71, 149] (Fig. 4). MDSCs can cause clonal expansion of pre-existing natural Treg or conversion of effector T cells into Treg cells [61]. Studies revealed that the induction of Treg cells by MDSCs is mediated through two pathways of TGF-β dependent and TGF-β independent [80, 149–151].
Macrophages are divided into two groups including classically activated macrophages (M1), which are activated by bacterial products and immune stimuli such as IFN-γ and produce low levels of IL-10 and high levels of IL-12 and have antitumor activity. In contrast, alternatively activated macrophages (M2), which are activated by glucocorticoid hormones, IL-10, IL-13, and IL-4, produce low levels of IL-12 and high levels of IL-10 and enhance tumor growth [71, 152]. MDSC-derived IL-10 decreases IL-12 production and increases IL-10 production by macrophages, which polarizes TAMs toward the M2 phenotype [8, 23]. These macrophages, in turn, influence on MDSCs and increase IL-10 production from these cells, providing a feedback cycle [153]. Consequently, by reducing IL-12 and increasing IL-10, tumor immunity will be deviated toward a tumor-promoting type 2 phenotype and will facilitate the tumor growth [153].
MDSCs also interact with NKT cells, which can lead to the expansion or reduction of MDSCs, depending on the type of NKT cells. Type I or iNKT cells limit the expansion of MDSCs, so activated iNKT cells can increase the expression of CD86, CD11b, and CD11c on MDSCs, by unknown mechanisms, and convert these cells into immunogenic antigen-presenting cells [154, 155]. On the other hand, type II NKT cells can induce the accumulation of MDSCs by the production of IL-13 and through IL-4R-STAT6 signaling pathway [64, 156, 157].
Interaction of MDSCs with other immune cells
Studies have found that MDSCs can induce anergy of NK cells through STAT5 activity, NK cell receptor NKp30 (NCR3), membrane-bound TGF-β, and ARG1 [71, 80]. MDSCs suppress the functions of NK cells via down-regulation of NKG2D on the surface of NK cells and inhibition of production of IFN-γ by these cells [32, 158–160]. In contrast, Nausch et al. reported that MDSCs express Rae-1, which activate NK cells through binding to NKG2D. These activated NK cells, in turn, can eliminate MDSCs [148]. This contradiction may be related to the subsets of MDSCs [32], that further studies are needed to clarify.
TLR signaling in DCs induces the production of IL-12, a cytokine involved in the activation of T cells. MDSCs inhibit the production of IL-12 from DCs via the production of IL-10, thereby decreasing DC-mediated activation of T cells [23, 161]. Mast cells are another innate immune cells which have interaction with MDSCs. Danelli et al. reported that interaction between mast cells and MDSCs caused enhancement of MDSC activity [162].
MDSCs can also recruit Th17 cells into tumor sites by secretion of TGF-β, IL-6, and IL-23. The recruited Th17, in turn, promotes the expansion of MDSCs through the IL-17 production. IL-17 is involved in the recruitment of MDSCs and stimulates the secretion of G-CSF, involved in MDSC expansion, from the cancer-associated fibroblasts (CAFs) [163, 164]. Additionally, IL-17 is involved in the up-regulation of COX2, MMP-9, ARG-1, and IDO in MDSCs, therefore, increased the inhibitory activity of these cells [165] (Fig. 4).
Inflammation and its role in the increasing of MDSC’s suppressive activity
Chronic inflammation increases the risk of cancer through induction of cell proliferation and alterations in cell trafficking, increasing neoangiogenesis, triggering of genetic changes, and several other mechanisms. [166] In addition, recent studies indicate that chronic inflammation also has a role in the expansion and activation of MDSCs. MDSCs, in turn, promote tumor progression by suppression of antitumor immunity [8]. Among the proinflammatory cytokines, IL-6 and IL-1β are present in the microenvironment of most tumors and increase the accumulation and suppressive activity of MDSCs [167, 168]. PGE2, which is found in the inflammatory environment, has a role in the MDSC induction and enhancement of their inhibitory function [169]. Similarly, S100A8, S100A9, and C5a stimulate the accumulation of MDSCs. In summary, inflammatory factors are involved in the expansion and activation of MDSCs. MDSCs, in turn, produce proinflammatory factors such as S100A8, S100A9, and IL-6 and maintain the inflammatory environment and, thus, maintain their population [8, 74].
MDSCs in tumor-bearing hosts
MDSCs accumulate largely within the tumor region, spleen, bone marrow, peripheral blood, and to lesser extent in the lymph nodes of tumor-bearing mice [17, 61, 62, 68, 74, 170]. But, tumor resection leads to reduction of MDSCs and restoration of the protective immunity [171]. Furthermore, elimination of MDSCs is associated with the improvement of immune responses [88]. The frequency of MDSCs in the blood of patients with different types of cancers is increased up to tenfold [39, 40, 43, 172] and is associated with poor prognosis [173–175]. There are similar reports regarding the frequency of MDSCs and its association with disease progression in the breast cancer and gastrointestinal tumors [43, 176, 177]. Tumor-derived soluble factors (TDSFs) are involved in the stimulation of myelopoiesis, recruitment of IMCs to the tumor site, activation of IMCs, and converting them into MDSCs by activation of different transcription factors. These MDSCs in turn contribute to tumor progression [71] (Fig. 2). Mundy-Bosse et al. reported that increased psychological stress in breast cancer patients was associated with increased number of MDSCs and reduction of the immune response in these patients [178]. Probably, hormones that are produced in stressful conditions are involved in regulation of function and expansion of MDSCs which needs further investigations.
MDSC’s role in proliferation and metastasis of tumor cells
In addition to suppression of immune surveillance, MDSCs can also directly promote tumor proliferation and metastasis through non-immunological functions. MDSC-secreted MMPs destroy extracellular matrix components in tissue surrounding a tumor and, thus, facilitate tumor metastasis [87, 89, 179]. MDSC-secreted MMP-9 stimulates VEGF production and increases its bioavailability [179], therefore promotes the growth of new blood vessels and facilitates tumor metastasis and invasion [173]. Toh et al. found that MDSCs induced epithelial-mesenchymal transition (EMT) in cancer cells by hepatocyte growth factor (HGF), TGF-β, and epidermal growth factor (EGF) signaling pathway, thereby facilitate the dissemination of tumor [180]. Yang et al. reported that the expansion of MDSCs in tumor-bearing mice promotes tumor angiogenesis [179]. However, the treatment of tumor-bearing mice with neutralizing anti-BV8 significantly reduces angiogenesis through the reduction of the numbers of infiltrated MDSCs [100]. Furthermore, some MDSCs are able to differentiate into endothelial-like cells and, thereby, contribute to tumor angiogenesis. In this condition, they even express some of endothelial cell markers such as CD31 and VEGFR2 [25, 179].
