
Abstract
Background
HDAC6, a structurally and functionally distinct member of the HDAC family, is an integral part of multiple cellular functions such as cell proliferation, apoptosis, senescence, DNA damage and genomic stability, all of which when deregulated contribute to carcinogenesis. Among several HDAC family members known so far, HDAC6 holds a unique position. It differs from the other HDAC family members not only in terms of its subcellular localization, but also in terms of its substrate repertoire and hence cellular functions. Recent findings have considerably expanded the research related to the substrate pool, biological functions and regulation of HDAC6. Studies in HDAC6 knockout mice highlighted the importance of HDAC6 as a cell survival player in stressful situations, making it an important anticancer target. There is ample evidence stressing the importance of HDAC6 as an anti-cancer synergistic partner of many chemotherapeutic drugs. HDAC6 inhibitors have been found to enhance the effectiveness of conventional chemotherapeutic drugs such as DNA damaging agents, proteasome inhibitors and microtubule inhibitors, thereby highlighting the importance of combination therapies involving HDAC6 inhibitors and other anti-cancer agents.
Conclusions
Here, we present a review on HDAC6 with emphasis on its role as a critical regulator of specific physiological cellular pathways which when deregulated contribute to tumorigenesis, thereby highlighting the importance of HDAC6 inhibitors as important anticancer agents alone and in combination with other chemotherapeutic drugs. We also discuss the synergistic anticancer effect of combination therapies of HDAC6 inhibitors with conventional chemotherapeutic drugs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Acetylation, a major form of protein post-translational modification is responsible for regulating various cellular processes [1]. Lysine acetylation regulates the enzymatic activity, subcellular localization and protein–protein interaction of many cytoplasmic and nuclear proteins. It also affects a multitude of vital cellular processes such as pluripotency, cellular signaling, protein turnover, cell differentiation and cell survival, all of which when deregulated contribute to carcinogenesis [2,3,4]. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) catalyze acetylation and deacetylation, respectively. Acetylome-based studies in various cells have revealed that HATs and HDACs not only target histones but also control the acetylation status of many non-histone proteins [5].
Based on sequence identity and catalytic activity, 18 mammalian HDACs have been divided into four major classes, out of which class I, II and IV HDACs are Zinc dependent and class III HDACs are NAD+ dependent and known as sirtuins. Members of class I HDACs (HDAC1, 2, 3 and 8) are primarily located in the nucleus and show substrate specificity toward histone. Class II HDACs are further divided into subclasses IIa and IIb. Members of the Class IIa subclass include HDAC4, 5, 7 and 9 and that of class IIb HDAC6 and 10. Class IIa HDACs shuttle between the nucleus and the cytoplasm. Class IIb HDACs are primarily cytoplasmic and have non-histone proteins as primary substrates. NAD+ dependent Class III HDACs comprise seven members, SIRT1 to SIRT7. HDAC11 is the only member of class IV HDACs [6].
Extensive studies during the past two decades revealed that HDACs, as important epigenetic modulators, play key roles in multiple stages of tumor initiation and progression, and that deregulation of HDAC activity plays a causative role in tumorigenesis. Numerous studies in the anti-cancer drug discovery field have shown that HDAC inhibitors (HDACi) may serve as effective chemotherapeutic agents. Several pan-HDACi are currently being used for the treatment of various types of cancer. SAHA (vorinostat) [7] and romidepsin (class I specific; depsipeptide or FK228) [8], have been approved by the US FDA in 2006 for the treatment of cutaneous T cell lymphoma [9, 10]. Belinostat and panobinostat, have also been granted FDA approval for the treatment of peripheral T‐cell lymphoma and multiple myeloma, respectively [11]. However, the usage of pan-HDAC inhibitors comes along with various side effects, which calls for the development of effective subtype-selective HDAC inhibitors.
Accumulating evidence indicates that HDAC subtypes are not redundant in their activity and that each HDAC subtype differs from the other in terms of substrate repertoire and, hence, the molecular pathways they regulate. HDAC6 stands out among all the HDACs known so far, owing to the fact that it is structurally and functionally distinct from the other members of its family. It differs significantly from the other HDACs in terms of its subcellular localization, substrate repertoire and, hence, the cellular/biological processes it regulates. HDAC6 is a regulator of multiple vital cellular processes and pathways, which are essential not only for physiological homeostasis, but also account for the various stages of multistep tumor formation including initiation, promotion, progression and metastasis, when deregulated. Its unique structure, substrate specificity and cellular functionality make it a good anticancer target and it forms the basis for the success of HDAC6 inhibitors as subtype-selective anticancer inhibitors [12, 13]. In this review we will discuss the uniqueness of HDAC6 with relevance to its biological functions and substrate proteins, which makes it an important anti-cancer target. We will also discuss recent advances in combination therapies of HDAC6 inhibitors with other chemotherapeutic drugs.
2 HDAC6: Unique member of the HDAC family
Zinc-dependent HDAC6 is a member of the class IIb subclass of the HDAC family. HDAC6’s unique structure and cytoplasmic localization make it different from other members of the HDAC family. HDAC6, unlike other HDACs, is primarily cytoplasmic in localization. Serine-Glu containing tetradecapeptide (SE14) motifs [7] and a leucine-rich nuclear export signal (NES) at the N terminus of HDAC6 play a crucial role in retainment of human HDAC6 in the cytoplasm. p300 mediated acetylation of the nuclear localization signal (NLS) of HDAC6 also contributes to its cytoplasmic retention by inhibiting the interaction of the NLS of HDAC6 with importin α [14].
HDAC6 has two fully functional catalytic deacetylase domains spanning amino acid residues 87–404 (DD1) and 482–800 (DD2) [15] that make it structurally distinct from other members of the HDAC family (Fig. 1). The role of these two catalytic domains of HDAC6 in the overall activity of HDAC6 has been a subject of controversy. An initial report by Grozingeret et al.[16] assumed that both catalytic domains of HDAC6 possess independent activities and contribute independently to the activity of HDAC6. However, this study was challenged by other studies that claimed that the activity of HDAC6 relies solely on DD2 [15] or the interplay between both domains [17]. Zsofia Kutil et al. used acetylome peptide microarrays and peptide libraries for mapping the substrate specificity of the DD1 and DD2 domains of human HDAC6 [18]. They found that the DD2 domain of HDAC6 has a broad substrate specificity, whereas the activity of DD1 is highly specific for peptides bearing a C-terminal acetyllysine residue. Their findings showed that DD1 is solely responsible for the deacetylation of substrates harboring the acetylated lysine at their C terminus, whereas DD2 exclusively deacetylates peptides with an internal acetylated lysine residue [18]. A zinc ion, present at the bottom of the catalytic pocket of HDAC6 is required for the deacetylation function of HDAC6.

Schematic structure of HDAC6. HDAC6 is a unique member of the HDAC family. HDAC6 consists of two fully functional catalytic deacetylase domains spanning amino acid residues 87–404 (DD1) and 482–800 (DD2), making it structurally distinct from other members of the HDAC family. HDAC6 also possesses a ZnF ubiquitin-binding domain and a dynein-binding domain
Apart from its deacetylase domains, HDAC6 also possesses a ZnF ubiquitin-binding domain and a dynein binding domain that is absent in other members of the HDAC family. HDAC6-mediated cellular processes are not only dependent on its lysine deacetylase activity, but also on its ubiquitin and dynein binding ability. The ubiquitin binding Zinc finger domain present in the C-terminal region of HDAC6 helps it to bind ubiquitinated proteins. The dynein binding domain of HDAC6 helps it to bind dynein and to transport the cargo of ubiquitinated proteins along microtubules (MTs). Thus, the ubiquitin-binding domain and dynein-binding domain help HDAC6 in protecting cells from stress resulting from the accumulation of cytotoxic protein aggregates in response to proteasome inhibition. HDAC6 binds and mediates the transport of ubiquitinated and misfolded proteins along MT tracks to pericentriolar structures and facilitates the formation of aggresomes, followed by activation of autophagy for clearance of cytotoxic protein aggregates.
Almost all members of the HDAC family are involved in the deacetylation of histones. However, HDAC6’s unique cytoplasmic substrate repertoire makes that it stand out from other members of the HDAC family. HDAC6 regulates the function of non-histone cytoplasmic proteins like α-tubulin, Hsp90, cortactin, retinoic acid-inducible gene I, β-catenin and many more. Although HDAC6 has been reported to interact and deacetylase histones in vitro, in vivo interaction of HDAC6 with histones has not been reported. Apart from cytoplasmic proteins, HDAC6 has also been found to interact with some nuclear proteins. Examples include the immunologically relevant transcription factor forkhead boxp3 (FOXP3) and the DNA repair factor KU70. HDAC6 is recruited to chromatin by active, phosphorylated RNA polymerase II to reset acetylation/deacetylation cycles, permitting transcription.
3 HDAC6: Role in the maintenance of normal cellular physiology
HDAC6 plays an essential role in many cellular processes that are important for the maintenance of the normal physiology of a cell. It is an important regulator of cellular proliferation, apoptosis, and cellular motility. It also plays an important role in the aggresome pathway and heat shock response.
3.1 HDAC6: Regulator of cellular proliferation
HDAC6 is an important regulator of many signaling pathways that are associated with cellular proliferation. HDAC6 regulates the Ras/MAPK/ERK, PI3K/Akt and Wnt signaling pathways, all of which are associated with cellular proliferation and are activated in most tumors. ERK1 and ERK2 are the final effectors of the ERK/MAPK pathway. ERK1 and ERK2, when activated phosphorylate a plethora of substrates, through which they regulate cell growth. Both ERK1 (Extracellular signal-regulated kinase 1) and ERK2 (Extracellular signal-regulated kinase 2) have been found to interact with HDAC6 in vivo. HDAC6-mediated deacetylation of ERK1 plays an important role in stimulating its activity [19]. HDAC6 is itself also phosphorylated at serine 1035 by ERK (Fig. 2a) [20].

Role of HDAC6 in cellular proliferation. (a) HDAC6 is an important regulator of the Ras/MAPK/ERK, (b) PI3K/Akt and (c) Wnt signaling pathways, all of which when activated are associated with enhanced cellular proliferation. (d) HDAC6-mediated tubulin deacetylation plays an important role in G1 to S phase transition of the cell cycle and (e) spindle formation during mitotic metaphase
The ERK1/2 is activated by the binding of growth factors such as epidermal growth factor (EGF), platelet-derived growth factor (PDGF) and G-protein-coupled receptors (GPCRs) to the epidermal growth factor receptor (EGFR). The EGFR and further activation of its downstream pathways play an important role in cellular proliferation, especially in lung cancer [21, 22]. Therefore, EGFR signaling, which accounts for the cellular proliferation of most tumors, can be modulated by affecting the synthesis and degradation of EGFR. HDAC6 via deacetylation of one of the substrates, α-tubulin [23,24,25] plays an important role in cell endocytosis and EGFR trafficking and degradation. HDAC6 deficiency results in the accumulation of acetylated α-tubulin, thereby resulting in deregulation of microtubule-dependent endocytic vesicle trafficking and, hence, accelerating EGFR degradation (Fig. 2a), further reducing cellular proliferation.
HDAC6 also regulates the PI3K/Akt pathway via modulation of the acetylation status of HSP90, which plays an important role in stabilizing Akt. HSP90 binds to Akt and hence protects it from the action of phosphatases, thereby maintaining the phosphorylation status of Akt and hence its activity. Binding of Akt to HSP90, in turn, protects HSP90 from proteasomal degradation. HDAC6-mediated deacetylation of HSP90 increases the binding between HSP90 and Akt. Thus, by deacetylating Hsp90, HDAC6 modulates the stability of Akt and hence its function, thereby regulating the PI3K/Akt signaling pathway, which in turn affects many oncogenic processes such as cell survival, differentiation, migration and angiogenesis. HDAC6 inhibition enhances Hsp90 acetylation, thereby reducing the binding between HSP90 and Akt, and hence reducing cellular proliferation (Fig. 2b).
HDAC6 also regulates oncogenic Wnt signaling pathways via deacetylation of β-catenin, thereby mediating its nuclear localization where it upregulates oncogenes such as c-Myc [26]. Phosphorylation of HDAC6 by PKCα has been found to enhance HDAC6’s ability to deacetylate beta-catenin (Fig. 2c). Also, in osteosarcoma cells RHO-associated coiled-coil kinases (ROCK1/-2) phosphorylate tubulin polymerization-promoting protein-1 (TPPP1/p25), thereby preventing its binding to HDAC6. Consequently, in absence of TPPP1/p25, the binding activity of HDAC6 is increased which then leads to deacetylation of β-catenin [26]. Subsequently, the cells enter the S-phase, ultimately resulting in continued cellular proliferation.
HDAC6 is also an important regulator of the PTEN/Akt pathway. PTEN is a tumor-suppressor [27]. Activation of PTEN through K163 acetylation, in response to HDAC6 inhibition, is an important mechanism underlying the antitumor effects of pan-HDAC inhibitors [28]. PTEN inhibits cellular growth and proliferation by removing a phosphate group from PIP3, thereby preventing Akt activation [29].Thus, HDAC6 inhibition-mediated PTEN activation results in Akt dephosphorylation and inactivation. Also, HDAC6 has been found to mediate its carcinogenic effect in endometrial cancer through the PTEN/Akt/mTOR pathway [30]. HDAC6 overexpression studies in stable HDAC6 overexpression cell clones resulted in a decrease in PTEN expression levels and an increase in the levels of p-Akt and p-mTOR, while no significant changes were observed in total Akt and mTOR levels [30].
HDAC6 also regulates distinct phases of the cell cycle. HDAC6-mediated tubulin acetylation/deacetylation plays an important role in cell cycle progression (G1/S transition) and mitosis. HDAC6-mediated tubulin acetylation/deacetylation affects the interaction between Bcl-3, a protooncogene, and the protein encoded by the cylindromatosis (CYLD) gene [31,32,33,34]. CYLD controls cell cycle progression by delaying the G1 to S phase transition via its interaction with Bcl-3, a protooncogene, thereby blocking Bcl-3-dependent NF-ҡB signaling [14]. Deacetylated microtubules prevent the interaction between CYLD and Bcl-3. Therefore, CYLD is not able to inhibit Bcl-3 by using its deubiquitinase activity. As a consequence, Bcl-3 is free to move to the nucleus leading to transcriptional activation of NF-κB, thereby promoting S phase progression.
Accumulation of acetylated microtubules upon HDAC6 silencing or knockdown results in translocation of CYLD to the perinuclear region, thereby promoting the interaction between CYLD and Bcl-3. CYLD then inhibits Bcl-3 by using its deubiquitinase activity, resulting in an increase in the levels of Bcl-3 in the cytoplasm with a simultaneous decrease in the nucleus. Bcl-3 deficiency in the nucleus prevents NF-ĸB transcriptional activity, thereby reducing cyclin D1 expression and delaying the G1/S transition of the cell cycle (Fig. 2d) [35].
HDAC6 also interacts with and is inhibited by the deubiquitinating enzyme CYLD at the perinuclear region, thereby significantly delaying the G1-to-S phase transition, and in the midbody where it regulates the rate of cytokinesis in a deubiquitinase-independent manner (Fig. 2e) [31]. Moreover, HDAC6 regulates the c-Raf-protein phosphatase (PP)1-ERK signaling pathway, and inhibition of HDAC6 activity has been found to contribute to early M-phase cell cycle transition arrest via sustained ERK activation in prostate cancer [36]. Regulation of α-tubulin acetylation status by HDAC6-mediated deacetylation can also affect cellular proliferation by enhancing mitosis.
3.2 HDAC6: Regulator of cellular motility
HDAC6 is an important regulator of cellular migration. HDAC6 regulates cellular motility via its substrates tubulin and cortactin. α-Tubulin was identified as the first physiological substrate of HDAC6 [37,38,39]. Microtubules (MTs), the key regulators of cellular movement, are assembled by α-tubulin. α-tubulin acetyltransferases (αTAT) [40,41,42,43] acetylate α-tubulin at lysine 40, whereas HDAC6 [39] and SIRT2 deacetylate it. HDAC6-mediated reversible deacetylation of α-tubulin affects MT stabilization and hence regulates MT-dependent cell motility (Fig. 3a). HDAC6-mediated α-tubulin deacetylation leads to microtubule depolymerization [38]. ERK1 has been found to phosphorylate HDAC6 at S1035, thereby enhancing its tubulin deacetylase activity, resulting in increased cellular migration. Thus, the EGF-Ras-Raf-MEK-ERK signaling cascade promotes cell migration via HDAC6 phosphorylation, thereby increasing its tubulin deacetylase activity. Overexpression of HDAC6 leads to tubulin hypoacetylation and, hence, greater motility [39], whereas inhibition of HDAC6 leads to stabilization of microtubules. Thus, the regulation of the acetylation status of α-tubulin by HDAC6 affects cellular motility.

Role of HDAC6 in cellular motility. HDAC6 regulates microtubule and actin polymerization-based cellular motility via (a) deacetylation of tubulin and (b) cortactin, respectively
HDAC6 is also a regulator of actin polymerization-based cellular mobility. Actin polymerization and crosslinking are mediated by cortactin. Actin-dependent cellular motility is regulated by HDAC6 via modulation of the acetylation status of cortactin [44]. Cortactin is acetylated on lysines within its F-actin binding repeats by acetyltransferase p300/CBP associated factor (PCAF). Cortactin is a F-actin binding protein. It is present in the leading edge of migrating cells, which involves dynamic actin assembly [45]. HDAC6 is translocated to actin enriched membrane ruffles, where it interacts with cortactin and deacetylates it [44]. Deacetylated cortactin enhances actin polymerization and cellular motility due to its increased ability to bind to F-actin (Fig. 3b). In contrast, highly acetylated cortactin is unable to activate Rac1 or Arp2/3 and, thus, does not translocate to the periphery of the cell. Therefore, binding between cortactin and F-actin is reduced due to less cortactin in the cell periphery, resulting in reduced cell motility. Cortactin is found to be overexpressed in many carcinomas and HDAC6 inhibition results in hyperacetylation of cortactin, thereby resulting in impaired cellular motility. Cells without cortactin have impaired invasion and migration abilities[46]. HDAC6 plays an important role in the regulation of endothelial cell migration and angiogenesis via cortactin deacetylation [47]. Thus, HDAC6 regulates actin-based cell motility by altering the ability of cortactin to bind F-actin via modulating the acetylation status of cortactin. Therefore, HDAC6 regulates microtubule and actin polymerization-based cellular motility by modulating the acetylation status of α-tubulin and cortactin, respectively.
Regulation of microtubule and actin polymerization-based cellular motility by HDAC6 is influenced by the interaction of HDAC6 with GRK2. GRK2 is a stimulator of HDAC6 [48]. G protein-coupled receptor kinase (GRK2) directly associates with and phosphorylates HDAC6, thereby stimulating its α-tubulin deacetylase activity, leading to increased motility (Fig. 3a) [48]. GRK2 phosphorylation specifically at the S670 position increases its ability to regulate HDAC6. HDAC6 colocalizes in the lamellipodia of migrating cells, resulting in local tubulin deacetylation and enhanced motility [48, 49]. GRK2 downregulation triggered by RNA interference in HeLa cells has been found to lead to a higher accumulation of acetylated tubulin in sync with decreased motility resulting from decreased GRK2 levels [48]. In agreement with a dependency of GRK2-mediated enhanced motility on α-tubulin acetylation–deacetylation cycling, migration of both parental and HeLa-wt5 cells was inhibited in the presence of α-tubulin-K40A mutant, a construct that enforces permanent hypoacetylation of MTs [41, 48], thereby highlighting a role of HDAC6 mediated α-tubulin deacetylation as a mechanism for GRK2 mediated enhanced motility.