Additionally, MDSCs can differentiate into fibrocytes, a cell type that can be differentiated into myofibroblasts. Myofibroblasts can drive cancer invasion and metastasis in the tumor context [181]. KLF4 is a transcription factor involved in the promotion of tumor growth and monocyte differentiation [182, 183]. Recently, Shi et al. demonstrated that KLF4 have a very important role in the differentiation of MDSCs into fibrocytes, and deficiency of KLF4 in MDSCs is associated with reduction of tumor metastasis and reduction of MDSC-derived fibrocytes and myofibroblasts. These results suggest that KLF4 can be a new therapeutic target for tumor metastasis [181].
Significance of miRNAs in MDSC biology
MicroRNAs are small non-coding RNAs of approximately 22 nucleotides in length that play an important role in post-transcriptional regulation of gene expression [184]. Recently, the role of microRNAs (miRNAs) in the regulation of immune system is further appreciated and studies showed that miRNAs have important roles in differentiation, function, and development of various immune cells [185, 186]. MicroRNAs that interact with MDSCs can be divided into three groups.
The first group of miRNAs is involved in the expansion and activation of MDSCs. Expression of these miRNAs is increased in tumor condition. TGF-β1 is a tumor-derived factor that is responsible for miR-494 up-regulation in MDSCs. Induction of miR-494 expression in MDSCs results in the inhibition of PTEN, increased Akt activity, MDSC survival, MDSC chemotaxis, as well raised expression of arginase and MMPs such as MMP2, MMP13, and MMP14. It is noteworthy that targeting and silencing of this miRNA lead to the reduction of MDSC suppressive activity and inhibition of tumor growth and metastasis. So, it is believed that miR-494 plays an important role in the expansion, survival, and function of MDSCs [187, 188]. TGF-β in MDSCs induces the up-regulation of miR-21 and miR-155, which inhibit PTEN and SHIP-1, respectively, and ultimately causes the activation of STAT3 signaling pathway and expansion of MDSCs [188, 189] (Fig. 5). In contrast, Wang et al. recently reported that miR-155 deficiency leads to the increased recruitment of MDSCs to tumor milieu and also increases the inhibitory activity of these cells, thereby promotes tumor growth [190]. This contradiction may be related to the tumor type that further studies are needed to clarify.

Significance of miRNAs in MDSC biology. a miRNAs involved in the expansion and activation of MDSCs. b miRNAs involved in the regulation of MDSC function. c miRNAs that their expression is altered in tumor cells by MDSC-secreted factors
The second group is miRNAs involved in the regulation of MDSC’s function. Expression of these miRNAs is decreased in tumor condition. MiR-20a and miR-17-5p reduce the production of H2O2 and ROS in MDSCs through the inhibition of STAT3 signaling and, therefore, regulate the suppressive activity of these cells. Expression of these miRNAs is reduced in MDSCs in tumor condition, which leads to increment of the suppressive activity of these cells (Fig. 5). Therefore, it seems that these miRNAs can be used as a drug to regulate the function of MDSCs [188, 191]. It is necessary to note that miR-20a and miR-17-5p play minor role in the regulation of M-MDSC functions. They are mostly involved in the regulation of G-MDSC functions [191]. It has been reported that MDSCs accumulate in peripheral lymphoid organs in the miRNA-146a-deficient mice [192]. Probably, this miRNA has a role in the regulation of MDSC expansion, but its exact mechanism is not clear. MiR-223 can inhibit differentiation of bone marrow cells into CD11b+Gr1+MDSC through targeting myocyte enhancer factor 2C (MEF2C). Expression of miR-223 is regulated by tumor-associated factors and down-regulation of this miRNA was reported in tumor-associated MDSCs [193, 194].
Unlike the two above groups that their expression is changed in MDSCs, the third group includes miRNAs with altered expression in tumor cells by MDSC-secreted factors. MDSCs inhibit corepressor C-terminal binding protein 2 (CtBP2) in cancer cells via induction of miR-101 in these cells and, thereby, enhance stemness of cancer cells and promote metastasis and tumor growth [188, 195]. In a study conducted on ovarian cancer, it was reported that ROS could induce the expression of ERBB2 and ERBB3 receptors by down-regulating miR-199a and miR-125b and thus accelerated tumor growth [196] (Fig. 5). Although the investigation revealed the importance of ROS in tumor growth, but the source of ROS in this study was not surveyed. So, in order to clarify the issue, it is required to determine the relationship between MDSC levels and down-regulation of miR-199a and miR-125b [10].
MDSC as a therapeutic target in cancer
Taking together, these findings suggest that elimination of MDSCs or limitation of their function is necessary for inhibition of tumor growth and increasing of the efficacy of immunotherapeutic interventions. Currently, different therapeutic strategies in order to cope with suppressive activity of MDSCs are being investigated [8, 60, 197–199] (Table 2). These methods are divided into four categories.
Inhibition of MDSC expansion and recruitment
Several agents are used in order to neutralize factors involved in expansion and recruitment of MDSCs to peripheral lymph nodes and tumor sites. As mentioned previously, inhibition of SCF signaling has been effective in inhibition of MDSC expansion [71, 72]. The amount of MMP 9 is increased with tumor progression, and MMP 9 is involved in the expansion and recruitment of MDSCs to tumor sites, thus MMP 9 can be considered as a suitable target for targeting MDSCs. It has been reported that amino-bisphosphonate can inhibit the production of MMP 9 and reduce MDSC recruitment [200]. The inhibition of VEGF receptor signaling leads to reduction of MDSC infiltration [217], and application of avastin, an antibody against VEGF, can reduce CD11b+ VEGFR+ population [200, 203].
Angiotensin-converting enzyme (ACE) is a peptidase involved in the metabolism of several bioactive peptides. Studies show that ACE is also involved in myelopoiesis, and accumulation of immature myeloid precursors in spleen and bone marrow can be seen in the absence of ACE [218, 219]. Recently, Shen et al. reported that expression of ACE in myeloid cells facilitates the maturation of these cells and limits MDSC expansion [218]. Further investigations will be required to determine what bioactive peptide(s) is (are) modulated by ACE and through what mechanism this bioactive peptide(s) limits the expansion of MDSCs. The answers to these questions may provide a suitable strategy for targeting MDSCs.