Sufficient literature supports that HDAC6 is involved in modulating microtubule dynamics and dynamic F-actin assembly by deacetylating ɑ-tubulin and cortactin respectively, thus promoting cellular motility. Chuang et al. reported two HDAC6 phosphorylation sites, pSer22 and pSer412, as Pin1 (peptidyl-prolyl cis/trans isomerase NIMA-interacting 1) substrates. HDAC6-Pin1 interaction is involved in HDAC6-mediated cellular motility through alteration of protein conformation and function. The authors reported that overexpression of Pin1 renders HDAC6 to a stable structure and increases HDAC6 transcript levels and hence cellular migration. Thus, anti-Pin1 may serve as a promising strategy to downregulate HDAC6 expression and may be anti-metastatic in lung cancer. Pin1 depletion abrogated HDAC6-induced cell migration and invasion in H1299 lung cancer cells. Therefore, Pin1 regulates HDAC6-mediated cellular motility via alteration of protein conformation and function [50, 51].
Liu et al. revealed that enhanced matrix metallopeptidase 9 (MMP9) activity via the HDAC6/PTPN1/ERK1/2 axis, may be one of the mechanisms underlying enhanced melanoma invasion and metastasis. They showed that HDAC6 interacts directly with Tyrosine-protein phosphatase non-receptor type 1(PTPN1) and, thereby, increases its protein level. PTPN1 has been found to promote enhanced proliferation and migration in melanoma via activation of extracellular signal-regulated kinase 1/2 (ERK1/2). Also MMP9 has been found to be a target of PTPN1/ERK1/2 pathway [52].
3.3 HDAC6: Regulator of apoptosis
HDAC6 regulates apoptosis by modulating the acetylation status of the DNA repair factor Ku70, which is involved in non-homologous end-joining DNA repair. Apart from its role in binding DNA double-strand breaks in non homologous end-joining DNA repair, Ku70 is also known to bind proapoptotic Bax protein in the cytoplasm, thus inhibiting Bax-induced cell death [53,54,55].The binding of Ku70 and Bax is regulated by the acetylation status of Ku70. Acetylation of Ku70 prevents it from interacting with Bax, hence Bax is free and can induce apoptosis. However, upon deacetylation by HDAC6, Ku70 causes Bax sequestration, which leads to the inhibition of apoptosis (Fig. 4a) [55]. Also, acetylation of Ku70 leads to its dissociation from the anti-apoptotic protein FLIP, leading to its proteasomal degradation and, hence, induction of apoptosis [56]. Thus, HDAC6 inhibits apoptosis and helps the cell to survive via deacetylating Ku70 (Fig. 4b).

Role of HDAC6 in apoptosis. HDAC6 regulates apoptosis by deacetylating (a and b) Ku70, (c) survivin and (d) GRP78
HDAC6’s role as an anti-apoptotic protein was further strengthened by the finding that survivin, an oncogenic protein, serves as one of the substrates of HDAC6 deacetylase activity. Survivin, when acetylated by the CREB-binding protein (CBP), translocates to the nucleus and is, thereby. not able to execute its anti-apoptotic effect [57]. However, survivin upon deacetylation by HDAC6, is transported from the nucleus to the cytoplasm, where it inactivates caspase proteins, thus inhibiting apoptosis and activating oncogenesis (Fig. 4c) [58]. Thus, survivin levels may increase in the cytoplasm upon HDAC6-mediated deacetylation. Survivin is highly expressed in breast cancer and HDAC6 has been found to be responsible for abrogating CBP-mediated survivin acetylation in the ER-positive breast cancer cell line MCF-7.
HDAC6 also regulates cellular apoptotic responses by deacetylating GRP78. Acetylation of GRP78 leads to its dissociation from PERK, resulting in activation of UPR which, in turn, leads to cell death. However, HDAC6-mediated deacetylation of GRP78 prevents cell death (Fig. 4d). Thus, to summarize, HDAC6 regulates apoptosis via deacetylation of survivin, Ku70 and GRP78. HDAC6 inhibition can lead to apoptosis.
3.4 HDAC6: Role in the aggresomal pathway
Defects in the proteasomal degradation machinery of the cell results in the accumulation of misfolded proteins which, in turn, results in activation of the aggresomal pathway of protein degradation. Aggresomes reduce the toxicity of scattered protein microaggregates, accumulated in response to proteasome inhibition. The sequestration of misfolded proteins into aggresomes is a cytoprotective response that is followed by the elimination of excessive amounts of misfolded proteins by autophagy. Therefore, targeting the aggresome pathway of protein degradation has become one of the strategies for cancer treatment.
HDAC6 plays a crucial role in the activation of the aggresomal pathway [59, 60]. In unstressed cells, HDAC6 forms a basal complex with p97/VCP, Hsp90 and HSF1, where VCP/P97 sequesters HDCA6 and HSF1 is maintained in its inactive form by Hsp90. Sequestering of HDAC6 by VCP/P97 prevents binding of HDAC6 with ubiquitinated proteins, making sure that ubiquitinated proteins undergo proteasomal degradation (Fig. 5a). However, upon failure of the ubiquitin proteasomal pathway of protein degradation, accumulation of ubiquitinated proteins takes place which causes HDAC6 to bind ubiquitinated proteins via its ZnF-UBD, resulting in dissociation of HDAC6 from the p97/VCP complex [60, 61]. HDAC6 binds ubiquitinated proteins via its ZnF-UBD and delivers them to the motor protein dynein which, in turn, transports the cargo of ubiquitinated proteins along the microtubules to the microtubule organizing centre (MTOC). HDAC6 deacetylase activity is not involved in the transport of misfolded proteins by HDAC6 along the microtubules to MTOC. At the MTOC, HDAC6-mediated deacetylation of cortactin leads to aggresome formation around misfolded proteins [61]. Chaperones are recruited to aggresomes, which facilitate the clearance of aggregated proteins. Thus, both the C terminal zinc-finger ubiquitin-binding domain as well as the deacetylase activity of HDAC6 are required for aggresome formation.

Role of HDAC6 in the autophagy/aggresomal pathway. (a) Under normal conditions, protein aggregates are degraded by the UPS of cells. (b) The aggresomal pathway of protein degradation is activated when the proteasomal degradation machinery of the cell becomes defective
Autophagy refers to the process of targeting aggresomal protein aggregates to lysosomes for further degradation. Protein degradation by autophagy helps avoid the accumulation of aggresomes containing polyubiquitinated protein aggregates. A role of HDAC6 in autophagy was first demonstrated by the Kopito laboratory, who showed that autophagic clearance of mutant huntingtin (Htt) aggregates was HDAC6 dependent [62]. The role of HDAC6 in autophagy was underscored when the function of HDAC6 was linked to autophagic clearance of polyglutamine huntingtin aggregates in Hela cells transfected with HDAC6 [62]. Also, studies on parkin-mediated K63 linked polyubiquitination coupled misfolded proteins to dynein through HDAC6 interaction, resulting in aggresome formation and its clearance by autophagy [63].
HDAC6 regulates autophagy by deacetylating LC3B-II, which is a key regulator of autophagy [64]. It also plays an adapter role in autophagy in a deacetylase-independent manner. The polyubiquitin binding protein p62 is required for both formation and degradation of aggresomes by autophagy [65]. E3 ubiquitin ligase TRIM50 interacts with HDAC6 and p62 and facilitates the clearance of ubiquitinated proteins into the aggresomes [66, 67]. p62 recruits autophagosomes to protein aggregates [68,69,70]. Autophagosomes engulf aggresomes, which then fuse to lysosomes for protein degradation by lysosomal hydrolases [67]. HDAC6 regulates the fusion of autophagosomes and lysosomes by recruiting and deacetylating cortactin, which recruits actin filaments to tether the two vesicles [67]. Thus, HDAC6 along with p97/VCP, TRIM50 and p62 controls aggresome formation and autophagosome maturation for ubiquitin selective quality control autophagy (Fig. 5b). Lack of HDAC6 causes failure of autophagosome maturation and protein aggregate accumulation rather than autophagy activation inhibition.
3.5 HDAC6: Role in activation of heat shock response
Defects in the proteasomal degradation machinery of the cell not only activates the aggresomal pathway and autophagy, but also induces heat shock response, and HDAC6 plays an important role in this response.
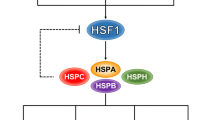
In a non-stressed cell, HDAC6 is associated with a protein complex containing VCP/p97, Hsp90 and heat-shock factor 1 (HSF) (Fig. 6a) [61]. Upon accumulation of misfolded proteins, the ubiquitin-binding domain of HDAC6 induces dissociation of HDAC6 from the Hsp90-HSF1 complex. This, in turn, results in dissociation of the Hsp90/HSF1 complex and, hence, release and activation of HSF1 which is one of the client proteins of Hsp90. Segregase activity of VCP/P97 plays a role in the dissociation of the Hsp90/HSF1 complex. Liberated HSF1 activates heat shock protein (Hsp) genes, including the gene encoding HSP90 that helps in reducing protein aggregate toxicity by excessive expression of chaperones, thereby providing a survival advantage to oncogenic transformed cells (Fig. 6b). A role of HDAC6 in the activation of HSF1 was further strengthened when HSF-1-Null mouse embryonic fibroblast (MEFs) were found to inhibit DMBA-TPA treatment-induced skin tumors similar to HDAC6 deficient cells and HDAC6 null mice. Elimination of HSF1 was found to protect mice from tumors induced by mutations of Ras or the p53 tumor suppressor protein [71].

Role of HDAC6 in activation of heat shock response. (a) Under normal conditions, protein aggregates are degraded by the UPS of cells. (b) When the proteasomal degradation machinery of a cell becomes defective, the aggresomal pathway of protein degradation is activated by HDAC6, which causes the release of HSF1 which, in turn, activates heat shock response. (c) HDAC6 also activates heat shock response by deacetylating Hsp90
Hsp90, whose expression is induced during heat shock response, is one of the substrates of HDAC6. Acetylation of Hsp90 regulates its ability to bind co-chaperones and its client proteins. The chaperone activity of Hsp90 is activated upon its deacetylation by HDAC6 [72, 73].Followed by HDAC6 mediated deacetylation, Hsp90 interacts with its client proteins like Bcr-Abl, glucocorticoid receptor (GR) or androgen receptor (AR). Thus, Hsp90 via its chaperone activity, ensures a favorable conformation of these proteins (Fig. 6c) [74, 75]. The stability of oncoproteins such as Bcr-Abl and Her2 is thus dependent on the chaperone activity of Hsp90.
HDAC6 knockdown studies have reaffirmed HDAC6’s critical role in the modulation of Hsp90 activity. Hsp90 loses its chaperone activity upon HDAC6 inhibition, thereby resulting in the degradation of its client proteins via the proteasomal degradation pathway [72]. Many of the client proteins of Hsp90 are oncogenic and play a critical role in cell growth and survival and they are, thereby, important for cancer progression[76]. LAQ824 and LBH589 mediated HDAC6 inhibition has been found to result in increased Hsp90 acetylation, thereby decreasing its activity resulting in polyubiquitination of its target proteins Bcr-Abl, c-Raf and Akt in human leukemia cells, thus preventing cancer [72]. HDAC6 knockdown mediated hyperacetylation of Hsp90 also leads to dissociation of Hsp90 from p23, a co-chaperone and, hence, loss of its chaperone activity. HDAC6 inhibition also affects Hsp90-mediated nuclear localization and transcriptional activation of GR. Hsp90 hyperacetylation upon HDAC6 knockdown in prostate cancer C4-2 cells resulted in inhibition of prostate-specific antigen (PSA) expression. It also impairs ligand-independent nuclear localization of endogenous androgen receptors and inhibits cell growth in the absence and presence of dihydrotestosterone (DHT). Since several oncoproteins rely on Hsp90 for stability, HDAC6 inhibition can lead to their ubiquitin-mediated degradation and, hence, cancer prevention.
3.6 HDAC6: Role in transcriptional repression
HDAC6 is primarily cytoplasmic, but it can shuttle to the nucleus and affect the activities of various transcriptional regulators directly as well as indirectly by deacetylating Hsp90 [74, 75, 77, 78]. Cellular proliferation prevents the nuclear translocation of HDAC6. However, when cell proliferation is arrested, a fraction of the HDAC6 protein translocates to the nucleus through its nuclear localization signal at the N-terminus.
HDAC6 directly controls the activities of the transcription factors Runx2 and nuclear factor-kB. RunX2 recruits HDAC6 from the cytoplasm to chromatin in osteoblasts to repress the p21 promoter (Fig. 7a) [79]. Transcription factors such as NFκB, p50 and p65 recruit HDAC6 from the cytoplasm to the nucleus to repress the expression of the gene encoding a subunit of H+-K+-ATPase (Fig. 7b) [80].HDAC6 results in transcriptional repression via its interaction with corepressors such as HDAC11, sumoylated HAT p300, and ligand-dependent nuclear receptor corepressor (LCoR). Sumoylation of the CRD1 domain of p300 mediates its repressor activity. HDAC6 binds to the SUMO-CRD1 domain of p300 resulting in transcriptional repression [81]. Binding of HDAC6 to the SUMO-CRD1 domain of p300 is involved in the repression of p53-dependent transcription (Fig. 7c) [82, 83]. p300 also acetylates HDAC6, and p300-mediated acetylation of HDAC6 inhibits its tubulin deacetylation activity [84] and suppresses HDAC6-induced Sp1 transcriptional activity [84]. HDAC6 retention in the cytoplasm is affected by acetylation in the N terminal nuclear localization signal (NLS) region. Mutation of this region prevents the interaction between HDAC6 and nuclear protein importin-α and, thereby, affects nuclear importing and histone deacetylation [14]. p300 modulates HDAC6 nuclear import by blocking the HDAC6/importin-α interaction [14].

Role of HDAC6 in regulating gene expression via interaction with transcription factors. (a) HDAC6 is recruited to chromatin by RunX2 and results in transcriptional repression of p21 in osteoblasts. (b) LCoR and HDAC6 are recruited to promoters of estrogen receptor alpha target genes in response to estrogen where they repress transactivation of estrogen inducible genes. (c) HDAC6 binds to the SUMO-CRD1 domain of p300 resulting in transcriptional repression. (d) HDAC6 is recruited to chromatin by NFκB resulting in transcriptional repression of the gene encoding a subunit of H+-K+- ATPase
LCoR is involved in ligand-dependent repressor activity of nuclear receptors [85] as well as ETO-2 [86]. HDAC6 interacts with LCoR and enhances its repression activity [85, 87]. In response to estrogen, LCoR and HDAC6 are recruited to the promoter of ER-α target genes in MCF-7 cells where they repress the transactivation of estrogen inducible genes (Fig. 7d) [87]. HDAC6 or LCOR knockdown leads to enhanced expression of some estrogen target genes such as Greb1 and Fos, suggesting that HDAC6 functions as a cofactor of LCoR.
Also, HDAC6 physically interacts with phosphorylated RNA polymerase II (Pol II) and its distribution pattern parallels the Pol II, but not the H3K36me3 signal. This result indicates that HDAC6 can be recruited to chromatin by active Pol II. HDAC inhibitor treatment causes increased acetylation of active genes, with the increased levels being correlated with HDAC binding, suggesting that HDAC recruitment to active genes functions by removing acetyl groups. This result implicates that HDAC may play a role in maintaining an adequate level of acetylation for active genes during transcription and in inhibiting promiscuous transcription initiation.
3.7 HDAC6: Role in metabolic response
HDAC6 plays an important role in hepatic glucocorticoid-induced gluconeogenesis via regulation of GR nuclear translocation. HDAC6 regulates Hsp90 deacetylation-mediated activation of glucocorticoid receptors [74, 75] and, hence, mediates transcription of gluconeogenic genes via glucocorticoid receptors in the liver (Fig. 8) [88]. Dexamethasone-induced expression of several hepatic genes such as G6P, FBP and PEPCK has been found to be reduced in HDAC6 knockout(KO) mice and HDAC6-specific inhibitor-treated cells. Also, dexamethasone-induced hepatic glucose output and GR translocation were found to be impaired in HDAC6 KO mice and tubacin-treated cells. Hence HDAC6 is a key regulator of hepatic glucocorticoid-induced gluconeogenesis via the regulation of GR nuclear translocation. It is a potential therapeutic target for glucocorticoid-induced diabetes [88].

Role of HDAC6 in hepatic glucocorticoid-induced gluconeogenesis via regulation of GR nuclear translocation. HDAC6 mediated Hsp90 deacetylation regulates activation of the glucocorticoid receptor, which is involved in gluconeogenesis
3.8 HDAC6: Regulator of DNA damage response
Under normal conditions, p53 is maintained at a low level. However, in response to cellular stresses such as DNA damage, p53 is quickly induced to accumulate in the cell nucleus. DNA damage-mediated sequential post-translational modifications including phosphorylation (Ser-15, Ser-20, and Ser-46) and acetylation (Lys-373/382) contribute to the induction of p53. For example, NH2-terminal phosphorylation of p53 promotes dissociation of MDM2 from p53, and -COOH terminal acetylation of p53 attenuates ubiquitination mediated by MDM2. Thus, these modifications repress the ubiquitin-dependent proteasomal degradation of p53, by which p53 becomes stable.
Upon repairable DNA damage, p53 transactivates its cell cycle-related target genes including p21 and 14–3-3 s to arrest cell cycle progression at the G1/S and/or G2/M boundaries to save time to repair damaged DNA, after which cells with repaired DNA re-enter the normal cell cycle. When cells undergo severe DNA damage, p53 instead promotes irreversible apoptosis through transactivating its pro-apoptotic target genes such as BAX, NOXA and PUMA and eliminates cells with seriously damaged DNA. Therefore, a proper DNA damage response, which monitors and ensures genomic integrity, has been considered to be a critical barrier to tumorigenesis, with p53 at the crossroad between cell survival and death following DNA damage.p53 becomes acetylated after DNA damage, and the acetylated form has been reported to exhibit increased transcriptional activity, promote coactivator recruitment, and enhance site-specific DNA binding. Acetylation of p53 is also believed to increase p53 stability by preventing the ubiquitination of key lysine residues and its subsequent proteasomal degradation.
HDAC6 is a novel p53 deacetylase (Fig. 9) with Lys381/382 as its target site. HDAC6 has been found to bind to the C terminal region of p53 via its deacetylase domain in colorectal cancer (CRC) cells. The C-terminal lysine residues within p53 are also targeted by HDAC1, HDAC2 and SIRT1. Romidepsin, a specific inhibitor of HDAC1/2, results in increased p53 acetylation at Lys320/372, but not at Lys381/382. The pan-HDACi SAHA, which inhibits primarily class I HDACs and HDAC6, increases p53 acetylation at Lys320/372/381/382. SIRT1 preferentially deacetylates p53 at Lys382. These data suggest that Lys382 can be targeted by HDAC6 and SIRT1 and Lys381 by HDAC6 in CRC cells [89]. HDAC6 levels are inversely correlated with p53 acetylation at lysines 381/382, which is associated with p53 functional activation. p53 acetylation strongly correlates with protein stability and is required for its functional activation as a tumor suppressor. HDAC6 makes p53 inactive by deacetylation and, hence, negatively regulates p53 activity. Decreased levels of nuclear HDAC6 also elicited changes in the expression of p53 target genes that promote cell cycle arrest and apoptosis. These HDAC6 effects on p53 stability and activity are attributed to HDAC6-mediated p53 deacetylation at lysines 381/382. HDAC6 knockdown studies confirmed these observations. Decreased levels of HDAC6 activity by A452 and depletion of HDAC6 have been found to contribute to the accumulation of p53 acetylation (Lys381/382) and its transcriptional activation. A452 increased the expressions of the p53 target genes p21, Bax and Bak, and p53 transactivation activity in wtp53 HCT116 and RKO cells [90]. Treatment of CRC cells with A452 caused a decrease in nuclear HDAC6 and interaction between HDAC6 and p53, and an increase in acetylation of p53 at Lys381/382. Thus, HDAC6 is considered a novel deacetylase of p53 at Lys381/382.

HDAC6 as a regulator of DNA damage response. HDAC6 is a novel deacetylase of p53. p53 becomes acetylated after DNA damage, resulting in increased transcriptional activity and stability. HDAC6 negatively regulates p53 activity by deacetylating it, thereby rendering it inactive, resulting in inhibition of transcription of target genes
Park et al. reported a role of HDAC6 in regulating the survival of mesenchymal stem cells (MSCs) via modulation of p53 acetylation at K120. HDAC6 was found to negatively regulate pro-apoptotic acetylation of p53, specifically at lysine residue 120 (K120) in MSCs. The authors showed that the loss of HDAC6 promotes acetylation of p53 at K120 and triggers caspase-dependent apoptosis by elevating the level of pro-apoptotic BCL-2 family member Bax and reducing the pro-survival protein Bcl-2 in MSCs. In addition, they found that HDAC6 inhibition results in elevated ROS levels, which is primarily caused by impaired mitochondrial oxidative metabolism [91].
Also, Runt-related transcription factor 2 (RUNX2) has been found to inhibit p53-dependent apoptosis via its collaboration with HDAC6 upon DNA damage [92]. RUNX2 plays an important role in osteoblast differentiation. Ozaki et al. found that RUNX2 acts as a negative regulator for p53 in response to DNA damage. In response to DNA damage upon Adriamycin (ADR) exposure, RUNX2, as well as p53 were induced at the protein and mRNA levels in U2OS cells along with a significant upregulation of various p53 target genes. RUNX2 is involved in p53-mediated transcriptional activation of genes. Chromatin immunoprecipitation studies revealed that in response to ADR, the RUNX2/p53 complex is recruited to p53 target promoters. RUNX2 depletion in p53-deficient H1299 cells had no detectable effect on the expression of p53-target genes irrespective of ADR treatment, which shows that RUNX2-mediated downregulation of p53 target genes is dependent on p53. Also, p53 target genes and, hence, ADR mediated apoptosis was found to be enhanced in response to ADR upon RUNX2 knockdown. Following ADR exposure, HDAC6 was also recruited to p53 target promoters, which shows that HDAC6 is part of the RUNX2/p53 complex. Also, upon tubacin treatment, a HDAC6-specific chemical inhibitor, ADR-mediated p53 target gene expression was further enhanced, confirming that HDAC6 deacetylase activity plays an important role in RUNX2-mediated downregulation of p53 target genes. Therefore, the study conducted by Ozaki et al. strongly emphasize that RUNX2 in collaboration with HDAC6 inhibits the DNA damage-induced transcriptional as well as pro-apoptotic activity of p53, which makes it an attractive therapeutic target for the treatment of cancer [93]. RUNX2-mediated induction of anti-apoptotic Bcl-2 has been found to contribute to the acquisition of an apoptosis-resistant phenotype in human prostate cancer. Although these studies have suggested an oncogenic property of RUNX2, the underlying molecular mechanism(s) is poorly understood. Collectively, these findings provide strong evidence that RUNX2 prohibits p53-mediated apoptosis in response to DNA damage through a collaboration with HDAC6 and may imply that RUNX2 is a potential therapeutic target for cancer treatment [93].
Yang et al. reported a role for HDAC6 in regulating radiosensitivity of glioma stem cells (GSCs). They found that HDAC6 inhibition in GSCs resulted in downregulation of the expression and activity of components of SHH signaling, glioma-associated oncogene homolog 1 (Gli1)and Patched (Ptch1 and Ptch2) receptors. Inhibition of HDAC6 activity resulted in decreased GSC proliferation, increased GSC apoptosis via inactivation of the SHH/Gli1 signaling pathway and decreased GSC DNA damage repair capacity via degradation of checkpoint kinase (CHK) 1 caused by X-linked inhibitor of apoptosis (XIAP) downregulation, resulting in elevated radiosensitivity [94].The role of HDAC6 in DNA damage response was further strengthened by the finding that MLH1 (MutL homolog 1), a key DNA mismatch repair protein, serves as a substrate of HDAC6. MLH1 is involved in the maintenance of genomic stability and DNA damage response. HDAC6 has been found to interact with MLH1 via its deacetylation domain both in vitro and in vivo. HDAC6 deacetylates the Lys-33 and Lys-241 sites of MLH1. MLH1 binding to MutSα is favored by acetylation of the Lys-33 and Lys-241residues. HDAC6-mediated deacetylation of MLH1 results in reduced affinity of MLH1 to the MutSα complex, thus disrupting the assembly of the MutSα–MutLα complex. Also, it was found that HDAC6-mediated deacetylation of MLH1 conferred 6-TG resistance via disruption of the MutS–MutL complex [92]. Zhang M et al. reported that HDAC6 interacts with the MMR proteins MSH2 and MSH6, and hence deacetylates them. Deacetylated MSH2 is then degraded by the proteasome. MSH2 is the major component of the DNA mismatch repair system and is involved in mismatch recognition. HDAC6-mediated deacetylation of MSH2 accounts for a significant decrease in sensitivity to DNA-damaging agents due to the downregulation of MMR activity [95].
4 Regulation of HDAC6 activity
Although, HDAC6 is itself involved in the regulation of a myriad of cellular processes, its deacetylase activity is tightly regulated at multiple levels such as (i) HDAC6 localization within the cell, (ii) various PTMs such as phosphorylation and acetylation (Fig. 10) [96] and (iii) direct or indirect interactions of HDAC6 with various partners, such as the membrane-associated protein dysferlin, invasion inhibitory protein (IIp)45 [97], tubulin polymerization-promoting protein/p25 (TPPP/p25) [98] or farnesyltransferase [99]. Phosphorylation of HDAC6 by various kinases has been found to enhance HDAC6 deacetylase activity. For example, glycogen synthase kinase 3(GSK3b) mediated phosphorylation of HDAC6 at position 22 (Ser22) [100, 101] and epidermal growth factor receptor (EGFR)–RAS–RAF–MEK–ERK signaling axis phosphorylation at position 1035 (Ser1035) [20] of HDAC6 has been found to significantly increase HDAC6-mediated tubulin deacetylase activity. EGFR-mediated phosphorylation of HDAC6 at Y570 has been found to result in reduced deacetylase activity and increased acetylation of α-tubulin [25]. Kinases, such as G protein-coupled receptor kinase 2 (GRK2), Aurora A, and protein kinase C isoform z (PKCz), have also been found to increase HDAC6-mediated tubulin deacetylase activity by phosphorylation at positions that remain undefined [49, 102]. Protein kinase CK2 (a serine/threonine kinase) mediated phosphorylation of HDAC6 at position 458 (Ser458) accelerates the process of aggresomal clearance of proteins[103]. Furthermore, upon Sendai virus infection, HDAC6 is phosphorylated and activated by PKCa, leading to the deacetylation and nuclear translocation of beta-catenin and, subsequently, the induction of antiviral immune response [104, 105].

Regulation of HDAC6 activity. Although HDAC6 is itself involved in the regulation of a myriad of cellular processes, its deacetylase activity is tightly regulated at multiple levels. Upper level: Phosphorylation-mediated regulation: Phosphorylation of HDAC6 by various kinases has been reported to alter HDAC6 deacetylase activity. Lower level: Acetylation-mediated regulation: Acetylation has been found to impair the deacetylase activity of HDAC6 affecting α-tubulin and transcriptional regulation
HDAC6 also undergoes acetylation-mediated regulation. Unlike phosphorylation, acetylation has been found to impair the deacetylase activity of HDAC6 against α-tubulin and transcriptional regulation. HDAC6 is acetylated by acetyltransferase p300 at 16 lysine residues, leading to inhibition of HDAC6-mediated tubulin deacetylation and suppression of Sp1 transcriptional activity [14]. Acetylation also plays role in the cytoplasmic retention of HDAC6. As a result of acetylation, interaction of HDAC6 with the nuclear import protein importin-α is blocked and, hence, HDAC6 is retained in the cytoplasm [14]. Apart from kinases and acetyltransferases, several other regulators have been found to directly interact with HDAC6 and to regulate its deacetylase activity. One such regulator is tubulin polymerization-promoting protein-1 (TPPP1/p25) [106]. TPPP1 has been found to regulate MT acetylation and β-catenin expression by binding to HDAC6 and inhibiting its deacetylase activity. TPPP1 overexpression in cells resulted in increased MT acetylation and, thereby, enhanced MT polymerization, whereas cells treated with TPPP1 RNAi/siRNA showed reduced MT acetylation. The TPPP1-HDAC6 interaction is impaired by Rho-associated coiled-coil kinase (ROCK) and cyclin-dependent kinase 1 (CDK1) mediated phosphorylation of TPPP1, resulting in increased HDAC6 activity followed by a decrease in cell motility and an increase in cell proliferation, respectively [98, 106, 107]. Other regulators such as paxillin [108, 109], CYLD, dysferlin, Mdp3 [110], p62 and RanBPM [111] directly interact with HDAC6 and, thereby, inhibit HDAC6 mediated functions such as deacetylation and aggresome formation. MicroRNAs can also regulate HDAC6 expression and function. miR-26, miR-433, miR-221, miR-206, miR-22 and miR-548 have been found to downregulate the expression and activity of HDAC6 [30, 112, 113].
Okuda et al. studied the role of S-nitrosylation, a PTM on HDAC6 in the regulation of protein acetylation. They found that HDAC6 is S-nitrosylated by endogenously produced NO. Thus, HDAC6 is a potential substrate of NO. S-nitrosylation of HDAC6 diminishes its deacetylase activity and, subsequently increases the acetylation of α-tubulin resulting in the accumulation of misfolded proteins and, ultimately, cell death [114].
5 HDAC6: An anti-cancer target
HDAC6 has emerged as a potential anti-cancer target (Fig. 11), owning to its well-known roles in tumorigenesis and tumor progression. HDAC6 is overexpressed in a variety of tumors including primary acute myeloid leukemia (AML) blasts [115, 116], colon cancer [117], some myeloblastic cell lines, breast cancer cells [118], ovarian cancer cells, human embryonic kidney cells (HEK), prostate epithelial cells (PrEC) and primary oral squamous cell carcinoma cell lines [119]. In cancer types such as oral squamous cell carcinoma, primary acute myeloid leukemia (AML), ovarian cancer, acute lymphoblastic leukemia (ALL) and hepatocellular carcinomas HDAC6 overexpression has been found to correlate with advanced tumor stages and enhanced tumor aggressiveness accompanied with lower survival rates. HDAC6 participates in the process of tumorigenesis by playing important roles in various processes, such as enhanced cellular proliferation, cancer cell migration and invasion [31, 48, 120]. Multiple HDAC6 knockdown and inhibition studies have been carried out suggesting a role of HDAC6 in cancer. HDAC6 is required for efficient oncogenic transformation, tumor growth and anchorage-independent proliferation, an important hallmark of tumor cells, which allows cells to survive by escaping anoikis. Multiple studies involving HDAC6 knockdown and inhibition have been carried out to check this fact. HDAC6 knockout MEFs have been found to be resistant to transformation, thus highlighting the importance of HDAC6 in oncogenic transformation. HDAC6 knockdown in MCF7, SKBR3 and SKOV3 cancer cells has been found to reduce their anchorage-independent growth [120]. In vivo studies in immunocompromised SCID mice also indicated that HDAC6 inactivation reduces the ability of the cells to form tumors. Mice injected with stably expressed HDAC6-specific shRNA showed fewer tumors than control mice (that were injected with stably expressed HDAC6 scrambled control) [120]. Also, HDAC6 null mice were found to be more resistant to chemical carcinogen-induced skin tumors [120]. HDAC6 activity was found to be significantly higher in inflammatory breast cancer (IBC) cells compared with non-IBC cells [121]. ACY 1215, a HDAC6-specific inhibitor, was found to inhibit the proliferation of IBC cells, both in vitro and in vivo, whereas non-IBC cells were found to be less sensitive to ACY 1215 treatment [121]. Keremu et al. showed that selective HDAC6 inhibition impaired tumor growth and downregulated PD-L1 production in vivo by modulating STAT3 Tyr-705 phosphorylation, suggesting that HDAC6 inhibitors may serve as potential anti-tumor agents in osteosarcoma [122].

HDAC6 as an anti-cancer target. Role of HDAC6 in inducing angiogenesis, activating oncogenic proliferative signaling pathways and tumor growth, activation of anti-apoptotic pathways via deacetylation of substrates, activation of aggresomal pathways, and enhancing cellular migration and metastasis makes it a good anticancer drug target
Inactivation of oncogenic signaling pathways and delay in cell cycle progression account for decreases in cellular proliferation and tumor growth upon HDAC6 inhibition. Cancer developmental steps involve sustained activation of growth factor signaling pathways leading to continuous cellular proliferation. As discussed above, HDAC6 is an important mediator of oncogenic signaling pathways which result in enhanced cellular proliferation. Concordantly, HDAC6 knockdown and inhibition have been found to decrease cellular proliferation via modulation of these oncogenic signaling pathways. ERK1/2 phosphorylation and levels of activated Ras and Akt were found to be decreased in HDAC6 knockout mice compared to wild-type control mice. HDAC6 inhibition promoted Akt and ERK dephosphorylation, and reduced cellular proliferation. A decrease in the levels of phosphorylated Akt and ERK [123,124,125,126] upon HDAC6 inhibition is at least partly due to hyperacetylation of Hsp90, one of the substrates of HDAC6. Hsp90 affects the PI3K/Akt pathway by affecting the functional stability of Akt [127, 128]. HDAC6 inhibition enhances Hsp90 acetylation, thereby reducing the interaction between Hsp90 and Akt, rendering Akt inactive and reducing cellular proliferation. It has also been found that HDAC6 overexpression in ovarian cancer, breast cancer and other tumor models may be due to oncogenic Ras signaling resulting in a substrate-independent proliferation of cancer cells.
HDAC6 overexpression has been found to be associated with the presence of an activated K-Ras mutant in colon cancer patients. Mutant K-Ras induces HDAC6 expression by a MAP kinase-dependent pathway. HDAC6 inhibition has been found to decrease epithelial cell proliferation by blocking epidermal growth factor-induced beta-catenin nuclear localization and, hence, decreasing c-Myc expression [26]. Inactivation of the EGFR pathway has also been found to account for a decrease in cellular proliferation upon HDAC6 knockdown. Accumulation of acetylated α-tubulin upon HDAC6 inhibition results in deregulation of microtubule-dependent endocytic vesicle trafficking and, thereby, enhanced EGFR degradation [24, 129, 130]. HDAC6 is involved in promoting the development of colon cancer by activating the MAPK/ERK signaling pathway. Thus, HDAC6 inhibition results in the inhibition of cell proliferation and migration via downregulating the MAPK/ERK signaling pathway in colon cancer. Zhang et al. reported that SET7 inhibits the growth of colon cancer cells by interacting with HDAC6 and, thereby, inactivating it. Thus, SET7 plays a role in tumor suppression in colon cancer via increasing levels of acetylated α-tubulin by inactivating HDAC6[131].
HDAC6 also enhances cellular proliferation via its control over cell cycle progression. Therefore, decreased cellular proliferation upon HDAC6 knockdown is exerted partly via a delay in cell cycle progression. As discussed earlier, HDAC6 regulates the cell cycle by promoting interaction between CYLD and Bcl3 via deacetylating α- tubulin [31, 33]. Upon HDAC6 knockdown, levels of acetylated α-tubulin increase and, hence, acetylated microtubules accumulate, resulting in translocation of CYLD to the perinuclear region. This results in reduced interaction between CYLD and Bcl3 [31, 35, 132]. Consequently, Bcl3 translocation to the nucleus is decreased resulting in inhibition of the transcriptional activity of NF-κB and reduced expression of cyclin D1, thereby delaying G1/S transition [31]. Thus, HDAC6 inhibition reduces cell proliferation by delaying the cell cycle progression.
One of the hallmarks of cancer is enhanced cellular migration and metastasis, and HDAC6 is required for enhanced tumor cell movement, invasion and metastasis. HDAC6 participates in cell motility via its substrates actin and tubulin. Overexpression and inhibition studies of HDAC6 have also highlighted the role of HDAC6 as a mediator of cellular mobility. HDAC6 inhibition promotes α-tubulin acetylation, which enhances microtubule stability and reduces cell migration [133]. TSA or tubacin-mediated HDAC6 inhibition resulted in increased microtubule acetylation and decreased microtubule dynamics, leading to a decrease in cellular mobility [134]. HDAC6-specific inhibitors have been found to decrease MT dynamics and reduce motility in neuroblastoma. NK84 (Tubacin derivative) treated ovarian cancer cells showed inhibition in migration, further emphasizing the importance of HDAC6 in the metastasis of cancer cells. HDAC6 is an estrogen upregulated gene. Therefore, expression of HDAC6 in estrogen-positive breast cancer MCF 7 cells was found to be increased, which further enhanced cellular motility by promoting binding to its substrate, α-tubulin, thereby enhancing microtubule activity [118]. Tamoxifen (TAM), and another anti-estrogen agent, ICI 182,780, in combination with tubacin prevented both estradiol-induced HDAC6 accumulation and HDAC6-mediated tubulin deacetylation, resulting in reduced motility. NIH3T3 cells overexpressing HDAC6 moved significantly faster than control cells in response to serum, and treatment with tubacin was found to decrease their motility [34]. HDAC6 overexpression in MCF-7 breast cancer cells increased their motility [118]. Cell motility studies in neuroblastoma showed that HDAC6 inhibitor treatment resulted in decreased MT dynamics and, therefore, reduced motility [135]. Metastasis depends on proteolytic degradation of the extracellular matrix, initiated by invadopodium formation. HDAC6 regulates metastasis by modulating the formation of invadopodia[136], which are actin-driven membrane protrusions that possess matrix degradative activity [137].The role of HDAC6 in invadopodia formation, invasion and metastasis was studied in breast cancer [136]. siRNA-mediated HDAC6 silencing in highly invasive MDA-MB-231 breast cancer cells confirmed the role of HDAC6 for 2D matrix proteolysis. HDAC6-mediated Hsp90 deacetylation destabilizes breast cancer metastasis suppressor 1, thereby decreasing its metastasis suppressor activity [77]. Breast cancer metastasis suppressor 1, one of the client proteins of Hsp90 [77] has also been found to interact with HDAC6. Numerous reports have shown that HDAC6 inhibition is effective in the treatment of cancer. Ovarian cancer cells treated with the HDAC6 inhibitor NK84 (Tubacin derivative) showed migration inhibition. siRNA or TSA treatment-mediated HDAC6 inhibition in MDA-MB-231 cells resulted in a decrease in their invasive ability in a 3D type 1 collagen matrix [136]. Similar results were obtained in hepatocellular carcinoma [138]. Inhibition of HDAC6 leads to stabilization of microtubules and accumulation of misfolded ubiquitinated proteins, promoting apoptosis in tumor cells. HDAC6 upregulation-mediated decreased tubulin acetylation has been found to enhance the motility of fibroblasts [37] and breast cancer cells [118, 139]. Pham et al. reported a role of HDAC6 in driving rhabdomyosarcoma (RMS) tumor growth, self-renewal and migration/invasion. HDAC6 is involved in promoting tumor growth by modulating cell cycle progression and tumor cell differentiation and has been found to regulate the migratory and self-renewal capacities of RMS tumor cells by altering the microtubule and actin-dependent cytoskeletal dynamics to promote RMS cell migration via RAC1. Thus, targeting the HDAC6-RAC1 axis may be a promising therapeutic approach for RMS [140].