Induction of differentiation in MDSCs
Induction of differentiation in MDSCs and converting them into mature non-suppressive cells is one of the promising strategies that can be used for targeting MDSCs. In this regard, it has been shown that All-trans retinoic acid (ATRA) can induce the differentiation of MDSCs into mature dendritic cell, granulocytes, and macrophages via up-regulation of glutathione synthesis and reduction of ROS in these cells [220–222]. Studies indicate that vitamin D3 is another agent that can promote myeloid cell maturation and reduce the number of MDSCs in cancer patient [209].
Inhibition of MDSC’s suppressive activity
Inhibition of MDSC’s suppressive activity or interfering with their functions is another approach which is used for targeting MDSCs. COX2 is a contributing agent for up-regulation of ARG1 in MDSCs by production of PGE2; as a result, COX2 inhibitors are exerted for inhibition of MDSC activity [99, 169]. Phosphodiesterase-5 (PDE5) inhibitors, like sildenafil, down-regulate the expression of arginase1, IL-4Rα, and iNOS in MDSCs and, thereby, restrain their activities [49, 151, 173]. Nitroaspirin, a non-steroidal anti-inflammatory drug (NSAID), can block the suppressive activity of MDSCs through inhibition of production of iNOS, ARG1, and ROS in these cells [213]. IL4-Rα is involved in the activation of MDSCs and, thus, can be a pertinent target for limiting of MDSC suppressive activity. So, IL4-Rα antagonists can be considered as effective agents [94].
The neutral lipid metabolic pathway has an important role in the biology of MDSCs. This pathway is regulated by lysosomal acid lipase (LAL) and accumulation of MDSCs is observed in LAL-deficient mice [223]. In addition, it has been reported that mutations in the LAL gene are associated with carcinogenesis [224]. Recently, Zhao et al. reported that LAL is involved in the regulation of MDSC’s functions by modulating the mammalian target of rapamycin (mTOR) pathway. The mTOR pathway is strongly activated in lal−/− MDSCs, and these cells are able to directly stimulate growth, metastasis, and proliferation of tumor cells. As a result, the mTOR pathway can be a new target for blocking MDSC activity; however, it needs to be further investigated [223].
Dopamine, a neurotransmitter, regulates the function of M-MDSCs and inhibits tumor growth. D1-like receptors that are expressed in M-MDSCs are activated by dopamine and other D1-like receptor agonists and inhibit NO production in these cells. D1-like receptors signaling inhibits activation of ERK and JNK, which occur in response to IFN-γ stimulation, therefore decrease the production of NO from M-MDSCs and attenuate the inhibitory activity of these cells [225]. The results of this study show that dopamine and D1-like receptor agonists can be effective in enhancing the antitumor immunity which requires further studies.
Induction of apoptosis in MDSCs
Some chemotherapeutic drugs are applied for elimination of MDSCs. Gemcitabine is a drug that reduces the number of MDSCs and enhances antitumor immune activity [158]. Administration of doxorubicin can selectively eliminate MDSCs. This drug is also effective in enhancement of the function of immune effector cells [9]. Zheng et al. reported that cimetidine can suppress lung tumor growth via regulation of caspase-dependent apoptosis pathway and induction of Fas and FasL in MDSCs [226].
The expression of TNF-related apoptosis-induced ligand receptors (TRAIL-Rs) is increased in MDSCs in response to endoplasmic reticulum (ER) stress and its signaling induces apoptosis in these cells. Targeting TRAIL-Rs in tumor-bearing mice can significantly reduce MDSC population but has no effect on the population of myeloid cells in naive mice. As a result, TRAIL-Rs can be an excellent targets in order to selectively eliminate MDSCs [227].
Conclusion
In recent years, extensive studies have been performed on the role of MDSCs in the tumor progression and have found that these cells are involved in tumor progression both directly and indirectly. Since increased frequency of MDSCs is associated with poor prognosis in cancer patients, thereby targeting these cells can be useful in tumor treatment. Therefore, study of MDSC inhibitory mechanisms and communication of these cells with other components of immune system and tumor cells is important in dealing with the function of these cells and to increase the efficacy of immunotherapeutic interventions. But an important issue in this viewpoint is the heterogeneity of these cells, particularly in human MDSCs, which express different surface markers in different cancers. Thus, precise identification of cell surface markers and exact definition of human MDSCs in different types of cancers as well developing therapeutic approaches used for targeting these cells can be efficient in the improvement of efficacy of immunotherapeutic interventions and cancer treatment.
References
Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70:68–77.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Jadidi-Niaragh F, Ghalamfarsa G, Memarian A, Asgarian-Omran H, Razavi SM, Sarrafnejad A, et al. Downregulation of IL-17-producing T cells is associated with regulatory T cell expansion and disease progression in chronic lymphocytic leukemia. Tumor Biol. 2013;34:929–40.
Jadidi-Niaragh F, Ghalamfarsa G, Yousefi M, Tabrizi MH, Shokri F. Regulatory T cells in chronic lymphocytic leukemia: implication for immunotherapeutic interventions. Tumor Biol. 2013;34:2031–9.
Jadidi-Niaragh F, Yousefi M, Memarian A, Hojjat-Farsangi M, Khoshnoodi J, Razavi SM, et al. Increased frequency of CD8+ and CD4+ regulatory T cells in chronic lymphocytic leukemia: association with disease progression. Cancer Investig. 2013;31:121–31.
Namdar A, Mirzaei HR, Jadidi-Niaragh F, Ashourpour M, Ajami M, Hadjati J, et al. Multiple low doses of 5-fluorouracil diminishes immunosuppression by myeloid derived suppressor cells in murine melanoma model. Iran J Immunol. 2015;12:176.
Butt A, Mills K. Immunosuppressive networks and checkpoints controlling antitumor immunity and their blockade in the development of cancer immunotherapeutics and vaccines. Oncogene. 2014;33:4623–31.
Ostrand-Rosenberg S. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother. 2010;59:1593–600.
Alizadeh D, Katsanis E, Larmonier N. Chemotherapeutic targeting of myeloid-derived suppressor cells. Oncoimmunology. 2014;3:e27359.