HDAC6 is a positive regulator of angiogenesis. One of the mechanisms via which HDAC6 promotes angiogenesis is by regulating the polarization and migration of vascular endothelial cells in a microtubule end-binding protein 1-dependent manner [141]. HDAC6 and HDAC10 play important roles in Hsp-mediated regulation of VEGFR in cancer cells [142]. Hypoxia has been found to upregulate HDAC6 mRNA and protein levels in endothelial cells (ECs) [47]. HDAC6 associates with HIF-1a and increases its stability and transcriptional activity in cancer cells [143]. HDAC6 increases hypoxia-inducible factor (HIF)-1 stability in cancer cells via direct deacetylation, and indirectly via modulation of the HSP90 chaperone function [144]. HIF-1 in turn stimulates its transcriptional activity toward target genes promoting angiogenesis such as vascular endothelial growth factor (VEGF) [143]. HDAC6-mediated HSP90 deacetylation ensures adequate binding to VEGF receptor (VEGFR)-1 or VEGFR-2, which transduces angiogenic signaling upon VEGF-A stimulation [142]. HDAC6 silencing in endothelial cells not only decreased sprouting and migration in vitro, but also reduced the formation of functional vascular networks in matrigel plugs in vivo [47]. HDAC6 deficient mice showed a reduced formation of perfused vessels in matrigel plugs [47]. Surprisingly, hypoxia-induced suppression of HDAC6 has been found to promote angiogenesis in hepatocellular carcinoma (HCC) by significantly upregulating HIF-1/VEGF-A expression levels [145]. Therefore, HDAC6’s role in migration, oncogenic signaling pathways, anti-apoptotic pathways, cellular stress surveillance factor, and aggresomal pathways makes it a good anticancer drug target.
6 HDAC6 inhibitors
HDACs are well-known anticancer targets. Pan-HDAC inhibitors have turned out to be very effective for cancer treatment. However, side effects associated with pan-HDAC inhibitors limit their usage. Therefore, numerous studies have been reported for the development and study of subtype-specific HDAC inhibitors owning to comparatively fewer side effects associated with their usage. Each HDAC subtype differs from the other in terms of its substrate specificity and, hence, the molecular pathway it participates in. Among the different HDAC subtypes, HDAC6 is of particular interest as it differs from the other HDACs, not only in terms of unique cytoplasmic substrates, but also in terms of its structure. As discussed earlier in this review, HDAC6 contains two catalytic domains and a ubiquitin-binding domain, which other HDAC members lack. These unique properties of HDAC6 make it structurally and functionally different from other members of the HDAC family. Also selectively targeting HDAC6 over other subtypes maximizes the pharmacological effects and also minimizes the side effects associated with pan-HDAC inhibitors. HDAC6 inactivation confers resistance to oncogenic transformation and tumor formation [120]. Thus, a great deal of effort has been devoted to studying the effect of HDAC6-selective inhibitors in various tumors for anti-cancer therapy with the hope to minimize the side effects associated with the usage of pan-inhibitors. Therefore, the development of HDAC6 inhibitors is a trending area of research and many studies are being carried out in this area. Research carried out in our laboratory found that the presence of features such as electron donor groups, sterically bulky scaffold and a hydrophobic ring is crucial for potent and selective HDAC6 inhibitory activity [146]. Another unique function of the HDAC6-specific inhibitors is that they increase acetylated tubulin expression without having much effect on acetylated histone levels. Table 1 shows a list of various HDAC6 inhibitors. All the listed inhibitors have been validated for their HDAC6-specific enzymatic activity via enzymatic assays, acetylated tubulin expression via Western blotting, or both. Out of all the HDAC6 inhibitors mentioned in Table 1, only ricolinostat (ACY-1215) and KA2507 have entered clinical trials as tabulated in Table 2, whereas all the rest are in the preclinical stages. In addition, HDAC6 plays an important role in protecting cells from stress-inducing chemotherapeutic drugs. This is the rationale why HDAC6 inhibitors have been found to enhance the effectiveness of stress-inducing chemotherapeutic agents used for tumor treatment as discussed in the subsequent sections.
7 HDAC6: An important player in cell survival under stressful conditions -combination therapy
Chemotherapy is one of the major strategies for cancer treatment, and functions by targeting the physiological characteristics of cancer cells, including proliferation, angiogenesis, apoptosis, invasion and migration [6]. However, drug resistance [7] and severe side effects [8] still hinder the effects of chemotherapy. Therefore, recently combinatorial therapies have gained importance as they not only enhance the therapeutic effects but also decrease the required dosages of each drug, thereby reducing the severity of adverse effects [9]. Thus, the search for combinations of chemotherapeutic agents that can achieve synergistic antitumor effects remains an important strategy.
Cancer cells may harbor several epigenetic alterations, including an increased expression and activity of histone deacetylases (HDACs). Dysregulation of HDACs is critical to the development and progression of the majority of tumors. Thus, HDAC inhibitors seem to be one of the promising anti-cancer drugs. The use of HDACi’s as monotherapy has shown positive pre-clinical results, but clinical trials showed only limited success due to associated side effects. However, combinatorial regimens of HDACi’s with other anti-cancer drugs have shown synergistic effects both in pre-clinical and clinical studies. At the same time, these combinations have enhanced the efficacy, and reduced the toxicity and tumor resistance to therapy.
HDACi’s are broadly classified into five classes: hydroxamates, cyclic peptides, short-chain fatty (aliphatic) acids, benzamides and sirtuin inhibitors. HDACi’s are also classified based on their ability to inhibit HDAC classes as pan-HDAC inhibitors (pan-HDACi’s) that act on all HDAC classes (not including sirtuins), and selective HDAC inhibitors, that target a specific HDAC. So far, pan-HDACi’s have been more widely studied and used rather than selective HDACi’s. Initially, several pan-HDACi’s were approved by the US Food and Drug Administration (FDA) for clinical treatment such as Suberoylanilidehydroxamic acid (SAHA; Vorinostat) used for the treatment of relapsed and refractory cutaneous T-cell lymphoma (CTCL), romidepsin or belinostat for various hematological tumors and many other HDACi’s for different malignancies. Vorinostat and Trichostatin A were the first generation HDACi’s.
Although pan-HDACi’s are currently approved by the FDA, their usage is limited as single agents against solid tumors in clinical trials compared to the hematological malignancies due to associated secondary effects such as thrombocytopenia, nausea, vomiting, anorexia and fatigue. To broaden the therapeutic window, combination of HDACi’s with other chemotherapeutic drugs (such as topoisomerase inhibitors, PARP inhibitors, proteasome inhibitors, radiotherapy, antimetabolites and mTOR inhibitors) has emerged as a new therapeutic strategy in the treatment of both hematological and solid tumors in both pre-clinical and clinical studies. Combination studies involving SAHA with cisplatin (alkylating agent) have shown a synergistic effect on cell proliferation, induction of apoptosis and cell cycle perturbation, and being beneficial for patients with larynx cancer. Another well-known HDACi Panobinostat in combination with Bortezomib and dexamethasone (proteasome inhibitor) has shown a synergistic effect in a phase III trial in MM patients. Also, a study conducted in our lab showed that Panobinostat synergized with Topotecan/Etoposide (Topoisomerase inhibitors) and resulted in cell death via induction of the intrinsic apoptotic pathway in cervical cancer cells [188, 189]. One more phase I study in patients with advanced solid malignancies involving a combination of pazopanib (Tyrosine Kinase Pathway Inhibitor) with pan-HDACi abexinostat resulted in a synergistic effect in patients with advanced solid malignancies. Moreover, combination of Quisinostat (pan-HDACi) and Flavopiridol (CDKi) turned out to be a promising therapeutic strategy for both cutaneous and uveal metastatic melanomas. Several such combination studies are currently in pre-clinical and clinical trials. Contrary to the synergism in combinatorial regimens, numerous studies did not have not have the same outcomes. For example, a phase I/II study involving combination treatment with SAHA and Rituximab-CHOP resulted in increased toxicity, particularly neutropenia and sepsis in patients with advanced-stage diffuse large B-cell lymphoma, whereas a phase II study involving a combination of belinostat and paclitaxel/carboplatin did not show any synergism and enhanced progression-free survival (PFS) in patients with carcinoma of unknown primary origin. The underlying reason for these contrary results is not clear but may involve drug interference. The pan-HDACi’s discussed so far show side effects such as fatigue, thrombocytopenia and gastrointestinal problems. Considering these adverse effects, researchers hypothesized that specific-HDACi’s with a higher selectivity and specificity may exhibit better therapeutic indices and fewer adverse side effects.
A pre-clinical combination study involving CS055 (Chidamide- a novel selective HDACi) and doxorubicin resulted in a synergistic antitumor effect in peripheral T-cell lymphoma cell lines (PTCL). Also, the combination of nicotinamide (SIRT1 inhibitor) and doxorubicin significantly enhanced inhibition of cell proliferation, induced apoptosis and reduced resistance to treatment in breast cancer cells. Furthermore, in a pre-clinical study, combination treatment with Sirtinol (selective inhibitor of SIRT2) and dichloroacetate acid (Tyrosine Kinase Pathway Inhibitor) showed a synergistic effect in proliferation inhibition and apoptosis induction in vitro, and a reduction of tumor volume in vivo in mice. Additionally, a pre-clinical study in lung cancer cells revealed a synergistic interaction of Tenovin-6 (inhibitor of both SIRT1 and SIRT2) and metformin (an anti-diabetic drug and mTOR signaling pathway inhibitor), resulting in cell growth inhibition and apoptosis induction. Thus, the above examples reiterate that subtype-selective HDACi’s may have synergistic anticancer effects. In particular, HDAC6 inhibitors have emerged as potential selective HDACi’s because of their involvement in a variety of cellular processes associated with cancer development [190]. Ricolinostat, a HDAC6-selective inhibitor, is currently in a phase I/II clinical trial. Amengual et al. studied the effect of a first-in-Class Selective HDAC6 Inhibitor (ACY-1215) in patients with relapsed and refractory lymphoid malignancies. ACY-1215 was well tolerated with no observed dose-limiting toxicities. However, mild and easily controlled toxicities of gastrointestinal symptoms were observed [191]. KA2507, a potent and selective small-molecule inhibitor of HDAC6 is currently under phase I and phase II trials for adult solid tumors and biliary tract cancer, respectively, as a single agent [156].
Also, HDAC6 deficient mice have been generated and found to be viable. Studies in HDAC6 deficient mice have shown that the HDAC6’s role is of minimal importance in an unstressed condition. However, its role becomes more important when a cell is subjected to stress. HDAC6 deficient mice were found to exhibit increased susceptibility to various stressful stimuli when challenged, highlighting its protective role under stressful conditions. It has also been shown that HDAC6 can sense, respond and protect cells from stress generated by chemotherapeutic drugs such as proteasome inhibitors, topoisomerase inhibitors and DNA damaging agents. Inhibition of the general proteasome machinery of the cell results in the accumulation of protein aggregates which, in turn, leads to induction of three types of protective cellular responses, i.e., autophagy, aggresome formation and heat shock response. As discussed above, HDAC6 is involved in the activation of all these three responses, which makes it an important cellular stress surveillance factor [59, 61].
Stress granules (SG) increase in numbers when the proteasome pathway is defective [192]. HDAC6 is important for stress granule formation, as pharmaceutical inhibition of HDAC6 has been found to abolish SG formation. HDAC6 is a critical component of stress granules [193]. Deacetylase activity as well as the ubiquitin-binding ability of HDAC6 is essential for SG formation. HDAC6 interacts with the SG protein Ras-GTPase activating protein SH3 domain-binding protein 1 (G3BP1) via its deacetylase domain [61]. Both HDAC6 and G3BP colocalize to the SGs under stress conditions [193]. Disruption of MT or motor proteins also disrupts SG formation. The absence of SG formation upon application of nocodazole and dynein ATPase inhibitor suggests that HDAC6 recruits SGs to motor proteins via binding of SG protein G3BP1, to allow movement of SG components along the MTs. Thus, HDAC6 coordinates SG formation by facilitating the motor protein-driven movement of SG components along the MTs. Therefore, HDAC6 not only protects cells from proteasome inhibitor-mediated cellular stress by autophagy, aggresome formations and heat shock response, but also via SG formation. Thus, HDAC6 prevents cells from undergoing proteasome inhibitor-mediated cell death by activating alternate pathways of degradation of ubiquitinated proteins. Also, cells lacking HDAC6 are more sensitive to proteasome inhibitor-mediated cell death. HDAC6 inhibition also sensitizes cells to MT inhibitors and the DNA damaging effect of topoisomerase II inhibitors. Therefore, HDAC6 inhibitors augment the anti-cancer effect of stress-inducing chemotherapeutic drugs. Several examples have been published where HDAC6 inhibition has been found to enhance cell death mediated by chemotherapeutic drugs. In the following section, we will review all combination studies of HDAC6 inhibitors with other chemotherapeutic drugs with relevance to cancer. Table 2 (https://www.clinicaltrials.gov/) summarizes HDAC6 inhibitors that are in clinical trials with other chemotherapeutic drugs.
7.1 Combination studies of HDAC6 inhibitors and proteasome inhibitors
Accumulating evidence indicates that HDAC6 and proteasomal inhibition is synergistic for cancer treatment as it impairs both proteasome-dependent and proteasome-independent aggresomal pathways of protein degradation. Misfolded proteins accumulated in transformed cells are cleared off by the proteasomal or aggresomal pathway. The aggresomal pathway is activated when the basal proteasomal machinery of the cell is unable to take care of a load of ubiquitinated protein and autophagy ensures clearance of the toxic protein aggregates in the aggresomes. As discussed above, both the ubiquitin-binding domain as well as deacetylase domain of HDAC6 is essential for the aggresomal and the autophagy process. Therefore, inhibition of both proteasomal and aggresomal pathways results in aggregation of misfolded proteins and, hence, apoptosis of cancer cells due to increased ubiquitin proteasome system stress resulting from enhanced metabolic activity compared to non-transformed cells.
Kastle et al. demonstrated involvement of HDAC6 in the induction of nuclear factor erythroid 2–related factor 2 (Nrf-2) and hemeoxygenase 1 (HO-1) upon proteasome inhibition. Several studies suggest that proteasome inhibition-mediated HO-1 elevation is mediated by Nrf-2 and by activation of the p38-MAPK pathway. HDAC6 siRNA studies by Kastle et al. revealed p38 as a substrate of HDAC6. HDAC6 knockdown resulted in attenuation of p38 phosphorylation after proteasome inhibition. HDAC6 was found to deacetylate p38, thereby facilitating phosphorylation of p38 which, in turn, led to Nrf-2 phosphorylation, consequently causing its dissociation from Keap-1 and its translocation to the nucleus, resulting in the transcription of Nrf-2 target genes, such as HO-1 [194]. Also as discussed above, HDAC6 plays a significant role in Hsp activation and de novo synthesis of Hsps post proteasome inhibitor treatment. Therefore, the involvement of HDAC6 in various processes which help in protecting cells from undergoing proteasome inhibition-mediated cell death reinforces that combination therapy of HDAC6 inhibitors and proteasome inhibitors may be quite effective for cancer treatment.
Several studies have highlighted the effectiveness of combination therapy of HDAC6 inhibitors and proteasome inhibitors for cancer treatment. Several HDAC6 inhibitors have been found to synergize with Bortezomib (BTZ) in Multiple Myeloma (MM) cells. Tubacin and BTZ combination treatment synergistically induced apoptosis in MM cells via the caspase 8-dependent pathway [195]. A452, a HDAC6-selective inhibitor, was found to significantly decrease the activation of BTZ-resistant markers, such as ERK and nuclear factor kappa B (NF-κB), in acquired BTZ-resistant MM cells. Combination treatment of A452 with BTZ or CFZ was also found to synergistically inhibit the activation of NF-κB and signal transducer and activator of transcription 3 (STAT3). Overall, A452 and BTZ/CFZ when used in combination result in synergistic cell death in BTZ-resistant U266 cells [196]. NexA could also overcome BTZ resistance in MM cells, and NexA in combination with BTZ showed a stronger efficacy [197]. Huang et al. studied the effect of combination treatment of HDAC6 inhibitor, N-hydroxy-4-((5-(4-methoxybenzoyl)- 1H-indol-1-yl)methyl)benzamide (MPT0G413) and BTZ using in vitro (RPMI-8226 and NCI-H929 cells) and in vivo models, thereby providing a framework for the clinical evaluation of combined therapies for MM. Combination treatment of MPT0G413 and BTZ enhanced apoptosis and anti-MM activity synergistically in human MM cells via inhibition of the proteasomal and aggresomal pathways of protein degradation and subsequent activation of apoptosis pathways. Combination treatment blocked the proteasomal and aggresomal pathways resulting in an increase in the expression of ubiquitinated proteins and decreased aggresome formation (LC3B). Combination therapy in a xenograft mouse model (in vivo studies) showed a potent reduction in tumor volume compared to individual treatments via apoptosis. Combination treatment also inhibited the adherence of MM cells to BMSCs, thereby inhibiting cell growth and decreasing VEGF and IL-6 secretion in the bone marrow microenvironment [198]. The combination of a novel HDAC6-selective inhibitor, MPT0G413, and proteasome inhibitor BTZ turned out to be synergistic in MM cells [198]. WT161, a potent HDAC6-selective inhibitor, and BTZ when used in combination, were also found to be synergistic in MM and to trigger a significant accumulation of polyubiquitinated proteins followed by apoptosis [195]. The combination was also effective in BTZ-resistant cells [199]. In addition to that, a pre-clinical study using ACY-1215 in combination with BTZ in a MM mouse model showed delayed tumor growth and increased mouse survival [200]. BTZ also increased the pharmacodynamics and pharmacokinetic properties of ACY-1215 [52]. Combination of HDAC6 inhibitors and BTZ has also been found to be effective in other cancer types. Synergistic cell death was observed in ovarian cancer cells that were treated with BTZ and Tubacin derivative NK84. Pancreatic cancer cells showed increased apoptosis upon combination treatment of HDAC6 siRNA along with BTZ [201]. In an in vivo xenograft mouse model of diffuse large B cell lymphoma (DLBCL), ACY-1215 and BTZ treatment significantly delayed tumor growth and prolonged overall survival. Dual targeting of protein degradation pathways via simultaneous treatment with ACY-1215 and BTZ turned out to be synergistic in lymphoma. In addition to BTZ, other proteasome inhibitors have also been found to synergize with HDAC6 inhibitors. Ricolinostat, when used in combination with CFZ, triggered synergistic anti-MM effects, even in BTZ-resistant cells. CFZ was found to increase the accumulation of ubiquitinated proteins and protein aggregates in the cytoplasm, as well as the engulfment of aggregated ubiquitinated proteins by autophagosomes, which was blocked by ricolinostat. Electron microscopy imaging showed increased autophagy, which was triggered by CFZ, and was inhibited by ACY-1215 addition. Finally, an in vivo mouse xenograft study confirmed a decrease in tumor volume, following treatment with CFZ in combination with ricolinostat. These results suggest that ricolinostat inhibits aggresome formation, caused by CFZ-induced inhibition of the proteasome pathway, resulting in enhanced apoptosis in MM cells [202].
7.2 Combination studies of HDAC6 inhibitors and microtubule inhibitors
Microtubules (MTs) are validated drug targets for the treatment of cancer [203, 204]. Taxol inhibits microtubule instability and hence cell division by stabilizing microtubules, resulting in acetylation of α-tubulin. The dynamic instability of microtubules is important for their role in cell division. Reversible acetylation of the amino group of Lys40 of α-tubulin regulates MT turnover [205]. HDAC6 regulates the stability of MTs by deacetylating α-tubulin. Several HDAC6i’s have been found to synergize with paclitaxel (MT targeting drug) in ERα-positive breast cancer cells. The combination of thiol-based HDAC6 inhibitors with paclitaxel caused synergistic inhibition in colorectal and breast cancer cells [206]. In vitro studies conducted by Yoo et al. showed that a second-generation HDAC6-selective inhibitor, ACY-241 (citarinostat), and a novel inhibitor, A452, exhibited synergistic anticancer effects with paclitaxel in AT-rich interaction domain 1A-mutated ovarian cancer [207]. Ricolinostat was found to enhance the anti-tumor effect of eribulin (an inhibitor of MT dynamics) through acetylation of α-tubulin in MDA-MB-231 and Hs578T cells [208]. Studies conducted by Tu et al. showed that co-treatment of MPT0G211 and vincristine may alter MT dynamics, thereby triggering acute lymphoblastic leukemia cells to arrest in the mitotic phase followed by induction of the apoptotic pathway [209]. When a taxane was used in combination with ACY1215, a synergistic cytotoxic effect accompanied by increased apoptosis was observed in various prostate cancer models [210].
7.3 Combination studies of HDAC6 inhibitors and DNA damaging agents
As discussed in detail above, HDAC6 is an important regulator of DNA damage response. HDAC 6 overexpression has been found to decrease the sensitivity of cells towards cell death induced by DNA damaging agents. Zhang M et al. reported that HDAC6 interacts with the MMR proteins MSH2 and MSH6 and, hence, deacetylates them. Deacetylated MSH2 is then degraded by the proteasome. MSH2 is a major component of the DNA mismatch repair system. It is involved in mismatch recognition. HDAC6-mediated deacetylation of MSH2 accounts for a significant decrease in sensitivity to DNA-damaging agents due to downregulation of MMR activity. Also, HDAC6-mediated deacetylation of MLH1 has been found to confer 6-TG resistance via disruption of the MutS–MutL complex [92].
Several studies support the notion that HDAC6 inhibition induces DNA damage and sensitizes transformed cells to DNA damaging agents. Kim et al. suggested HDAC6 selective inhibition as a promising approach for the treatment of Temozolomide (TMZ)-resistant GBM. TMZ is a DNA damaging agent. HDAC6-selective inhibitors increased the levels of MSH2 protein in TMZ-resistant GBM cells. They studied the effect of combination treatment of Temozolomide (TMZ) with the HDAC6 inhibitors A452 and ACY-1215 in TMZ sensitive and TMZ-resistant GBM cells. Combination treatment with both inhibitors synergistically induced apoptosis in TMZ-resistant cells by increasing MMR proteins and, hence, activating MMR pathways in TMZ-resistant T98G cells, but not in TMZ-sensitive U87 GBM cells. The HDAC6 inhibitors A452, and ACY-1215 also decreased the expression of MGMT (DNA repair protein) in TMZ-resistant GBM cells [99], thereby highlighting that HDAC6 inhibitors can overcome resistance and hence synergize with DNA damaging agents [95]. Tubacin enhanced cell death mediated by the topoisomerase II inhibitors etoposide and doxorubicin in MCF 7 and LnCaP cells [211]. Also, HDAC6 inhibitor ACY 1215 enhanced the cell death mediated by oxaliplatin in colorectal cancer cells [212]. Ozaki et al. reported that tubacin, when used in combination with ADR, enhances ADR-mediated p53 target gene expression upregulation [93]. Combination treatment involving ACY241 with the chemotherapy drug oxaliplatin led to significantly enhanced tumor-associated T cell effects or functionality in lung cancer-bearing mice and patient-derived tumors, suggesting a better therapeutic intervention in NSCLC [213]. ACY1215 when used alone was found to suppress the proliferation and induce the apoptosis of gallbladder cancer cells. In addition to that, ACY1215 also increased the chemotherapy effect of gemcitabine (inhibitor of DNA synthesis) and oxaliplatin [214]. HDAC6 depletion sensitized NSCLC cells to cisplatin, one of the first-line chemotherapeutic agents used to treat this disease [199]. Sun et al. showed that WT161, a histone deacetylase 6 (HDAC6) inhibitor, when used in combination with cisplatin had a synergistic inhibitory effect on retinoblastoma cells [215]. Hydroxybenzoic acid (4-HBA), a HDAC6 inhibitor, successfully reversed ADM resistance in human breast cancer cells. It also potentiated the anticancer effect of ADM in a MCF-7 breast cancer animal model with low toxicity [216]. MPT0G211 combined with doxorubicin-induced DNA damage response in human acute myeloid leukemia cells and ectopic expression of HDAC6, successively reversed apoptosis triggered by the combined treatment [209]. Won et al. found that A452 in combination with irinotecan was more potent than either drug alone in activating the apoptotic pathway in colorectal cancer as evidenced by activated caspase-3 and PARP, increased Bak and pp38 expression, decreased Bcl-xL, pERK and pAkt expression, and induced apoptosis [217]. WT161, a selective HDAC6 inhibitor, suppressed the growth and induced apoptosis of osteosarcoma cells in a dose- and time-dependent manner. Also, WT161 in combination with 5-FU showed synergistic inhibition on osteosarcoma cells both in vitro and in vivo [180]. Yang et al. reported a role of HDAC6 in regulating the radiosensitivity of glioma stem cells (GSCs). They found that HDAC6 inhibition in GSCs resulted in downregulation of the expression and activity of components of SHH signaling, glioma-associated oncogene homolog 1 (Gli1), Patched (Ptch1 and Ptch2) receptors. Inhibition of HDAC6 activity resulted in decreased proliferation and increased apoptosis of GSCs via inactivation of the SHH/Gli1 signaling pathway, and decreased DNA damage repair capacity of GSCs via degradation of checkpoint kinase (CHK) 1 caused by X-linked inhibitor of apoptosis (XIAP) downregulation, thereby resulting in increased radiosensitivity [94].
7.4 Combination studies of HDAC6 inhibition and miscellaneous inhibitors
HDAC6 inhibitors have been found to synergize with several kinase inhibitors in several cancer types. Bobrowicz et al. suggested that PI3K inhibitor when used in combination with HDAC6 inhibitor may turn out to be a better therapeutic option for patients with cutaneous T-cell lymphoma CTCL. They evaluated the effect of HDAC6 inhibitors in combination with PI3K inhibitors on two CTCL cell lines (HUT78 and HH). Combination studies revealed a synergistic anti-tumor effect on the CTCL cell lines. In combination studies on primary T-cells, CD4 + cells isolated from patients, generally apoptosis-resistant, also exhibited anti-tumor effects to some extent [218]. Imatinib (tyrosine kinase inhibitor) when used in combination with 7b (a HDAC6 inhibitor) resulted in strong synergistic caspase-dependent apoptotic cell death and drastically reduced the proportion of leukemia stem cells. Also, HDAC6i potentiated the effect of imatinib and overcame TKI resistance in chronic myeloid leukemia (CML) cells [219]. In vitro and in vivo studies conducted by Qin et al. showed that ACY-1215 synergized with imatinib in suppressing the growth of CML cells [220]. Lee et al. studied the effect of combination treatment of HDAC6 inhibitors with a Bruton's tyrosine kinase (BTK) inhibitor ibrutinib in NHL (Non-Hodgkin lymphoma) cell lines. The combination treatment resulted in synergistic cell growth inhibition, decreased cell viability and a significant increase in apoptosis. Ibrutinib, an irreversible selective inhibitor of BTK, induced cellular toxicity and resulted in inhibition of growth in NHL cell lines. Combination treatment with HDAC6 selective inhibitors enhanced ibrutinib-induced cellular toxicity, resulting in significant synergistic growth inhibition, induction of apoptosis and increased DNA damage in NHL cell lines. Lee et al. also studied the molecular mechanism underlying A452-ibrutinib synergism. Combination treatment decreased ERK and Akt phosphorylation, altering the expression of its main downstream effectors (Bcl-2, p27 and cyclin D1). Combination treatment of HDAC6-selective inhibitors and ibrutinib leads to suppression of cytoprotective Akt, ERK and c-Myc. Combination treatment works via regulating the MAPK and Akt signaling pathways and anti-apoptotic factors including Bcl-2 and c-Myc. This study also demonstrated that combination treatment with A452 shows a higher anti-lymphoma activity compared to ACY-1215 [221]. Corno et al. investigated the interaction between ricolinostat and the MEK-inhibitor selumetinib (AZD6244) in prostate cancer models. Using cell lines that exhibited differential activation of survival pathways (PC3, DU145, 22Rv1), a synergistic interaction was observed in all cell models. The drug combination was particularly effective in 22Rv1 cells. RNAi-mediated HDAC6 knockdown in selumetinib-treated 22Rv1 cells resulted in increased apoptosis [210]. Park et al. evaluated the anticancer effect of ACY-241, a HDAC6-selective inhibitor, on erlotinib-resistant pancreatic cancer cells that overexpress HDAC6. ACY-241 treatment alone resulted in a significant reduction in the viability of erlotinib-resistant pancreatic cells, BxPC3-ER and HPAC-ER. Also, combined treatment with ACY-241 and erlotinib induced synergistic cell death by reducing Akt–mTOR activity and increasing phospho-AMPK signaling [222]. ACY-1215 has been found to sensitize BRAF-mutant A375 cells to vemurafenib-induced proliferation inhibition and apoptosis induction [223].
Some reports also suggest that HDAC 6 inhibitors synergize with COX inhibitors. Zhang et al. studied the effect of combination treatment of a HDAC6 inhibitor, Tubastatin, and a cyclooxygenase– 2 (COX-2) inhibitor, celecoxib, in CAL 27 and SACC-83 cells. The combination treatment resulted in synergistic antitumor effects [224]. Celecoxib was found to enhance the Tubastatin-induced anti-cancer effect via activation of the PTEN/Akt signaling pathway [224]. Celecoxib and Tubastatin alone activated PTEN and inactivated Akt and combination treatment with both inhibitors synergistically activated PTEN and inactivated Akt [162]. The synergistic activation of the PTEN/Akt signaling pathway could be reproduced in PTEN-deficient U-87 MG cells that had been stably transfected with wild-type PTEN, but not in PTEN-deficient U-87 MG cells that had been stably transfected with mutant PTEN-K163R, reinforcing that the celecoxib and Tubastatin combination works synergistically via activation of the PTEN/Akt signaling pathway [224].
HDAC 6 inhibitors have also been found to enhance the anticancer activity of other HDAC6 inhibitors and pan-HDAC inhibitors. Aceroside VIII selectively inhibited HDAC6 catalytic activity, whereas a combined treatment of aceroside VIII with A452 (HDAC6 inhibitor), led to a synergistic increase in levels of acetylated α-tubulin. Aceroside VIII was found to synergistically enhance apoptosis and growth inhibition by A452. Additional findings point to a mechanism via which HDAC6-selective inhibitors may enhance the efficacy of other histone deacetylase 6 inhibitors in HT29 colon cancer cells [175]. A452 in combination with SAHA (pan-HDAC inhibitor) was more potent than either drug alone in activating the apoptotic pathway as indicated by activated caspase-3 and PARP, increased Bak and pp38 expression, decreased Bcl-xL, pERK and pAkt expression and induced apoptosis [217].
Vekaria et al. reported the effects of combined inhibition of p97 and HDAC6 in mantle cell lymphoma (MCL) cells. Combined inhibition of p97 and HDAC6 with a p97 inhibitor, CB-5083, and a HDAC6 inhibitor, ACY-1215, resulted in synergistic cell death by inducing ER stress, depleting CDK4, CyclinD1, BRCA1 and ATR. Additionally, synergistic apoptotic activity was observed via dysregulation of proteostasis and impaired DNA repair. Co-treatment with CB-5083 and ACY-1215 resulted in reduced tumor volumes and, hence, improved survival in Z138C and Jeko-1 xenografted NSG mice. Thus, combined inhibition of p97 and HDAC6 abrogated the resolution of proteotoxic stress and impaired DNA repair in MCL cells [221]. Cosenza et al. suggested that HDAC6 inhibitor ricolinostat in combination with bendamustine may be a novel combination with a potential for use as an antitumor regimen in lymphoma. They investigated the anticancer effects of HDAC6 inhibitor ricolinostat alone and in combination with bendamustine in lymphoma cell lines {WSU-NHL, RL (follicular lymphoma, FL), Granta-519, Jeko-1 (mantle cell lymphoma, MCL), Hut-78 (cutaneous T cell lymphoma, CTCL), and Karpas-299 (anaplastic large cell lymphoma, ALCL)}. Combination treatment with ricolinostat and bendamustine synergistically enhanced the anti-tumor activity by decreasing cell viability via downregulating the Akt pathway and increased tubulin acetylation resulting in microtubule stabilization. Combination treatment reduced the proportion of cells in the G0/G1 and S phases and increased the “sub-G0/G1” peak, modulated Bcl-2 protein family members and activated caspase-3 causing PARP degradation. Combination treatment showed enhanced apoptosis via ROS generation, resulting in higher endoplasmic reticulum (ER) and unfolded protein response (UPR) stress [225]. Carew et al. proposed HDAC6 upregulation as a major underlying mechanism in drug resistance to bromodomain and extra terminal (BET) inhibitors. They identified HDAC6 inhibition as a novel approach to augment BET inhibitor therapy in MM. Their study showed that BET inhibitor treatment synergized with HDAC6 inhibition to suppress tumor growth via c-Myc and Bcl-2 suppression and showed synergistic anti-tumor activity in MM [226]. Studies conducted in a MM in vivo murine xenograft model revealed that pomalidomide when used in combination with ACY-241 resulted in enhanced tumor growth inhibition via increased suppression of pro-survival factors such as survivin, Myc and IRF4 [227]. Miyake et al. demonstrated that co-administration of RCS with Adv (Adavosertib) suppressed cyclin-dependent kinase 1(Tyr15) phosphorylation and increased the expression of γ-H2A.X (a marker of DNA double-strand breaks) in CAL27 cells. Also RCS enhanced Adv-induced premature mitotic entry and cell death induction in the mitotic phase. Co-administration of RCS with Adv in HNSCC cells resulted in the suppression of Chk1 activity, thereby leading to synergistically enhanced apoptosis via mitotic catastrophe in a p53-dependent manner [228]. Adeegbe et al. demonstrated that the selective HDAC6 inhibitor ricolinostat synergized with the bromodomain inhibitor JQ1 and enhanced suppression of tumor growth of non-small cell lung cancer (NSCLC). Combination treatment enhanced immune-dependent suppression of tumor growth via T cell-mediated anti-tumor response. The tumor killing potential of tumor infiltrating T cells was enhanced significantly upon combination treatment in mouse models resulting in prolonged survival [229]. Cho et al. reported that a combination of ACY-241 and JQ1 resulted in synergistic growth inhibition, viability reduction and apoptosis induction in HNSCC cells through inactivation of Akt and NF-κB signaling in human papillomavirus (HPV)-positive and HPV-negative HNSCC cells. They also demonstrated that combined treatment of ACY-241 and JQ1 synergistically suppressed TNF-α-induced migration and invasion via dysregulating matrix metalloproteinase (MMP)-2, MMP-9 and MT1-MMP [230]. Also, combined treatment of human and mouse SCLC cell line–derived xenograft tumors with ricolinostat (HDAC6 inhibitor) and JQ1 resulted in significant inhibition of tumor growth [231].
Epigenetic modulator DNMTi (5-Azacytidine) when used in combination with HDAC6i (Nexturastat A) resulted in an amplified type I interferon response, leading to increased cytokine and chemokine expression and higher expression of the MHC I antigen presentation complex in human and mouse ovarian cancer cell lines. Treating mice bearing ID8 Trp53−/− ovarian cancer with HDAC6i/DNMTi led to an increase in tumor-killing cells and a reversal of the immunosuppressive tumor microenvironment with a decrease in MDSCs and PD-1hiCD4 T cells, corresponding with an increase in survival [232]. Fukumoto et al. showed that HDAC6 inhibition via ACY1215 synergized with anti-PD-L1 immune checkpoint blockade in ARID1A-inactivated ovarian cancer. Reduced tumor burden and improved survival were observed inARID1Aflox/flox/PIK3CAH1047ROCCC mice treated with ACY1215 and anti-PD-L1 immune checkpoint blockade as a result of activation and increased presence of IFNγ-positive CD8 T cells. Thus combined treatment limited tumor progression in a cytotoxic T-cell-dependent manner, as depletion of CD8+T cells abrogated this antitumor effect [233]. In vivo studies revealed that a combination of Suprastat and anti-PD1 immunotherapy enhanced the antitumor immune response, mediated by a decrease of pro-tumoral M2 macrophages and an increased infiltration of antitumor CD8+ effector and memory T-cells [177]. Kim et al. proposed that immunomodulatory drugs (IMiDs) when used in combination with A452, a HDAC6 inhibitor, could overcome resistance to IMiDs, the principal MM therapeutic drugs. Their findings highlighted that A452 alone reduced the viability of IMiD-resistant cells and synergistically reduced viability and induced apoptosis when combined with IMiDs. To determine if A452 overcomes IMiD resistance, they checked the change in the protein level of IMiDs direct and indirect targets. They found that the combination of A452 and IMiDs slightly increased CRBN and decreased Aiolos and Ikaros, the targets of CRBN. Moreover, A452 decreased c-Myc and IRF-4 when combined with IMiDs. These data suggest that A452 helps to overcome the resistance of IMiDs [234].
8 Conclusions
The distinct structural and functional features of HDAC6 make it unique among all the known conventional HDAC enzymes. Unlike other HDACs, HDAC6 is not limited to histone acetylation and deacetylation, but also interacts with several non-histone substrates, such as cortactin, α-tubulin and heat shock protein 90 (HSP90),which are associated with cell proliferation, mitosis, metastasis and invasion in tumors. Any alteration in HDAC6 expression level results in oncogenic cell transformation and tumor cell proliferation, mitosis, metastasis and invasion. There is ample evidence for HDACi’s as a promising class of antitumor drugs. Pan-HDACs being more specific result in a lower toxicity. Over the past few years, much attention has been given to the development of more selective HDACi’s which are more tolerable than pan-HDACi’s. HDAC6 is a promising target due to its distinct structural and functional features and its interaction with non-histone substrates, which are closely associated with oncogenic transformation. Also, HDAC6 is a regulator of many pathways that are frequently found to be deregulated in cancer. HDAC6 inhibitors being more specific, result in a lower toxicity. Various HDAC6-selective inhibitors have been studied so far. These studies show combinations of HDAC6 inhibitors and other anticancer drugs have synergistic anticancer effects. Thus, combination studies may yield very attractive therapeutic strategies. The present review discussed the structural features of HDAC6, its interaction with non-histone substrates and associated functions. We further discussed the role of HDAC6 in normal cell physiology and its role in numerous physiological pathways deregulated in cancer. We also discussed the therapeutic potential of selective HDAC6 inhibitors (HDAC6i) as emerging drugs in cancer treatment.
Data availability
Not applicable.