Khaled YS, Ammori BJ, Elkord E. Myeloid-derived suppressor cells in cancer: recent progress and prospects. Immunol Cell Biol. 2013;91:493–502.
Subiza JL, Vinuela JE, Rodriguez R, Gil J, Figueredo M, de la Concha EG. Development of splenic natural suppressor (NS) cells in Ehrlich tumor‐bearing mice. Int J Cancer. 1989;44:307–14.
Strober S. Natural suppressor (NS) cells, neonatal tolerance, and total lymphoid irradiation: exploring obscure relationships. Annu Rev Immunol. 1984;2:219–37.
Slavin S, Strober S. Induction of allograft tolerance after total lymphoid irradiation (TLI): development of suppressor cells of the mixed leukocyte reaction (MLR). J Immunol. 1979;123:942–6.
Roder J, Duwe A, Bell D, Singhal S. Immunological senescence. I. The role of suppressor cells. Immunol. 1978;35:837.
Bennett JA, Rao VS, Mitchell MS. Systemic bacillus Calmette-Guerin (BCG) activates natural suppressor cells. Proc Natl Acad Sci. 1978;75:5142–4.
Duwe AK, Singhal SK. The immunoregulatory role of bone marrow: I. Suppression of the induction of antibody responses to T-dependent and T-independent antigens by cells in the bone marrow. Cell Immunol. 1979;43:362–71.
Ribechini E, Greifenberg V, Sandwick S, Lutz MB. Subsets, expansion and activation of myeloid-derived suppressor cells. Med Microbiol Immunol. 2010;199:273–81.
Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013;13:739–52.
Gabrilovich DI, Bronte V, Chen S-H, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67:425.
Yang R, Roden RB. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67:426.
Choi J, Suh B, Ahn Y-O, Kim TM, Lee J-O, Lee S-H, et al. CD15+/CD16low human granulocytes from terminal cancer patients: granulocytic myeloid-derived suppressor cells that have suppressive function. Tumor Biol. 2012;33:121–9.
Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32:19–25.
Yazdani Y, Mohammadnia-Afrouzi M, Yousefi M, Anvari E, Ghalamfarsa G, Hasannia H, et al. Myeloid-derived suppressor cells in B cell malignancies. Tumor Biol 2015:1–15.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74.
Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–31.
Melani C, Chiodoni C, Forni G, Colombo MP. Myeloid cell expansion elicited by the progression of spontaneous mammary carcinomas in c-erbB-2 transgenic BALB/c mice suppresses immune reactivity. Blood. 2003;102:2138–45.
Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Investig. 2007;117:1155.
Bronte V, Serafini P, Apolloni E, Zanovello P. Tumor-induced immune dysfunctions caused by myeloid suppressor cells. J Immunother. 2001;24:431–46.
Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4:941–52.
Greten TF, Manns MP, Korangy F. Myeloid derived suppressor cells in human diseases. Int Immunopharmacol. 2011;11:802–7.
Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1–dependent mechanism that is up-regulated by interleukin-13. Blood. 2010;116:5738–47.
Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–506.
Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172:989–99.
Youn J-I, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–802.
Hestdal K, Ruscetti F, Ihle J, Jacobsen S, Dubois C, Kopp W, et al. Characterization and regulation of RB6-8C5 antigen expression on murine bone marrow cells. J Immunol. 1991;147:22–8.
Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13:828–35.
Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell–suppressive activity. Blood. 2008;111:4233–44.
Burke CF, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: Immune-suppressive cells that facilitate tumor progression and promote and deter cancer-associated inflammation. Tumor Immunology and Immunotherapy 2014;95
Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res. 2007;13:721s–6.
Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–89.
Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Investig. 2006;116:2777.
Srivastava MK, Bosch JJ, Thompson JA, Ksander BR, Edelman MJ, Ostrand-Rosenberg S. Lung cancer patients’ CD4+ T cells are activated in vitro by MHC II cell-based vaccines despite the presence of myeloid-derived suppressor cells. Cancer Immunol Immunother. 2008;57:1493–504.
Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin–cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59.
Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60:1419–30.
Kusmartsev S, Su Z, Heiser A, Dannull J, Eruslanov E, Kübler H, et al. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2008;14:8270–8.
Gros A, Turcotte S, Wunderlich JR, Ahmadzadeh M, Dudley ME, Rosenberg SA. Myeloid cells obtained from the blood but not from the tumor can suppress T-cell proliferation in patients with melanoma. Clin Cancer Res. 2012;18:5212–23.
Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182:5693–701.
Mandruzzato S, Solito S, Falisi E, Francescato S, Chiarion-Sileni V, Mocellin S, et al. IL4Rα + myeloid-derived suppressor cell expansion in cancer patients. J Immunol. 2009;182:6562–8.
Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203:2691–702.
Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M, et al. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor–based antitumor vaccine. J Clin Oncol. 2007;25:2546–53.
Oberlies J, Watzl C, Giese T, Luckner C, Kropf P, Müller I, et al. Regulation of NK cell function by human granulocyte arginase. J Immunol. 2009;182:5259–67.
Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14:6674–82.
Zhao F, Hoechst B, Duffy A, Gamrekelashvili J, Fioravanti S, Manns MP, et al. S100A9 a new marker for monocytic human myeloid‐derived suppressor cells. Immunology. 2012;136:176–83.
Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Krüger C, Manns MP, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4+ CD25+ Foxp3+ T cells. Gastroenterology. 2008;135:234–43.
Vuk-Pavlović S, Bulur PA, Lin Y, Qin R, Szumlanski CL, Zhao X, et al. Immunosuppressive CD14+ HLA‐DRlow/− monocytes in prostate cancer. Prostate. 2010;70:443–55.
Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65:3044–8.
Liu C-Y, Wang Y-M, Wang C-L, Feng P-H, Ko H-W, Liu Y-H, et al. Population alterations of L-arginase-and inducible nitric oxide synthase-expressed CD11b+/CD14−/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J Cancer Res Clin Oncol. 2010;136:35–45.
Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009;69:1553–60.
Pak A, Wright MA, Matthews JP, Collins SL, Petruzzelli GJ, Young M. Mechanisms of immune suppression in patients with head and neck cancer: presence of CD34 (+) cells which suppress immune functions within cancers that secrete granulocyte-macrophage colony-stimulating factor. Clin Cancer Res. 1995;1:95–103.
Talmadge JE. Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin Cancer Res. 2007;13:5243–8.
Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor‐induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–79.
Kusmartsev S, Nagaraj S, Gabrilovich DI. Tumor-associated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. J Immunol. 2005;175:4583–92.
Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol. 2003;170:270–8.
Terabe M, Matsui S, Park J-M, Mamura M, Noben-Trauth N, Donaldson DD, et al. Transforming growth factor-β production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte–mediated tumor immunosurveillance abrogation prevents tumor recurrence. J Exp Med. 2003;198:1741–52.
Merchav S, Apte R, Tatarsky I, Ber R. Effect of plasmacytoma cells on the production of granulocyte-macrophage colony-stimulating activity (GM-CSA) in the spleen of tumor-bearing mice. Exp Hematol. 1987;15:995–1000.
Fu Y, Watson G, Jimenez JJ, Wang Y, Lopez DM. Expansion of immunoregulatory macrophages by granulocyte-macrophage colony-stimulating factor derived from a murine mammary tumor. Cancer Res. 1990;50:227–34.
Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999;162:5728–37.
Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004;64:6337–43.
Parmiani G, Castelli C, Pilla L, Santinami M, Colombo M, Rivoltini L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann Oncol. 2007;18:226–32.
Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, et al. Hierarchy of immunosuppressive strength among myeloid‐derived suppressor cell subsets is determined by GM‐CSF. Eur J Immunol. 2010;40:22–35.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–68.
Kao J, Ko EC, Eisenstein S, Sikora AG, Fu S, Chen S-H. Targeting immune suppressing myeloid-derived suppressor cells in oncology. Crit Rev Oncol Hematol. 2011;77:12–9.
Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis. 2008;29:2035–43.
Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181:4666–75.
Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9:1225–35.
Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002;168:689–95.
Kusmartsev S, Gabrilovich DI. Immature myeloid cells and cancer-associated immune suppression. Cancer Immunol Immunother. 2002;51:293–8.
Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol. 2005;174:4880–91.
Sinha P, Clements VK, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. 2005;174:636–45.
Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid‐derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013;138:105–15.
Corzo CA, Condamine T, Lu L, Cotter MJ, Youn J-I, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–53.
Kutomi G, Tamura Y, Tanaka T, Kajiwara T, Kukita K, Ohmura T, et al. Human endoplasmic reticulum oxidoreductin 1‐α is a novel predictor for poor prognosis of breast cancer. Cancer Sci. 2013;104:1091–6.
Tanaka T, Kajiwara T, Torigoe T, Okamoto Y, Sato N, Tamura Y. Cancer-associated oxidoreductase ERO1-α drives the production of tumor-promoting myeloid-derived suppressor cells via oxidative protein folding. J Immunol. 2015;194:2004–10.
Park S-J, Nakagawa T, Kitamura H, Atsumi T, Kamon H, Sawa S-I, et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol. 2004;173:3844–54.
Pan P-Y, Wang GX, Yin B, Ozao J, Ku T, Divino CM, et al. Reversion of immune tolerance in advanced malignancy: modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood. 2008;111:219–28.
Young MRI, Wright MA, Coogan M, Young ME, Bagash J. Tumor-derived cytokines induce bone marrow suppressor cells that mediate immunosuppression through transforming growth factor β. Cancer Immunol Immunother. 1992;35:14–8.
Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGFβ signaling in mammary carcinomas recruits Gr-1+ CD11b + myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35.
Youn JI, Gabrilovich DI. The biology of myeloid‐derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010;40:2969–75.
Nefedova Y, Nagaraj S, Rosenbauer A, Muro-Cacho C, Sebti SM, Gabrilovich DI. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res. 2005;65:9525–35.
Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809.
Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol. 2007;81:28–37.
Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205:2235–49.
Farren MR, Carlson LM, Lee KP. Tumor-mediated inhibition of dendritic cell differentiation is mediated by down regulation of protein kinase C beta II expression. Immunol Res. 2010;46:165–76.
Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPβ transcription factor. Immunity. 2010;32:790–802.
Sander LE, Sackett SD, Dierssen U, Beraza N, Linke RP, Müller M, et al. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J Exp Med. 2010;207:1453–64.
Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69:2506–13.
Munera V, Popovic PJ, Bryk J, Pribis J, Caba D, Matta BM, et al. Stat 6-dependent induction of myeloid derived suppressor cells after physical injury regulates nitric oxide response to endotoxin. Ann Surg. 2010;251:120–6.
Bunt SK, Clements VK, Hanson EM, Sinha P, Ostrand-Rosenberg S. Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J Leukoc Biol. 2009;85:996–1004.
Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. 2005;202:931–9.
Shojaei F, Wu X, Zhong C, Yu L, Liang X-H, Yao J, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450:825–31.
LeCouter J, Zlot C, Tejada M, Peale F, Ferrara N. Bv8 and endocrine gland-derived vascular endothelial growth factor stimulate hematopoiesis and hematopoietic cell mobilization. Proc Natl Acad Sci U S A. 2004;101:16813–8.
Sawanobori Y, Ueha S, Kurachi M, Shimaoka T, Talmadge JE, Abe J, et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood. 2008;111:5457–66.
Crook KR, Jin M, Weeks MF, Rampersad RR, Baldi RM, Glekas AS, et al. Myeloid-derived suppressor cells regulate T cell and B cell responses during autoimmune disease. J Leukoc Biol. 2015;97:573–82.
Choudhury B, Srivastava S, Choudhury HH, Purkayastha A, DuttaGupta S, Ghosh SK. Arginase and C-reactive protein as potential serum-based biomarker of head and neck squamous cell carcinoma patients of north east India. Tumor Biol. 2014;35:6739–48.
Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–91.
Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–49.
Rodriguez P, Zea A, Ochoa A. Mechanisms of tumor evasion from the immune response. Cancer Chemother Biol Response Modif. 2002;21:351–64.
Rodríguez PC, Ochoa AC, editors. T cell dysfunction in cancer: role of myeloid cells and tumor cells regulating amino acid availability and oxidative stress. Semin Cancer Biol 2006. Academic Press.
Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2, 3-dioxygenase. Immunity. 2005;22:633–42.
Gey A, Tadie JM, Caumont‐Prim A, Hauw‐Berlemont C, Cynober L, Fagon JY, et al. Granulocytic myeloid‐derived suppressor cells inversely correlate with plasma arginine and overall survival in critically ill patients. Clin Exp Immunol. 2015;180:280–8.
Gout P, Buckley A, Simms C, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x (c)-cystine transporter: a new action for an old drug. Leukemia. 2001;15:1633–40.