Abbreviations
- αTAT:
-
α Tubulin acetyltransferases
- ADR:
-
Adriamycin
- Adv:
-
Adavosertib
- ALCL:
-
Anaplastic large cell lymphoma
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- AR:
-
Androgen receptor
- BTK:
-
Bruton's tyrosine kinase
- BTZ:
-
Bortezomib
- CBP:
-
CREB-binding protein
- CC:
-
Cholangiocarcinoma
- CDK1:
-
Cyclin-dependent kinase 1
- CDKi:
-
Cyclin-dependent kinase inhibitor
- CRD-1:
-
Cell cycle regulatory domain 1
- CHK:
-
Checkpoint kinase
- CLL:
-
Chronic Lymphoid Leukemia
- COX-2:
-
Cyclooxygenase– 2
- CYLD:
-
Cylindromatosis
- DHT:
-
Dihydrotestosterone
- DLBCL:
-
Diffuse large B cell lymphoma
- ECs:
-
Endothelial cells
- EGF:
-
Epidermal growth factor
- EGFR:
-
Epidermal growth factor receptor
- ER:
-
Estrogen Receptor
- ERKs:
-
Extracellular signal-regulated kinases
- FOXP3:
-
Forkhead boxp3
- GC:
-
Gynecological cancer
- Gli1:
-
Glioma-associated oncogene homolog 1
- GPCRs:
-
G protein coupled receptors
- GR:
-
Glucocorticoid receptor
- GRK2:
-
G protein-coupled receptor kinase
- GRP:
-
Glucose-regulated protein
- GSCs:
-
Glioma stem cells
- GSK3b:
-
Glycogen synthase kinase 3
- G3BP1:
-
GTPase activating protein SH3 domain binding protein 1
- HATs:
-
Histone acetyltransferases
- HCC:
-
Hepatocellular carcinoma
- HDAC6i:
-
HDAC6 inhibitors
- HDACs:
-
Histone deacetylases
- HEK:
-
Human embryonic kidney cells
- HIF-1:
-
Hypoxia-inducible factor
- HO-1:
-
Hemeoxygenase 1
- HSF1:
-
Heat-shock factor 1
- Hsp90:
-
Heat shock protein 90
- IBC:
-
Inflammatory breast cancer
- IIp:
-
Invasion inhibitory protein
- IMiDs:
-
Immunomodulatory drugs
- MAPK:
-
Microtubule associated protein kinase
- MBC:
-
Metastatic breast cancer
- MCL:
-
Mantle cell lymphoma
- MEFs:
-
Mouse embryonic fibroblast
- MLH1:
-
MutL homolog 1
- MM:
-
Multiple myeloma
- MMP9:
-
Matrix metallopeptidase 9
- MMR:
-
Mismatch repair
- MPT0G413:
-
N-hydroxy-4-((5-(4-methoxybenzoyl)- 1H-indol-1-yl)methyl)benzamide
- MSCs:
-
Mesenchymal stem cells
- MT:
-
Microtubule
- MTOC:
-
Microtubule-organizing center
- Mtor:
-
Mammalian target of rapamycin
- NAD:
-
Nicotinamide adenine dinucleotide
- NES:
-
Nuclear export signal
- NHL:
-
Non-Hodgkin lymphoma
- NLS:
-
Nuclear localization signal
- Nrf-2:
-
Nuclear factor erythroid 2–related factor 2
- NSCLC:
-
Non-Small Cell Lung Cancer
- PCAF:
-
p300/CBP associated factor
- PDGF:
-
Platelet-derived growth factor
- PI3K:
-
Phosphoinositide 3-kinases
- Pkc:
-
Protein kinase C
- PKCz:
-
Protein kinase C isoform z
- PrEC:
-
Prostate epithelial cells
- PTEN:
-
Phosphatase and tensin homolog
- PTPN1:
-
Tyrosine-protein phosphatase non-receptor type 1
- RMS:
-
Rhabdomyosarcoma
- ROCK:
-
Rho-associated coiled-coil kinase
- ROS:
-
Reactive oxygen species
- RUNX2:
-
Runt-related transcription factor 2
- SAHA:
-
Suberoylanilidehydroxamic acid
- SIRT:
-
Sirtuins
- TMZ:
-
Temozolomide
- TPPP/p25:
-
Tubulin polymerization-promoting protein/p25
- UPR:
-
Unfolded protein response
- VEGF:
-
Vascular endothelial growth factor
- XIAP:
-
X-linked inhibitor of apoptosis
References
A. Drazic, L.M. Myklebust, R. Ree, T. Arnesen, The world of protein acetylation. Biochem Biophys Acta BBA - Proteins Proteomics 1864, 1372–1401 (2016)
C. Choudhary, C. Kumar, F. Gnad, M.L. Nielsen, M. Rehman, T.C. Walther, J.V. Olsen, M. Mann, Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 (2009)
Y.C. Wang, S.E. Peterson, J.F. Loring, Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 24, 143–160 (2014)
C. Seidel, M. Schnekenburger, M. Dicato, M. Diederich, Antiproliferative and proapoptotic activities of 4-hydroxybenzoic acid-based inhibitors of histone deacetylases. Cancer Lett. 343, 134–146 (2014)
S. Spange, T. Wagner, T. Heinzel, O.H. Krämer, Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 41, 185–198 (2009)
A.J. de Ruijter, A.H. van Gennip, H.N. Caron, S. Kemp, A.B. van Kuilenburg, Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370, 737–749 (2003)
E.A. Olsen, Y.H. Kim, T.M. Kuzel, T.R. Pacheco, F.M. Foss, S. Parker, S.R. Frankel, C. Chen, J.L. Ricker, J.M. Arduino, M. Duvic, Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 25, 3109–3115 (2007)
R.L. Piekarz, R. Frye, M. Turner, J.J. Wright, S.L. Allen, M.H. Kirschbaum, J. Zain, H.M. Prince, J.P. Leonard, L.J. Geskin, C. Reeder, D. Joske, W.D. Figg, E.R. Gardner, S.M. Steinberg, E.S. Jaffe, M. Stetler-Stevenson, S. Lade, A.T. Fojo, S.E. Bates, Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 27, 5410–5117 (2009)
P.A. Marks, R. Breslow, Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 25, 84–90 (2007)
C. Campas-Moya, Romidepsin for the treatment of cutaneous T-cell lymphoma. Drugs Today Barc. 45, 787–795 (2009)
P.G. Richardson, R.L. Schlossman, M. Alsina, D.M. Weber, S.E. Coutre, C. Gasparetto, S. Mukhopadhyay, M.S. Ondovik, M. Khan, C.S. Paley, S. Lonial, PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib refractory myeloma. Blood 122, 2331–2337 (2013)
C. Seidel, M. Schnekenburger, M. Dicato, M. Diesderich, Histone deacetylase 6 in health and disease. Epigenomics 7, 103–108 (2015)
S.N. Batchu, A.S. Brijmohan, A. Advani, The therapeutic hope for HDAC6 inhibitors in malignancy and chronic disease. Clin. Sci. 130, 987–1003 (2016)
Y. Liu, L. Peng, E. Seto, S. Huang, Y. Qiu, Modulation of histone deacetylase 6 (HDAC6) nuclear import and tubulin deacetylase activity through acetylation. J. Biol. Chem. 287, 29168–29174 (2012)
H. Zou, Y. Wu, M. Navre, B.C. Sang, Characterization of the two catalytic domains in histone deacetylase 6. Biochem. Biophys. Res. Commun. 341, 45–50 (2006)
C.M. Grozinger, C.A. Hassig, S.L. Schreiber, Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. U.S.A. 96, 4868–4873 (1999)
Y. Zhang, B. Gilquin, S. Khochbin, P. Matthias, Two catalytic domains are required for protein deacetylation. J. Biol. Chem. 281, 2401–2404 (2006)
Z. Kutil, L. Skultetyova, D. Rauh, M. Meleshin, I. Snajdr, Z. Novakova, J. Mikesova, J. Pavlicek, M. Hadzima, P. Baranova, B. Havlinova, P. Majer, M. Schutkowski, C. Barinka, The unraveling of substrate specificity of histone deacetylase 6 domains using acetylome peptide microarrays and peptide libraries. FASEB J. 33, 4035–4045 (2019)
J.-Y. Wu, S. Xiang, M. Zhang, B. Fang, H. Huang, O.K. Kwon, Y. Zhao, Z. Yang, W. Bai, G. Bepler, X.M. Zhang, Histone deacetylase 6 (HDAC6) deacetylates extracellular signal-regulated kinase 1 (ERK1) and thereby stimulates ERK1 activity. J. Biol. Chem. 293, 1976–1993 (2018)
K.A. Williams, M. Zhang, S. Xiang, C. Hu, J.Y. Wu, S. Zhang, M. Ryan, A.D. Cox, C.J. Der, B. Fang, J. Koomen, E. Haura, G. Bepler, S.V. Nicosia, P. Matthias, C. Wang, W. Bai, X. Zhang, Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J. Biol. Chem. 288, 33156–33170 (2013)
M.L. Selenica, L. Benner, S.B. Housley, B. Manchec, D.C. Lee, K.R. Nash, J. Kalin, J.A. Bergman, A. Kozikowski, M.N. Gordon, D. Morgan, Histone deacetylase 6 inhibition improves memory and reduces total tau levels in a mouse model of tau deposition. Alzheimers Res. Ther. 6, 12 (2014)
L. Zhang, S. Sheng, C. Qin, The role of HDAC6 in Alzheimer’s disease. J. Alzheimers Dis. 33, 283–295 (2013)
Y.S. Gao, C.C. Hubbert, T.P. Yao, The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J. Biol. Chem. 285, 11219–11226 (2010)
W. Liu, L.X. Fan, X. Zhou, W.E. Sweeney, E.D. Avner, X. Li, HDAC6 regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation in renal epithelial cells. PLoS ONE 7, e49418 (2012)
Y. L. Deribe, P. Wild, A. Chandrashaker, J. Curak, M. H. H. Schmidt, Y. Kalaidzidis, N. Milutinovic, I. Kratchmarova, L. Buerkle, M. J. Fetchko, P. Schmidt, S. Kittanakom, K. R. Brown, I. Jurisica, B. Blagoev, M. Zerial, I. Stagljar, and I. Dikic, Regulation of epidermal growth factor receptor trafficking by lysine deacetylase HDAC6. Sci. Signal 2, ra84 (2009)
Y. Li, X. Zhang, R.D. Polakiewicz, T.P. Yao, M.J. Comb, HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J. Biol. Chem. 283, 12686–12690 (2008)
J. Gu, D. Wang, J. Zhang, Y. Zhu, Y. Li, H. Chen, M. Shi, X. Wang, B. Shen, X. Deng, Q. Zhan, G. Wei, C. Peng, GFRα2 prompts cell growth and chemoresistance through down-regulating tumor suppressor gene PTEN via Mir-17-5p in pancreatic cancer. Cancer Lett. 380, 434–431 (2016)
Z. Meng, L.F. Jia, Y.H. Gan, PTEN activation through K163 acetylation by inhibiting HDAC6 contributes to tumour inhibition. Oncogene 35, 2333–2344 (2016)
M. Tesio, A. Trinquand, E. Macintyre, V. Asnafi, Oncogenic PTEN functions and models in T-cell malignancies. Oncogene 35, 3887–3896 (2016)
Y. Zheng, X. Yang, C. Wang, S. Zhang, Z. Wang, M. Li, Y. Wang, X. Wang, HDAC6, modulated by miR-206, promotes endometrial cancer progression through the PTEN/AKT/mTOR pathway. Sci. Rep. 10, 3576 (2020)
S.A. Wickström, K.C. Masoumi, S. Khochbin, R. Fässler, R. Massoumi, CYLD negatively regulates cell-cycle progression by inactivating HDAC6 and increasing the levels of acetylated tubulin. EMBO J. 29, 131–144 (2010)
G.I. Aldana-Masangkay, K.M. Sakamoto, The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2011, 875824 (2011)
Y. Ishikawa, K. Tsunoda, M. Shibazaki, K. Takahashi, T. Akasaka, T. Masuda, C. Maesawa, Downregulation of cylindromatosis gene, CYLD, confers a growth advantage on malignant melanoma cells while negatively regulating their migration activity. Int. J. Oncol. 41, 53–60 (2012)
S.J. Haggarty, K.M. Koeller, J.C. Wong, C.M. Grozinger, S.L. Schreiber, Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. U.S.A. 100, 4389–4394 (2003)
R. Massoumi, K. Chmielarska, K. Hennecke, A. Pfeifer, R. Fässler, Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell 125, 665–677 (2006)
M.J. Chuang, S.T. Wu, S.H. Tang, X.M. Lai, H.C. Lai, K.H. Hsu, K.H. Sun, G.H. Sun, S.Y. Chang, D.S. Yu, P.W. Hsiao, S.M. Huang, T.L. Cha, The HDAC inhibitor LBH589 induces ERK-dependent prometaphase arrest in prostate cancer via HDAC6 inactivation and down-regulation. PLoS ONE 8, e73401 (2013)
C. Hubbert, A. Guardiola, R. Shao, Y. Kawaguchi, A. Ito, A. Nixon, M. Yoshida, X.F. Wang, T.P. Yao, HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458 (2002)
A. Matsuyama, T. Shimazu, Y. Sumida, A. Saito, Y. Yoshimatsu, D. Seigneurin-Berny, H. Osada, Y. Komatsu, N. Nishino, S. Khochbin, S. Horinouchi, M. Yoshida, In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 21, 6820–6831 (2002)
Y. Zhang, N. Li, C. Caron, G. Matthias, D. Hess, S. Khochbin, P. Matthias, HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 22, 1168–1179 (2003)
M. Conacci-Sorrell, C. Ngouenet, R.N. Eisenman, Myc-nick: a cytoplasmic cleavage product of Myc that promotes alpha-tubulin acetylation and cell differentiation. Cell 142, 480–493 (2010)
C. Creppe, L. Malinouskaya, M.L. Volvert, M. Gillard, P. Close, O. Malaise, S. Laguesse, I. Cornez, S. Rahmouni, S. Ormenese, S. Belachew, B. Malgrange, J.P. Chapelle, U. Siebenlist, G. Moonen, A. Chariot, L. Nguyen, Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha-tubulin. Cell 136, 551–564 (2009)
S.W. L’Hernault, J.L. Rosenbaum, Chlamydomonas alpha-tubulin is posttranslationally modified in the flagella during flagellar assembly. J. Cell Biol. 97, 258–263 (1983)
N. Ohkawa, S. Sugisaki, E. Tokunaga, K. Fujitani, T. Hayasaka, M. Setou, K. Inokuchi, N-acetyltransferase ARD1-NAT1 regulates neuronal dendritic development. Genes Cells 13, 1171–1183 (2008)
X. Zhang, Z. Yuan, Y. Zhang, S. Yong, A. Salas-Burgos, J. Koomen, N. Olashaw, J.T. Parsons, X.J. Yang, S.R. Dent, T.P. Yao, W.S. Lane, E. Seto, HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell 27, 197–213 (2007)
H. Wu, J.T. Parsons, Cortactin, an 80/85-kilodalton pp60src substrate, is a filamentous actin-binding protein enriched in the cell cortex. J. Cell Biol. 120, 1417–1426 (1993)
N.S. Bryce, E.S. Clark, J.L. Leysath, J.D. Currie, D.J. Webb, A.M. Weaver, Cortactin promotes cell motility by enhancing lamellipodial persistence. Curr. Biol. 15, 1276–1285 (2005)
D. Kaluza, J. Kroll, S. Gesierich, T.P. Yao, R.A. Boon, E. Hergenreider, M. Tjwa, L. Rössig, E. Seto, H.G. Augustin, A.M. Zeiher, S. Dimmeler, C. Urbich, Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J. 30, 4142–4156 (2011)
V. Lafarga, I. Aymerich, O. Tapia, F. Mayor, P. Penela, A novel GRK2/HDAC6 interaction modulates cell spreading and motility. EMBO J. 31, 856–869 (2012)
P. Penela, V. Lafarga, O. Tapia, V. Rivas, L. Nogués, E. Lucas, R. Vila-Bedmar, C. Murga, and F. Mayor, Roles of GRK2 in cell signaling beyond GPCR desensitization: GRK2-HDAC6 interaction modulates cell spreading and motility. Sci. Signal 5, pt3 (2012)
H.H. Chuang, M.S. Huang, P.H. Wang, Y.P. Liu, M. Hsiao, C.J. Yang, Pin1 Is Involved in HDAC6-mediated Cancer Cell Motility. Int. J. Med. Sci. 15, 1573–1581 (2018)
H.H. Chuang, J.F. Hsu, H.L. Chang, P.H. Wang, P.J. Wei, D.W. Wu, M.S. Huang, M. Hsiao, C.J. Yang, Pin1 coordinates HDAC6 upregulation with cell migration in lung cancer cells. Int. J. Med. Sci. 17, 2635–2643 (2020)
J. Liu, W. Luan, Y. Zhang, J. Gu, Y. Shi, Y. Yang, Z. Feng, F. Qi, HDAC6 interacts with PTPN1 to enhance melanoma cells progression. Biochem. Biophys. Res. Commun. 495, 2630–2636 (2018)
C. Featherstone, S.P. Jackson, Ku, a DNA repair protein with multiple cellular functions? Mutat. Res. 434, 3–15 (1999)
H.Y. Cohen, S. Lavu, K.J. Bitterman, B. Hekking, T.A. Imahiyerobo, C. Miller, R. Frye, H. Ploegh, B.M. Kessler, D.A. Sinclair, Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol. Cell 13, 627–638 (2004)
C. Subramanian, J.A. Jarzembowski, A.W. Opipari, V.P. Castle, R.P. Kwok, HDAC6 deacetylates Ku70 and regulates Ku70-Bax binding in neuroblastoma. Neoplasia 13, 726–734 (2011)
E. Kerr, C. Holohan, K.M. McLaughlin, J. Majkut, S. Dolan, K. Redmond, J. Riley, K. McLaughlin, I. Stasik, M. Crudden, S. Van Schaeybroeck, C. Fenning, R. O’Connor, P. Kiely, M. Sgobba, D. Haigh, P.G. Johnston, D.B. Longley, Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death Differ. 19, 1317–1327 (2012)
H. Wang, M.P. Holloway, L. Ma, Z.A. Cooper, M. Riolo, A. Samkari, K.S. Elenitoba-Johnson, Y.E. Chin, R.A. Altura, Acetylation directs survivin nuclear localization to repress STAT3 oncogenic activity. J. Biol. Chem. 285, 36129–36137 (2010)
M.T. Riolo, Z.A. Cooper, M.P. Holloway, Y. Cheng, C. Bianchi, E. Yakirevich, L. Ma, Y.E. Chin, R.A. Altura, Histone deacetylase 6 (HDAC6) deacetylates survivin for its nuclear export in breast cancer. J. Biol. Chem. 287, 10885–10893 (2012)
C. Boyault, Y. Zhang, S. Fritah, C. Caron, B. Gilquin, S. H. Kwon, C. Garrido, T. P. Yao, C. Vourc’h, P. Matthias, and S. Khochbin, HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 21, 2172–2181 (2007)
Y. Kawaguchi, J.J. Kovacs, A. McLaurin, J.M. Vance, A. Ito, T.P. Yao, The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738 (2003)
C. Boyault, B. Gilquin, Y. Zhang, V. Rybin, E. Garman, W. Meyer-Klaucke, P. Matthias, C.W. Müller, S. Khochbin, HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 25, 3357–3366 (2006)
A. Iwata, B.E. Riley, J.A. Johnston, R.R. Kopito, HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 280, 40282–40292 (2005)
J.A. Olzmann, L.S. Chin, Parkin-mediated K63-linked polyubiquitination: a signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy 4, 85–87 (2008)
K.P. Liu, D. Zhou, D.Y. Ouyang, L.H. Xu, Y. Wang, L.X. Wang, H. Pan, X.H. He, LC3B-II deacetylation by histone deacetylase 6 is involved in serum-starvation-induced autophagic degradation. Biochem. Biophys. Res. Commun. 441, 970–975 (2013)
S. Pankiv, T.H. Clausen, T. Lamark, A. Brech, J.A. Bruun, H. Outzen, A. Øvervatn, G. Bjørkøy, T. Johansen, p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145 (2007)
C. Fusco, L. Micale, M. Egorov, M. Monti, E.V. D’Addetta, B. Augello, F. Cozzolino, A. Calcagnì, A. Fontana, R.S. Polishchuk, G. Didelot, A. Reymond, P. Pucci, G. Merla, The E3-ubiquitin ligase TRIM50 interacts with HDAC6 and p62, and promotes the sequestration and clearance of ubiquitinated proteins into the aggresome. PLoS ONE 7, e40440 (2012)
J.Y. Lee, H. Koga, Y. Kawaguchi, W. Tang, E. Wong, Y.S. Gao, U.B. Pandey, S. Kaushik, E. Tresse, J. Lu, J.P. Taylor, A.M. Cuervo, T.P. Yao, HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 29, 969–980 (2010)
W.J. Liu, L. Ye, W.F. Huang, L.J. Guo, Z.G. Xu, H.L. Wu, C. Yang, H.F. Liu, p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett. 21, 29 (2016)
G. Bjørkøy, T. Lamark, A. Brech, H. Outzen, M. Perander, A. Overvatn, H. Stenmark, T. Johansen, p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614 (2005)
M.L. Seibenhener, J.R. Babu, T. Geetha, H.C. Wong, N.R. Krishna, M.W. Wooten, Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell Biol. 24, 8055–8068 (2004)
C. Dai, L. Whitesell, A.B. Rogers, S. Lindquist, Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130, 1005–1018 (2007)
P. Bali, M. Pranpat, J. Bradner, M. Balasis, W. Fiskus, F. Guo, K. Rocha, S. Kumaraswamy, S. Boyapalle, P. Atadja, E. Seto, K. Bhalla, Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 280, 26729–26734 (2005)
B.T. Scroggins, K. Robzyk, D. Wang, M.G. Marcu, S. Tsutsumi, K. Beebe, R.J. Cotter, S. Felts, D. Toft, L. Karnitz, N. Rosen, L. Neckers, An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol. Cell 25, 151–159 (2007)
J.J. Kovacs, P.J. Murphy, S. Gaillard, X. Zhao, J.T. Wu, C.V. Nicchitta, M. Yoshida, D.O. Toft, W.B. Pratt, T.P. Yao, HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 18, 601–607 (2005)
P.J. Murphy, Y. Morishima, J.J. Kovacs, T.P. Yao, W.B. Pratt, Regulation of the dynamics of hsp90 action on the glucocorticoid receptor by acetylation/deacetylation of the chaperone. J. Biol. Chem. 280, 33792–33799 (2005)
C. Caron, C. Boyault, S. Khochbin, Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. BioEssays 27, 408–415 (2005)
D.R. Hurst, A. Mehta, B.P. Moore, P.A. Phadke, W.J. Meehan, M.A. Accavitti, L.A. Shevde, J.E. Hopper, Y. Xie, D.R. Welch, R.S. Samant, Breast cancer metastasis suppressor 1 (BRMS1) is stabilized by the Hsp90 chaperone. Biochem. Biophys. Res. Commun. 348, 1429–1435 (2006)
X. Kong, Z. Lin, D. Liang, D. Fath, N. Sang, J. Caro, Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol. Cell Biol. 26, 2019–2028 (2006)
J.J. Westendorf, S.K. Zaidi, J.E. Cascino, R. Kahler, A.J. van Wijnen, J.B. Lian, M. Yoshida, G.S. Stein, X. Li, Runx2 (Cbfa1, AML-3) interacts with histone deacetylase 6 and represses the p21(CIP1/WAF1) promoter. Mol. Cell Biol. 22, 7982–7992 (2002)
Z. Yu, W. Zhang, B.C. Kone, Histone deacetylases augment cytokine induction of the iNOS gene. J. Am. Soc. Nephrol. 13, 2009–2017 (2002)
D. Girdwood, D. Bumpass, O.A. Vaughan, A. Thain, L.A. Anderson, A.W. Snowden, E. Garcia-Wilson, N.D. Perkins, R.T. Hay, P300 transcriptional repression is mediated by SUMO modification. Mol. Cell 11, 1043 (2003)
L. Ling, P.E. Lobie, RhoA/ROCK activation by growth hormone abrogates p300/histone deacetylase 6 repression of Stat5-mediated transcription. J. Biol. Chem. 279, 32737 (2004)
H. Ma, C. Nguyen, K.S. Lee, M. Kahn, Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene 24, 3619–3631 (2005)
Y. Han, H.M. Jeong, Y.H. Jin, Y.J. Kim, H.G. Jeong, C.Y. Yeo, K.Y. Lee, Acetylation of histone deacetylase 6 by p300 attenuates its deacetylase activity. Biochem. Biophys. Res. Commun. 383, 88–92 (2009)
I. Fernandes, Y. Bastien, T. Wai, K. Nygard, R. Lin, O. Cormier, H.S. Lee, F. Eng, N.R. Bertos, N. Pelletier, S. Mader, V.K. Han, X.J. Yang, J.H. White, Ligand-dependent nuclear receptor corepressor LCoR functions by histone deacetylase-dependent and -independent mechanisms. Mol. Cell 11, 139–150 (2003)
J.M. Amann, J. Nip, D.K. Strom, B. Lutterbach, H. Harada, N. Lenny, J.R. Downing, S. Meyers, S.W. Hiebert, ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol. Cell Biol. 21, 6470–6483 (2001)
A. Palijan, I. Fernandes, Y. Bastien, L. Tang, M. Verway, M. Kourelis, L.E. Tavera-Mendoza, Z. Li, V. Bourdeau, S. Mader, X.J. Yang, J.H. White, Function of histone deacetylase 6 as a cofactor of nuclear receptor coregulator LCoR. J. Biol. Chem. 284, 30264–30274 (2009)
R. Winkler, V. Benz, M. Clemenz, M. Bloch, A. Foryst-Ludwig, S. Wardat, N. Witte, M. Trappiel, P. Namsolleck, K. Mai, J. Spranger, G. Matthias, T. Roloff, O. Truee, K. Kappert, M. Schupp, P. Matthias, U. Kintscher, Histone deacetylase 6 (HDAC6) is an essential modifier of glucocorticoid-induced hepatic gluconeogenesis. Diabetes 61, 513–523 (2012)
J.M. Solomon, R. Pasupuleti, L. Xu, T. McDonagh, R. Curtis, P.S. DiStefano, L.J. Huber, Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell Biol. 26, 28–38 (2006)
C. Blackburn, C. Barrett, J. Chin, K. Garcia, K. Gigstad, A. Gould, J. Gutierrez, S. Harrison, K. Hoar, C. Lynch, R.S. Rowland, C. Tsu, J. Ringeling, H. Xu, Potent histone deacetylase inhibitors derived from 4-(aminomethyl)-N-hydroxybenzamide with high selectivity for the HDAC6 isoform. J. Med. Chem. 56, 7201–7211 (2013)
S.-Y. Park, S. Phorl, S. Jung, K. Sovannarith, S. Lee, S. Noh, M. Han, R. Naskar, J.-Y. Kim, Y.-J. Choi, J.-Y. Lee, HDAC6 deficiency induces apoptosis in mesenchymal stem cells through p53 K120 acetylation. Biochem. Biophys. Res. Commun. 494, 51–56 (2017)
M. Zhang, C. Hu, N. Moses, J. Haakenson, S. Xiang, D. Quan, B. Fang, Z. Yang, W. Bai, G. Bepler, G.M. Li, X.M. Zhang, HDAC6 regulates DNA damage response via deacetylating MLH1. J. Biol. Chem. 294, 5813–5826 (2019)
T. Ozaki, D. Wu, H. Sugimoto, H. Nagase, A. Nakagawara, Runt-related transcription factor 2 (RUNX2) inhibits p53-dependent apoptosis through the collaboration with HDAC6 in response to DNA damage. Cell Death Dis. 4, e610 (2013)
W. Yang, Y. Liu, R. Gao, H. Yu, T. Sun, HDAC6 inhibition induces glioma stem cells differentiation and enhances cellular radiation sensitivity through the SHH/Gli1 signaling pathway. Cancer Lett. 415, 164–176 (2018)
G.W. Kim, D.H. Lee, S.K. Yeon, Y.H. Jeon, J. Yoo, S.W. Lee, S.H. Kwon, Temozolomide-resistant Glioblastoma Depends on HDAC6 Activity Through Regulation of DNA Mismatch Repair. Anticancer Res. 39, 6731–6741 (2019)
O.H. Krämer, S. Mahboobi, A. Sellmer, Drugging the HDAC6-HSP90 interplay in malignant cells. Trends Pharmacol. Sci. 35, 501–509 (2014)
Y. Wu, S.W. Song, J. Sun, J.M. Bruner, G.N. Fuller, W. Zhang, IIp45 inhibits cell migration through inhibition of HDAC6. J. Biol. Chem. 285, 3554–3560 (2010)
N. Tokési, A. Lehotzky, I. Horváth, B. Szabó, J. Oláh, P. Lau, J. Ovádi, Drugging the HDAC6-HSP90 interplay in malignant cells. J. Biol. Chem. 285, 17896–17906 (2010)
J. Zhou, C.C. Vos, A. Gjyrezi, M. Yoshida, F.R. Khuri, F. Tamanoi, P. Giannakakou, The protein farnesyltransferase regulates HDAC6 activity in a microtubule-dependent manner. J. Biol. Chem. 284, 9648–9655 (2009)
S. Chen, G.C. Owens, H. Makarenkova, D.B. Edelman, HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS ONE 5, e10848 (2010)
K. Leroy, Z. Yilmaz, J.P. Brion, Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 33, 43–55 (2007)
E.N. Pugacheva, S.A. Jablonski, T.R. Hartman, E.P. Henske, E.A. Golemis, HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 129, 1351–1363 (2007)
M. Watabe, T. Nakaki, Protein kinase CK2 regulates the formation and clearance of aggresomes in response to stress. J. Cell Sci. 124, 1519–1532 (2011)
J. Zhu, C.B. Coyne, S.N. Sarkar, PKC alpha regulates Sendai virus-mediated interferon induction through HDAC6 and β-catenin. EMBO J. 30, 4838–4849 (2011)
Y. Du, M.L. Seibenhener, J. Yan, J. Jiang, M.C. Wooten, aPKC phosphorylation of HDAC6 results in increased deacetylation activity. PLoS ONE 10, e0123191 (2015)
A.V. Schofield, C. Gamell, R. Suryadinata, B. Sarcevic, O. Bernard, Tubulin polymerization promoting protein 1 (Tppp1) phosphorylation by Rho-associated coiled-coil kinase (rock) and cyclin-dependent kinase 1 (Cdk1) inhibits microtubule dynamics to increase cell proliferation. J. Biol. Chem. 288, 7907–7917 (2013)
A.V. Schofield, C. Gamell, O. Bernard, Tubulin polymerization promoting protein 1 (TPPP1) increases β-catenin expression through inhibition of HDAC6 activity in U2OS osteosarcoma cells. Biochem. Biophys. Res. Commun. 436, 571–577 (2013)
N.O. Deakin, C.E. Turner, Paxillin inhibits HDAC6 to regulate microtubule acetylation, Golgi structure, and polarized migration. J. Cell Biol. 206, 395–413 (2014)
N. Kasai, A. Kadeer, M. Kajita, S. Saitoh, S. Ishikawa, T. Maruyama, Y. Fujita, The paxillin-plectin-EPLIN complex promotes apical elimination of RasV12-transformed cells by modulating HDAC6-regulated tubulin acetylation. Sci. Rep. 8, 2097 (2018)
Tala, X. Sun, J. Chen, L. Zhang, N. Liu, J. Zhou, D. Li, and M. Liu, Microtubule stabilization by Mdp3 is partially attributed to its modulation of HDAC6 in addition to its association with tubulin and microtubules. PLoS One 9, e90932 (2014)
L.M. Salemi, A.W. Almawi, K.J. Lefebvre, C. Schild-Poulter, Aggresome formation is regulated by RanBPM through an interaction with HDAC6. Biol. Open 3, 418–430 (2014)
A. P. Mansini, M. J. Lorenzo Pisarello, K. M. Thelen, M. Cruz-Reyes, E. Peixoto, S. Jin, B. N. Howard, C. E. Trussoni, G. B. Gajdos, N. F. LaRusso, M. J. Perugorria, J. M. Banales, and S. A. Gradilone, MicroRNA (miR)-433 and miR-22 dysregulations induce histone-deacetylase-6 overexpression and ciliary loss in cholangiocarcinoma. Hepatol. Baltim. Md. 68, 561–573 (2018)
T. Lwin, X. Zhao, F. Cheng, X. Zhang, A. Huang, B. Shah, Y. Zhang, L.C. Moscinski, Y.S. Choi, A.P. Kozikowski, J.E. Bradner, W.S. Dalton, E. Sotomayor, J. Tao, A microenvironment-mediated c-Myc/miR-548m/HDAC6 amplification loop in non-Hodgkin B cell lymphomas. J. Clin. Invest. 123, 4612–4626 (2013)
K. Okuda, A. Ito, T. Uehara, Regulation of histone deacetylase 6 activity via S-nitrosylation. Biol. Pharm. Bull. 38, 1434–1437 (2015)
C.A. Bradbury, F.L. Khanim, R. Hayden, C.M. Bunce, D.A. White, M.T. Drayson, C. Craddock, B.M. Turner, Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 19, 1751–1759 (2005)
B. Hackanson, L. Rimmele, M. Benkißer, M. Abdelkarim, M. Fliegauf, M. Jung, M. Lübbert, HDAC6 as a target for antileukemic drugs in acute myeloid leukemia. Leuk. Res. 36, 1055–1062 (2012)
S.L. Zhang, H.Y. Zhu, B.Y. Zhou, Y. Chu, J.R. Huo, Y.Y. Tan, D.L. Liu, Histone deacetylase 6 is overexpressed and promotes tumor growth of colon cancer through regulation of the MAPK/ERK signal pathway. Onco. Targets Ther. 12, 2409–2419 (2019)
S. Saji, M. Kawakami, S. Hayashi, N. Yoshida, M. Hirose, S. Horiguchi, A. Itoh, N. Funata, S.L. Schreiber, M. Yoshida, M. Toi, Significance of HDAC6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene 24, 4531–4539 (2005)
T. Sakuma, K. Uzawa, T. Onda, M. Shiiba, H. Yokoe, T. Shibahara, H. Tanzawa, Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int. J. Oncol. 29, 117–124 (2006)
Y.S. Lee, K.H. Lim, X. Guo, Y. Kawaguchi, Y. Gao, T. Barrientos, P. Ordentlich, X.F. Wang, C.M. Counter, T.P. Yao, The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 68, 7561–7569 (2008)
P. Putcha, J. Yu, R. Rodriguez-Barrueco, L. Saucedo-Cuevas, P. Villagrasa, E. Murga-Penas, S.N. Quayle, M. Yang, V. Castro, D. Llobet-Navas, D. Birnbaum, P. Finetti, W.A. Woodward, F. Bertucci, M.L. Alpaugh, A. Califano, J. Silva, HDAC6 activity is a non-oncogene addiction hub for inflammatory breast cancers. Breast Cancer Res. 17, 149 (2015)
A. Keremu, A. Aimaiti, Z. Liang, X. Zou, Role of the HDAC6/STAT3 pathway in regulating PD-L1 expression in osteosarcoma cell lines. Cancer Chemother. Pharmacol. 83, 255–264 (2019)
C.S. Chen, S.C. Weng, P.H. Tseng, H.P. Lin, Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J. Biol. Chem. 280, 38879–38887 (2005)
I.A. Kim, M. No, J.M. Lee, J.H. Shin, J.S. Oh, E.J. Choi, I.H. Kim, P. Atadja, E.J. Bernhard, Epigenetic modulation of radiation response in human cancer cells with activated EGFR or HER-2 signaling: potential role of histone deacetylase 6. Radiother. Oncol. 92, 125–132 (2009)
S.C. Tien, Z.F. Chang, Oncogenic Shp2 disturbs microtubule regulation to cause HDAC6-dependent ERK hyperactivation. Oncogene 33, 2938–2946 (2014)
Q.Y. Zhu, Z. Wang, C. Ji, L. Cheng, Y.L. Yang, J. Ren, Y.H. Jin, Q.J. Wang, X.J. Gu, Z.G. Bi, G. Hu, Y. Yang, C6-ceramide synergistically potentiates the anti-tumor effects of histone deacetylase inhibitors via AKT dephosphorylation and α-tubulin hyperacetylation both in vitro and in vivo. Cell Death Dis. 2, e117 (2011)
S. Aoyagi, T.K. Archer, Modulating molecular chaperone Hsp90 functions through reversible acetylation. Trends Cell Biol. 15, 565–567 (2005)
S. Tsutsumi, K. Beebe, L. Neckers, Impact of heat-shock protein 90 on cancer metastasis. Future Oncol. 5, 679–688 (2009)
Y.Z. Gu, Q. Xue, Y.J. Chen, G.H. Yu, M.D. Qing, Y. Shen, M.Y. Wang, Q. Shi, X.G. Zhang, Different roles of PD-L1 and FasL in immunomodulation mediated by human placenta-derived mesenchymal stem cells. Hum. Immunol. 74, 267–276 (2013)
J. Wen, J. Fu, Y. Ling, W. Zhang, MIIP accelerates epidermal growth factor receptor protein turnover and attenuates proliferation in non-small cell lung cancer. Oncotarget 7, 9118–9134 (2016)
S.L. Zhang, X. Du, L.N. Tan, F.H. Deng, B.Y. Zhou, H.J. Zhou, H.Y. Zhu, Y. Chu, D.L. Liu, Y.Y. Tan, SET7 interacts with HDAC6 and suppresses the development of colon cancer through inactivation of HDAC6. Am. J. Transl. Res. 12, 602–611 (2020)
F. Ikeda, I. Dikic, CYLD in ubiquitin signaling and tumor pathogenesis. Cell 125, 643–645 (2006)
M. Bazzaro, Z. Lin, A. Santillan, M.K. Lee, M.C. Wang, K.C. Chan, R.E. Bristow, R. Mazitschek, J. Bradner, R.B. Roden, Ubiquitin proteasome system stress underlies synergistic killing of ovarian cancer cells by bortezomib and a novel HDAC6 inhibitor. Clin. Cancer Res. 14, 7340–7347 (2008)
Y. Zilberman, C. Ballestrem, L. Carramusa, R. Mazitschek, S. Khochbin, A. Bershadsky, Regulation of microtubule dynamics by inhibition of the tubulin deacetylase HDAC6. J. Cell Sci. 122, 3531–3541 (2009)
L. Zhang, N. Liu, S. Xie, X. He, J. Zhou, M. Liu, D. Li, HDAC6 regulates neuroblastoma cell migration and may play a role in the invasion process. Cancer Biol. Ther. 15, 1561–1570 (2014)
M. Rey, M. Irondelle, F. Waharte, F. Lizarraga, P. Chavrier, HDAC6 is required for invadopodia activity and invasion by breast tumor cells. Eur. J. Cell Biol. 90, 128–135 (2011)
G.P. Gupta, J. Massagué, Cancer metastasis: building a framework. Cell 127, 679–695 (2006)
K. Kanno, S. Kanno, H. Nitta, N. Uesugi, T. Sugai, T. Masuda, G. Wakabayashi, C. Maesawa, Overexpression of histone deacetylase 6 contributes to accelerated migration and invasion activity of hepatocellular carcinoma cells. Oncol. Rep. 28, 867–873 (2012)
K. Azuma, T. Urano, K. Horie-Inoue, S. Hayashi, R. Sakai, Y. Ouchi, S. Inoue, Association of estrogen receptor alpha and histone deacetylase 6 causes rapid deacetylation of tubulin in breast cancer cells. Cancer Res. 69, 2935–2940 (2009)
T.Q. Pham, K. Robinson, L. Xu, M.N. Pavlova, S.X. Skapek, E.Y. Chen, HDAC6 promotes growth, migration/invasion, and self-renewal of rhabdomyosarcoma. Oncogene 40, 578–591 (2021)
D. Li, S. Xie, Y. Ren, L. Huo, J. Gao, D. Cui, M. Liu, J. Zhou, Microtubule-associated deacetylase HDAC6 promotes angiogenesis by regulating cell migration in an EB1-dependent manner. Protein Cell 2, 150–160 (2011)
J.H. Park, S.H. Kim, M.C. Choi, J. Lee, D.Y. Oh, S.A. Im, Y.J. Bang, T.Y. Kim, Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem. Biophys. Res. Commun. 368, 318–322 (2008)