Ishii I, Akahoshi N, Yu X, Kobayashi Y, Namekata K, Komaki G, et al. Murine cystathionine gamma-lyase: complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochem J. 2004;381:113–23.
Mansoor MA, Svardal AM, Ueland PM. Determination of the in vivo redox status of cysteine, cysteinylglycine, homocysteine, and glutathione in human plasma. Anal Biochem. 1992;200:218–29.
Arnér ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267:6102–9.
Bannai S. Transport of cystine and cysteine in mammalian cells. Biochimica et Biophysica Acta (BBA)-Reviews on. Biomembranes. 1984;779:289–306.
Sato H, Watanabe H, Ishii T, Bannai S. Neutral amino acid transport in mouse peritoneal macrophages. J Biol Chem. 1987;262:13015–9.
Angelini G, Gardella S, Ardy M, Ciriolo MR, Filomeni G, Di Trapani G, et al. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc Natl Acad Sci. 2002;99:1491–6.
Castellani P, Angelini G, Delfino L, Matucci A, Rubartelli A. The thiol redox state of lymphoid organs is modified by immunization: role of different immune cell populations. Eur J Immunol. 2008;38:2419–25.
Sakakura Y, Sato H, Shiiya A, Tamba M, Sagara J-I, Matsuda M, et al. Expression and function of cystine/glutamate transporter in neutrophils. J Leukoc Biol. 2007;81:974–82.
Zhang SM, Willett WC, Selhub J, Manson JE, Colditz GA, Hankinson SE. A prospective study of plasma total cysteine and risk of breast cancer. Cancer Epidemiol Biomark Prev. 2003;12:1188–93.
Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2, 3-dioxygenase. Nat Med. 2003;9:1269–74.
Friberg M, Jennings R, Alsarraj M, Dessureault S, Cantor A, Extermann M, et al. Indoleamine 2, 3‐dioxygenase contributes to tumor cell evasion of T cell‐mediated rejection. Int J Cancer. 2002;101:151–5.
Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–74.
Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190:3783–97.
Fischer TA, Palmetshofer A, Gambaryan S, Butt E, Jassoy C, Walter U, et al. Activation of cGMP-dependent protein kinase Iβ inhibits interleukin 2 release and proliferation of T cell receptor-stimulated human peripheral T cells. J Biol Chem. 2001;276:5967–74.
Duhé RJ, Evans GA, Erwin RA, Kirken RA, Cox GW, Farrar WL. Nitric oxide and thiol redox regulation of Janus kinase activity. Proc Natl Acad Sci. 1998;95:126–31.
Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol. 1998;160:5729–34.
Macphail SE, Gibney CA, Brooks BM, Booth CG, Flanagan BF, Coleman JW. Nitric oxide regulation of human peripheral blood mononuclear cells: critical time dependence and selectivity for cytokine versus chemokine expression. J Immunol. 2003;171:4809–15.
Gehad AE, Lichtman MK, Schmults CD, Teague JE, Calarese AW, Jiang Y, et al. Nitric oxide–producing myeloid-derived suppressor cells inhibit vascular E-selectin expression in human squamous cell carcinomas. J Investig Dermatol. 2012;132:2642–51.
Mundy-Bosse BL, Lesinski GB, Jaime-Ramirez AC, Benninger K, Khan M, Kuppusamy P, et al. Myeloid-derived suppressor cell inhibition of the IFN response in tumor-bearing mice. Cancer Res. 2011;71:5101–10.
Xia Y, Roman LJ, Masters BSS, Zweier JL. Inducible nitric-oxide synthase generates superoxide from the reductase domain. J Biol Chem. 1998;273:22635–9.
Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–80.
Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci. 2004;101:4003–8.
Kusmartsev SA, Li Y, Chen S-H. Gr-1+ myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J Immunol. 2000;165:779–85.
Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol. 2003;24:301–5.
Aulak KS, Miyagi M, Yan L, West KA, Massillon D, Crabb JW, et al. Proteomic method identifies proteins nitrated in vivo during inflammatory challenge. Proc Natl Acad Sci. 2001;98:12056–61.
Kono K, Salazar‐Onfray F, Petersson M, Hansson J, Masucci G, Wasserman K, et al. Hydrogen peroxide secreted by tumor‐derived macrophages down‐modulates signal‐transducing zeta molecules and inhibits tumor‐specific T cell‐and natural killer cell‐mediated cytotoxicity. Eur J Immunol. 1996;26:1308–13.
Hildeman DA, Mitchell T, Aronow B, Wojciechowski S, Kappler J, Marrack P. Control of Bcl-2 expression by reactive oxygen species. Proc Natl Acad Sci. 2003;100:15035–40.
Li Y, Brazzell J, Herrera A, Walcheck B. ADAM17 deficiency by mature neutrophils has differential effects on L-selectin shedding. Blood. 2006;108:2275–9.
Lorenzen I, Trad A, Grötzinger J. Multimerisation of A disintegrin and metalloprotease protein-17 (ADAM17) is mediated by its EGF-like domain. Biochem Biophys Res Commun. 2011;415:330–6.
Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S. Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J Immunol. 2009;183:937–44.
Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. 2011;208:1949–62.
Sakuishi K, Jayaraman P, Behar SM, Anderson AC, Kuchroo VK. Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol. 2011;32:345–9.
Anderson AC. Tim-3, a negative regulator of anti-tumor immunity. Curr Opin Immunol. 2012;24:213–6.
Nagaraj S, Gabrilovich DI. Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer Res. 2008;68:2561–3.
Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001;61:4756–60.
Priceman SJ, Sung JL, Shaposhnik Z, Burton JB, Torres-Collado AX, Moughon DL, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. 2010;115:1461–71.
Nausch N, Galani IE, Schlecker E, Cerwenka A. Mononuclear myeloid-derived “suppressor” cells express RAE-1 and activate natural killer cells. Blood. 2008;112:4080–9.
Huang B, Pan P-Y, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+ CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–31.
Yang R, Cai Z, Zhang Y, Yutzy WH, Roby KF, Roden RB. CD80 in immune suppression by mouse ovarian carcinoma–associated Gr-1+ CD11b + myeloid cells. Cancer Res. 2006;66:6807–15.
Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008;68:5439–49.
Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol. 2009;70:325–30.
Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179:977–83.
De Santo C, Salio M, Masri SH, Lee LY-H, Dong T, Speak AO, et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest. 2008;118:4036.
Ko H-J, Lee J-M, Kim Y-J, Kim Y-S, Lee K-A, Kang C-Y. Immunosuppressive myeloid-derived suppressor cells can be converted into immunogenic APCs with the help of activated NKT cells: an alternative cell-based antitumor vaccine. J Immunol. 2009;182:1818–28.
Sinha P, Clements VK, Ostrand-Rosenberg S. Interleukin-13–regulated M2 macrophages in combination with myeloid suppressor cells block immune surveillance against metastasis. Cancer Res. 2005;65:11743–51.
Terabe M, Swann J, Ambrosino E, Sinha P, Takaku S, Hayakawa Y, et al. A nonclassical non-Vα14Jα18 CD1d-restricted (type II) NKT cell is sufficient for down-regulation of tumor immunosurveillance. J Exp Med. 2005;202:1627–33.
Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b + myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–21.
Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-β1. J Immunol. 2009;182:240–9.
Liu C, Yu S, Kappes J, Wang J, Grizzle WE, Zinn KR, et al. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood. 2007;109:4336–42.
Hu C-E, Gan J, Zhang R-D, Cheng Y-R, Huang G-J. Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand J Gastroenterol. 2011;46:156–64.
Danelli L, Frossi B, Gri G, Mion F, Guarnotta C, Bongiovanni L, et al. Mast cells boost myeloid-derived suppressor cell activity and contribute to the development of tumor-favoring microenvironment. Cancer Immunol Res. 2015;3:85–95.
Kitamura T, Qian B-Z, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15:73–86.
Novitskiy SV, Pickup MW, Gorska AE, Owens P, Chytil A, Aakre M, et al. TGF-β receptor II loss promotes mammary carcinoma progression by Th17-dependent mechanisms. Cancer Discovery. 2011;1:430–41.
Condamine T, Ramachandran I, Youn J-I, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med. 2015;66:97–110.
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44.
Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. 2006;176:284–90.
Song X, Krelin Y, Dvorkin T, Bjorkdahl O, Segal S, Dinarello CA, et al. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1β-secreting cells. J Immunol. 2005;175:8200–8.
Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67:4507–13.
Haile LA, Von Wasielewski R, Gamrekelashvili J, Krüger C, Bachmann O, Westendorf AM, et al. Myeloid-derived suppressor cells in inflammatory bowel disease: a new immunoregulatory pathway. Gastroenterology. 2008;135:871–81. e5.
Salvadori S, Martinelli G, Zier K. Resection of solid tumors reverses T cell defects and restores protective immunity. J Immunol. 2000;164:2214–20.
Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006;66:9299–307.
Fujimura T, Mahnke K, Enk AH. Myeloid derived suppressor cells and their role in tolerance induction in cancer. J Dermatol Sci. 2010;59:1–6.
Jiang H, Gebhardt C, Umansky L, Beckhove P, Schulze TJ, Utikal J, et al. Elevated chronic inflammatory factors and myeloid‐derived suppressor cells indicate poor prognosis in advanced melanoma patients. Int J Cancer. 2015;136:2352–60.
Huang H, Zhang G, Li G, Ma H, Zhang X. Circulating CD14+ HLA-DR−/low myeloid-derived suppressor cell is an indicator of poor prognosis in patients with ESCC. Tumor Biol 2015:1–10.
Mundy-Bosse BL, Young GS, Bauer T, Binkley E, Bloomston M, Bill MA, et al. Distinct myeloid suppressor cell subsets correlate with plasma IL-6 and IL-10 and reduced interferon-alpha signaling in CD4+ T cells from patients with GI malignancy. Cancer Immunol Immunother. 2011;60:1269–79.
OuYang L-Y, Wu X-J, Ye S-B, Zhang R-X, Li Z-L, Liao W, et al. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J Transl Med. 2015;13:47.
Mundy-Bosse BL, Thornton LM, Yang H-C, Andersen BL, Carson WE. Psychological stress is associated with altered levels of myeloid-derived suppressor cells in breast cancer patients. Cell Immunol. 2011;270:80–7.
Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr + CD11b + cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–21.
Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong W-C, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS-Biol. 2011;9:1933.
Shi Y, Ou L, Han S, Li M, Pena M, Pena E, et al. Deficiency of Kruppel-like factor KLF4 in myeloid-derived suppressor cells inhibits tumor pulmonary metastasis in mice accompanied by decreased fibrocytes. Oncogenesis. 2014;3:e129.
Pandya AY, Talley LI, Frost AR, Fitzgerald TJ, Trivedi V, Chakravarthy M, et al. Nuclear localization of KLF4 is associated with an aggressive phenotype in early-stage breast cancer. Clin Cancer Res. 2004;10:2709–19.
Yu F, Li J, Chen H, Fu J, Ray S, Huang S, et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30:2161–72.
He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–31.
Gracias DT, Katsikis PD. MicroRNAs: key components of immune regulation. Crossroads between Innate and Adaptive Immunity III: Springer; 2011. p. 15–26.
O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010;10:111–22.
Liu Y, Lai L, Chen Q, Song Y, Xu S, Ma F, et al. MicroRNA-494 is required for the accumulation and functions of tumor-expanded myeloid-derived suppressor cells via targeting of PTEN. J Immunol. 2012;188:5500–10.
Suzuki H, Katsura A, Matsuyama H, Miyazono K. MicroRNA regulons in tumor microenvironment. Oncogene. 2014.
Li L, Zhang J, Diao W, Wang D, Wei Y, Zhang C-Y, et al. MicroRNA-155 and MicroRNA-21 promote the expansion of functional myeloid-derived suppressor cells. J Immunol. 2014;192:1034–43.
Wang J, Yu F, Jia X, Iwanowycz S, Wang Y, Huang S, et al. MicroRNA‐155 deficiency enhances the recruitment and functions of myeloid‐derived suppressor cells in tumor microenvironment and promotes solid tumor growth. Int J Cancer. 2015;136:E602–13.
Zhang M, Liu Q, Mi S, Liang X, Zhang Z, Su X, et al. Both miR-17-5p and miR-20a alleviate suppressive potential of myeloid-derived suppressor cells by modulating STAT3 expression. J Immunol. 2011;186:4716–24.
Zhao JL, Rao DS, Boldin MP, Taganov KD, O’Connell RM, Baltimore D. NF-κB dysregulation in microRNA-146a–deficient mice drives the development of myeloid malignancies. Proc Natl Acad Sci. 2011;108:9184–9.
Liu Q, Zhang M, Jiang X, Zhang Z, Dai L, Min S, et al. miR‐223 suppresses differentiation of tumor‐induced CD11b + Gr1+ myeloid‐derived suppressor cells from bone marrow cells. Int J Cancer. 2011;129:2662–73.
Chen S, Zhang Y, Kuzel TM, Zhang B. Regulating Tumor Myeloid-Derived Suppressor Cells by MicroRNAs. Cancer cell & microenvironment. 2015;2.
Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity. 2013;39:611–21.
He J, Xu Q, Jing Y, Agani F, Qian X, Carpenter R, et al. Reactive oxygen species regulate ERBB2 and ERBB3 expression via miR‐199a/125b and DNA methylation. EMBO Rep. 2012;13:1116–22.
Curiel TJ. Regulatory T, cells and treatment of cancer. Curr Opin Immunol. 2008;20:241–6.
Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86:1065–73.
Pan W, Sun Q, Wang Y, Wang J, Cao S, Ren X. Highlights on mechanisms of drugs targeting MDSCs: providing a novel perspective on cancer treatment. Tumor Biol. 2015;36:3159–69.
Melani C, Sangaletti S, Barazzetta FM, Werb Z, Colombo MP. Amino-biphosphonate-mediated MMP-9 inhibition breaks the tumor-bone marrow axis responsible for myeloid-derived suppressor cell expansion and macrophage infiltration in tumor stroma. Cancer Res. 2007;67:11438–46.
Veltman J, Lambers ME, van Nimwegen M, Hendriks R, Hoogsteden H, Hegmans J, et al. Zoledronic acid impairs myeloid differentiation to tumour-associated macrophages in mesothelioma. Br J Cancer. 2010;103:629–41.
Fricke I, Mirza N, Dupont J, Lockhart C, Jackson A, Lee J-H, et al. Vascular endothelial growth factor-trap overcomes defects in dendritic cell differentiation but does not improve antigen-specific immune responses. Clin Cancer Res. 2007;13:4840–8.
Kusmartsev S, Eruslanov E, Kübler H, Tseng T, Sakai Y, Su Z, et al. Oxidative stress regulates expression of VEGFR1 in myeloid cells: link to tumor-induced immune suppression in renal cell carcinoma. J Immunol. 2008;181:346–53.
Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15:2148–57.
Ko JS, Rayman P, Ireland J, Swaidani S, Li G, Bunting KD, et al. Direct and differential suppression of myeloid-derived suppressor cell subsets by sunitinib is compartmentally constrained. Cancer Res. 2010;70:3526–36.
van Cruijsen H, van der Veldt AA, Vroling L, Oosterhoff D, Broxterman HJ, Scheper RJ, et al. Sunitinib-induced myeloid lineage redistribution in renal cell cancer patients: CD1c + dendritic cell frequency predicts progression-free survival. Clin Cancer Res. 2008;14:5884–92.
Huang D, Ding Y, Zhou M, Rini BI, Petillo D, Qian C-N, et al. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010;70:1063–71.
Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514–22.
Lathers DM, Clark JI, Achille NJ, Young MRI. Phase 1B study to improve immune responses in head and neck cancer patients using escalating doses of 25-hydroxyvitamin D3. Cancer Immunol Immunother. 2004;53:422–30.
Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441–9.
Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer. 2010;10:464.
Weed DT, Vella JL, Reis IM, Adriana C, Gomez C, Sargi Z, et al. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res. 2015;21:39–48.
De Santo C, Serafini P, Marigo I, Dolcetti L, Bolla M, Del Soldato P, et al. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc Natl Acad Sci U S A. 2005;102:4185–90.
Ko H-J, Kim Y-J, Kim Y-S, Chang W-S, Ko S-Y, Chang S-Y, et al. A combination of chemoimmunotherapies can efficiently break self-tolerance and induce antitumor immunity in a tolerogenic murine tumor model. Cancer Res. 2007;67:7477–86.
Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res. 2010;16:4583–94.
Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70:3052–61.
Roland CL, Lynn KD, Toombs JE, Dineen SP, Udugamasooriya DG, Brekken RA. Cytokine levels correlate with immune cell infiltration after anti-VEGF therapy in preclinical mouse models of breast cancer. PLoS ONE. 2009;4:e7669.
Shen XZ, Okwan-Duodu D, Blackwell W-L, Ong FS, Janjulia T, Bernstein EA, et al. Myeloid expression of angiotensin-converting enzyme facilitates myeloid maturation and inhibits the development of myeloid-derived suppressor cells. Lab Investig. 2014;94:536–44.
Lin C, Datta V, Okwan-Duodu D, Chen X, Fuchs S, Alsabeh R, et al. Angiotensin-converting enzyme is required for normal myelopoiesis. FASEB J. 2011;25:1145–55.
Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007;67:11021–8.
Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001;166:5398–406.
Hengesbach LM, Hoag KA. Physiological concentrations of retinoic acid favor myeloid dendritic cell development over granulocyte development in cultures of bone marrow cells from mice. J Nutr. 2004;134:2653–9.
Zhao T, Du H, Ding X, Walls K, Yan C. Activation of mTOR pathway in myeloid-derived suppressor cells stimulates cancer cell proliferation and metastasis in lal−/− mice. Oncogene. 2015;34:1938–48.
Elleder M, Chlumská A, Hyánek J, Poupětová H, Ledvinová J, Maas S, et al. Subclinical course of cholesteryl ester storage disease in an adult with hypercholesterolemia, accelerated atherosclerosis, and liver cancer. J Hepatol. 2000;32:528–34.
Wu J, Zhang R, Tang N, Gong Z, Zhou J, Chen Y, et al. Dopamine inhibits the function of Gr-1+ CD115+ myeloid-derived suppressor cells through D1-like receptors and enhances anti-tumor immunity. J Leukoc Biol. 2015;97:191–200.
Zheng Y, Xu M, Li X, Jia J, Fan K, Lai G. Cimetidine suppresses lung tumor growth in mice through proapoptosis of myeloid-derived suppressor cells. Mol Immunol. 2013;54:74–83.
Condamine T, Kumar V, Ramachandran IR, Youn J-I, Celis E, Finnberg N, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest. 2014;124:2626.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Motallebnezhad, M., Jadidi-Niaragh, F., Qamsari, E.S. et al. The immunobiology of myeloid-derived suppressor cells in cancer. Tumor Biol. 37, 1387–1406 (2016). https://doi.org/10.1007/s13277-015-4477-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-4477-9