D.Z. Qian, S.K. Kachhap, S.J. Collis, H.M. Verheul, M.A. Carducci, P. Atadja, R. Pili, Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 66, 8814–8821 (2006)
H.W. Ryu, H.R. Won, D.H. Lee, S.H. Kwon, HDAC6 regulates sensitivity to cell death in response to stress and post-stress recovery. Cell Stress Chaperones 22, 253–261 (2017)
Z. Lv, X. Weng, C. Du, C. Zhang, H. Xiao, X. Cai, S. Ye, J. Cheng, C. Ding, H. Xie, L. Zhou, J. Wu, S. Zheng, Downregulation of HDAC6 promotes angiogenesis in hepatocellular carcinoma cells and predicts poor prognosis in liver transplantation patients. Mol. Carcinog. 55, 1024–1033 (2016)
M. Sharma, P. Jha, P. Verma, M. Chopra, Combined comparative molecular field analysis, comparative molecular similarity indices analysis, molecular docking and molecular dynamics studies of histone deacetylase 6 inhibitors. Chem. Biol. Drug Des. 93, 910–925 (2019)
J. Jochems, J. Boulden, B.G. Lee, J.A. Blendy, M. Jarpe, R. Mazitschek, J.H. Van Duzer, S. Jones, O. Berton, Antidepressant-like properties of novel HDAC6-selective inhibitors with improved brain bioavailability. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 39, 389–400 (2014)
J.-H. Lee, A. Mahendran, Y. Yao, L. Ngo, G. Venta-Perez, M.L. Choy, N. Kim, W.-S. Ham, R. Breslow, P.A. Marks, Development of a histone deacetylase 6 inhibitor and its biological effects. Proc. Natl. Acad. Sci. U. S. A. 110, 15704–15709 (2013)
J.H. Kalin, J.A. Bergman, Development and therapeutic implications of selective histone deacetylase 6 inhibitors. J. Med. Chem. 56, 6297–6313 (2013)
D.V. Smil, S. Manku, Y.A. Chantigny, S. Leit, A. Wahhab, T.P. Yan, M. Fournel, C. Maroun, Z. Li, A.-M. Lemieux, A. Nicolescu, J. Rahil, S. Lefebvre, A. Panetta, J.M. Besterman, R. Déziel, Novel HDAC6 isoform selective chiral small molecule histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 19, 688–692 (2009)
Y. Chen, M. Lopez-Sanchez, D.N. Savoy, D.D. Billadeau, G.S. Dow, A.P. Kozikowski, A series of potent and selective, triazolylphenyl-based histone deacetylases inhibitors with activity against pancreatic cancer cells and Plasmodium falciparum. J. Med. Chem. 51, 3437–3448 (2008)
A.P. Kozikowski, S. Tapadar, D.N. Luchini, K.H. Kim, D.D. Billadeau, Use of the nitrile oxide cycloaddition (NOC) reaction for molecular probe generation: a new class of enzyme selective histone deacetylase inhibitors (HDACIs) showing picomolar activity at HDAC6. J. Med. Chem. 51, 4370–4373 (2008)
E.S. Inks, B.J. Josey, S.R. Jesinkey, C.J. Chou, A novel class of small molecule inhibitors of HDAC6. ACS Chem. Biol. 7, 331–339 (2012)
C.-W. Yu, P.-T. Chang, L.-W. Hsin, J.-W. Chern, Quinazolin-4-one derivatives as selective histone deacetylase-6 inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 56, 6775–6791 (2013)
V. Zuco, M. De Cesare, R. Cincinelli, R. Nannei, C. Pisano, N. Zaffaroni, F. Zunino, Synergistic Antitumor Effects of Novel HDAC Inhibitors and Paclitaxel In Vitro and In Vivo. PLoS ONE 6, e29085 (2011)
A. M. Tsimberidou, P. A. Beer, C. A. Cartwright, C. Haymaker, H. H. Vo, S. Kiany, A. R. L. Cecil, J. Dow, K. Haque, F. A. Silva, L. Coe, H. Berryman, E. A. Bone, G. M. Nogueras-Gonzalez, D. Vining, H. McElwaine-Johnn, and I. I. Wistuba, Preclinical development and First-in-human study of KA2507, a selective and potent inhibitor of histone deacetylase 6, for patients with refractory solid tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 27, 3584–3594 (2021)
H. Song, X. Niu, J. Quan, Y. Li, L. Yuan, J. Wang, C. Ma, E. Ma, Discovery of specific HDAC6 inhibitor with anti-metastatic effects in pancreatic cancer cells through virtual screening and biological evaluation. Bioorganic Chem. 97, 103679 (2020)
P. Linciano, L. Pinzi, S. Belluti, U. Chianese, R. Benedetti, D. Moi, L. Altucci, S. Franchini, C. Imbriano, C. Sorbi, G. Rastelli, Inhibitors of histone deacetylase 6 based on a novel 3-hydroxy-isoxazole zinc binding group. J. Enzyme Inhib. Med. Chem. 36, 2080–2086 (2021)
N. Relitti, A.P. Saraswati, G. Chemi, M. Brindisi, S. Brogi, D. Herp, K. Schmidtkunz, F. Saccoccia, G. Ruberti, C. Ulivieri, F. Vanni, F. Sarno, L. Altucci, S. Lamponi, M. Jung, S. Gemma, S. Butini, G. Campiani, Synthesis, molecular modeling studies and biological investigation. Eur. J. Med. Chem. 212, 112998 (2021)
T. Liang, J. Xue, Z. Yao, Y. Ye, X. Yang, X. Hou, H. Fang, Design, synthesis and biological evaluation of 3, 4-disubstituted-imidazolidine-2, 5-dione derivatives as HDAC6 selective inhibitors. Eur. J. Med. Chem. 221, 113526 (2021)
Y. Li, J. Quan, H. Song, D. Li, E. Ma, Y. Wang, C. Ma, Novel pyrrolo[2,1-c][1,4]benzodiazepine-3,11-dione (PBD) derivatives as selective HDAC6 inhibitors to suppress tumor metastasis and invasion in vitro and in vivo. Bioorganic Chem. 114, 105081 (2021)
S. Li, C. Zhao, G. Zhang, Q. Xu, Q. Liu, W. Zhao, C. James Chou, Y. Zhang, Development of selective HDAC6 inhibitors with in vitro and in vivo anti-multiple myeloma activity. Bioorganic Chem. 116, 105278 (2021)
C. Sharma, Y.J. Oh, B. Park, S. Lee, C.-H. Jeong, S. Lee, J.H. Seo, Y.H. Seo, Development of Thiazolidinedione-Based HDAC6 Inhibitors to Overcome Methamphetamine Addiction. Int. J. Mol. Sci. 20, 6213 (2019)
Y. Song, J. Lim, Y.H. Seo, A novel class of anthraquinone-based HDAC6 inhibitors. Eur. J. Med. Chem. 164, 263–272 (2019)
L. Goracci, N. Deschamps, G.M. Randazzo, C. Petit, C. Dos Santos Passos, P.-A. Carrupt, C. Simões-Pires, A. Nurisso, A Rational Approach for the Identification of Non-Hydroxamate HDAC6-Selective Inhibitors. Sci. Rep. 6, 29086 (2016)
X.-H. Zhang, H.-Q. Kang, Y.-Y. Tao, Y.-H. Li, J.-R. Zhao, Ya-Gao, L.-Y. Ma, H.-M. Liu, Identification of novel 1,3-diaryl-1,2,4-triazole-capped histone deacetylase 6 inhibitors with potential anti-gastric cancer activity. Eur. J. Med. Chem. 218, 113392 (2021)
C. Seidel, M. Schnekenburger, A. Mazumder, M.-H. Teiten, G. Kirsch, M. Dicato, M. Diederich, 4-Hydroxybenzoic acid derivatives as HDAC6-specific inhibitors modulating microtubular structure and HSP90α chaperone activity against prostate cancer. Biochem. Pharmacol. 99, 31–52 (2016)
M. Leonhardt, A. Sellmer, O.H. Krämer, S. Dove, S. Elz, B. Kraus, M. Beyer, S. Mahboobi, Design and biological evaluation of tetrahydro-β-carboline derivatives as highly potent histone deacetylase 6 (HDAC6) inhibitors. Eur. J. Med. Chem. 152, 329–357 (2018)
H.-Y. Lee, A.-C. Tsai, M.-C. Chen, P.-J. Shen, Y.-C. Cheng, C.-C. Kuo, S.-L. Pan, Y.-M. Liu, J.-F. Liu, T.-K. Yeh, J.-C. Wang, C.-Y. Chang, J.-Y. Chang, J.-P. Liou, Azaindolylsulfonamides, with a more selective inhibitory effect on histone deacetylase 6 activity, exhibit antitumor activity in colorectal cancer HCT116 cells. J. Med. Chem. 57, 4009–4022 (2014)
Y.-M. Liu, H.-Y. Lee, M.-J. Lai, S.-L. Pan, H.-L. Huang, F.-C. Kuo, M.-C. Chen, J.-P. Liou, Pyrimidinedione-mediated selective histone deacetylase 6 inhibitors with antitumor activity in colorectal cancer HCT116 cells. Org. Biomol. Chem. 13, 10226–10235 (2015)
M. Kaliszczak, S. Trousil, O. Åberg, M. Perumal, Q.-D. Nguyen, E.O. Aboagye, A novel small molecule hydroxamate preferentially inhibits HDAC6 activity and tumour growth. Br. J. Cancer 108, 342–350 (2013)
K. Nepali, H.-Y. Lee, M.-J. Lai, R. Ojha, T.-Y. Wu, G.-X. Wu, M.-C. Chen, J.-P. Liou, Ring-opened tetrahydro-γ-carbolines display cytotoxicity and selectivity with histone deacetylase isoforms. Eur. J. Med. Chem. 127, 115–127 (2017)
M.-C. Chen, Y.-C. Lin, Y.-H. Liao, J.-P. Liou, C.-H. Chen, MPT0G612, a novel HDAC6 inhibitor, induces apoptosis and suppresses IFN-γ-induced programmed death-ligand 1 in human colorectal carcinoma cells. Cancers 11, 1617 (2019)
H.-Y. Lee, K. Nepali, F.-I. Huang, C.-Y. Chang, M.-J. Lai, Y.-H. Li, H.-L. Huang, C.-R. Yang, J.-P. Liou, (N-Hydroxycarbonylbenylamino)quinolines as selective histone deacetylase 6 inhibitors suppress growth of multiple myeloma in vitro and in vivo. J. Med. Chem. 61, 905–917 (2018)
H.-W. Ryu, D.-H. Lee, D.-H. Shin, S.H. Kim, S.H. Kwon, Aceroside VIII is a new natural selective HDAC6 inhibitor that synergistically enhances the anticancer activity of HDAC inhibitor in HT29 cells. Planta Med. 81, 222–227 (2015)
C.-W. Yu, P.-Y. Hung, H.-T. Yang, Y.-H. Ho, H.-Y. Lai, Y.-S. Cheng, J.-W. Chern, Quinazolin-2,4-dione-based hydroxamic acids as selective histone deacetylase-6 inhibitors for treatment of non-small cell lung cancer. J. Med. Chem. 62, 857–874 (2019)
S. Noonepalle, S. Shen, J. Ptáček, M.T. Tavares, G. Zhang, J. Stránský, J. Pavlíček, G.M. Ferreira, M. Hadley, G. Pelaez, C. Bařinka, A.P. Kozikowski, A. Villagra, Rational design of suprastat: A novel selective histone deacetylase 6 inhibitor with the ability to potentiate immunotherapy in melanoma models. J. Med. Chem. 63, 10246–10262 (2020)
X. Chen, X. Chen, R.R. Steimbach, T. Wu, H. Li, W. Dan, P. Shi, C. Cao, D. Li, A.K. Miller, Z. Qiu, J. Gao, Y. Zhu, Novel 2, 5-diketopiperazine derivatives as potent selective histone deacetylase 6 inhibitors: Rational design, synthesis and antiproliferative activity. Eur. J. Med. Chem. 187, 111950 (2020)
M. Pérez-Salvia, E. Aldaba, Y. Vara, M. Fabre, C. Ferrer, C. Masdeu, A. Zubia, E.S. Sebastian, D. Otaegui, P. Llinàs-Arias, M. Rosselló-Tortella, M. Berdasco, C. Moutinho, F. Setien, A. Villanueva, E. González-Barca, J. Muncunill, J.-T. Navarro, M.A. Piris, F.P. Cossio, M. Esteller, In vitro and in vivo activity of a new small-molecule inhibitor of HDAC6 in mantle cell lymphoma. Haematologica 103, e537–e540 (2018)
J. Sun, W. Wu, X. Tang, F. Zhang, C. Ju, R. Liu, Y. Liang, B. Yu, B. Lv, Y. Guo, D. Zeng, X. Tao, M. Wang, Z. Zhang, C. Zhang, and X.-B. Lv, HDAC6 inhibitor WT161 performs anti-tumor effect on osteosarcoma and synergistically interacts with 5-FU. Biosci. Rep. 41, BSR20203905 (2021)
F. Wang, L. Zheng, Y. Yi, Z. Yang, Q. Qiu, X. Wang, W. Yan, P. Bai, J. Yang, D. Li, H. Pei, T. Niu, H. Ye, C. Nie, Y. Hu, S. Yang, Y. Wei, L. Chen, SKLB-23bb, A HDAC6-Selective Inhibitor, Exhibits Superior and Broad-Spectrum Antitumor Activity via Additionally Targeting Microtubules. Mol. Cancer Ther. 17, 763–775 (2018)
M.K. Ediriweera, N.B. To, Y. Lim, S.K. Cho, Odd-chain fatty acids as novel histone deacetylase 6 (HDAC6) inhibitors. Biochimie 186, 147–156 (2021)
Y.W. Song, Y. Lim, S.K. Cho, 2,4-Di-tert-butylphenol, a potential HDAC6 inhibitor, induces senescence and mitotic catastrophe in human gastric adenocarcinoma AGS cells. Biochem. Biophys. Acta BBA - Mol. Cell Res. 1865, 675–683 (2018)
J. Dong, N. Zheng, X. Wang, C. Tang, P. Yan, H. Zhou, J. Huang, A novel HDAC6 inhibitor exerts an anti-cancer effect by triggering cell cycle arrest and apoptosis in gastric cancer. Eur. J. Pharmacol. 828, 67–79 (2018)
G. Yan, D. Li, X. Zhong, G. Liu, X. Wang, Y. Lu, F. Qin, Y. Guo, S. Duan, D. Li, Identification of HDAC6 selective inhibitors: pharmacophore based virtual screening, molecular docking and molecular dynamics simulation. J. Biomol. Struct. Dyn. 39, 1928–1939 (2021)
C. Zhao, J. Gao, L. Zhang, L. Su, Y. Luan, Novel HDAC6 selective inhibitors with 4-aminopiperidine-1- carboxamide as the core structure enhanced growth inhibitory activity of bortezomib in MCF-7 cells. Biosci. Trends 13, 91–97 (2019)
M. Dawood, M. Elbadawi, M. Böckers, G. Bringmann, T. Efferth, Molecular docking-based virtual drug screening revealing an oxofluorenyl benzamide and a bromonaphthalene sulfonamido hydroxybenzoic acid as HDAC6 inhibitors with cytotoxicity against leukemia cells. Biomed. Pharmacother. 129, 110454 (2020)
L. Wasim, M. Chopra, Panobinostat induces apoptosis via production of reactive oxygen species and synergizes with topoisomerase inhibitors in cervical cancer cells. Biomed. Pharmacother. 84, 1393–1405 (2016)
L. Wasim, M. Chopra, Synergistic anticancer effect of panobinostat and topoisomerase inhibitors through ROS generation and intrinsic apoptotic pathway induction in cervical cancer cells. Cell. Oncol. Dordr. 41, 201–212 (2018)
L. Hontecillas-Prieto, R. Flores-Campos, A. Silver, E. de Álava, N. Hajji, D.J. García-Domínguez, Synergistic enhancement of cancer therapy using HDAC inhibitors: opportunity for clinical trials. Front. Genet. 11, 578011 (2020)
J. E. Amengual, J. K. Lue, H. Ma, R. Lichtenstein, B. Shah, S. Cremers, S. Jones, and A. Sawas, First-in-class selective HDAC6 inhibitor (ACY-1215) Has a highly favorable safety profile in patients with relapsed and refractory lymphoma. Oncologist 26, 184 (3)
R. Mazroui, S. Di Marco, R.J. Kaufman, I.E. Gallouzi, Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell 18, 2603–2618 (2007)
S. Kwon, Y. Zhang, P. Matthias, The deacetylase HDAC6 is a novel critical component of stress granules involved in the stress response. Genes Dev. 21, 3381–3394 (2007)
M. Kästle, E. Woschee, T. Grune, Histone deacetylase 6 (HDAC6) plays a crucial role in p38MAPK-dependent induction of heme oxygenase-1 (HO-1) in response to proteasome inhibition. Free Radic. Biol. Med. 53, 2092–2101 (2012)
T. Hideshima, J.E. Bradner, J. Wong, D. Chauhan, P. Richardson, S.L. Schreiber, K.C. Anderson, Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. U.S.A. 102, 8567–8572 (2005)
S.W. Lee, S.-K. Yeon, G.W. Kim, D.H. Lee, Y.H. Jeon, J. Yoo, S.Y. Kim, S.H. Kwon, HDAC6-selective inhibitor overcomes bortezomib resistance in multiple myeloma. Int. J. Mol. Sci. 22, 1341 (2021)
X. Sun, Y. Xie, X. Sun, Y. Yao, H. Li, Z. Li, R. Yao, and K. Xu, The selective HDAC6 inhibitor Nexturastat A induces apoptosis, overcomes drug resistance and inhibits tumor growth in multiple myeloma. Biosci. Rep. 39, BSR20181916 (2019)
F.I. Huang, Y.W. Wu, T.Y. Sung, J.P. Liou, M.H. Lin, S.L. Pan, C.R. Yang, MPT0G413, A novel HDAC6-selective inhibitor, and bortezomib synergistically exert anti-tumor activity in multiple myeloma cells. Front. Oncol. 9, 249 (2019)
J.E. Amengual, P. Johannet, M. Lombardo, K. Zullo, D. Hoehn, G. Bhagat, L. Scotto, X. Jirau-Serrano, D. Radeski, J. Heinen, H. Jiang, S. Cremers, Y. Zhang, S. Jones, O.A. O’Connor, Dual targeting of protein degradation pathways with the selective HDAC6 inhibitor ACY-1215 and bortezomib is synergistic in lymphoma. Clin. Cancer Res. 21, 4663–4675 (2015)
L. Santo, T. Hideshima, A.L. Kung, J.C. Tseng, D. Tamang, M. Yang, M. Jarpe, J.H. van Duzer, R. Mazitschek, W.C. Ogier, D. Cirstea, S. Rodig, H. Eda, T. Scullen, M. Canavese, J. Bradner, K.C. Anderson, S.S. Jones, N. Raje, Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 119, 2579–2589 (2012)
S.T. Nawrocki, J.S. Carew, M.S. Pino, R.A. Highshaw, R.H. Andtbacka, K. Dunner, A. Pal, W.G. Bornmann, P.J. Chiao, P. Huang, H. Xiong, J.L. Abbruzzese, D.J. McConkey, Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 66, 3773–3781 (2006)
Y. Mishima, L. Santo, H. Eda, D. Cirstea, N. Nemani, A.J. Yee, E. O’Donnell, M.K. Selig, S.N. Quayle, S. Arastu-Kapur, C. Kirk, L.H. Boise, S.S. Jones, N. Raje, Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br. J. Haematol. 169, 423–434 (2015)
R.A. Stanton, K.M. Gernert, J.H. Nettles, R. Aneja, Drugs that target dynamic microtubules: a new molecular perspective. Med. Res. Rev. 31, 443–481 (2011)
E. Mukhtar, V.M. Adhami, H. Mukhtar, Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 13, 275–284 (2014)
J. Asthana, S. Kapoor, R. Mohan, and D. Panda, Inhibition of HDAC6 deacetylase activity increases its binding with microtubules and suppresses microtubule dynamic instability in MCF-7 cells. J. Biol. Chem. 288, 22516–22526–5438 (2013)
Y. Itoh, T. Suzuki, A. Kouketsu, N. Suzuki, S. Maeda, M. Yoshida, H. Nakagawa, N. Miyata, Design, synthesis, structure-selectivity relationship, and effect on human cancer cells of a novel series of histone deacetylase 6-selective inhibitors. J. Med. Chem. 50, 5425–5438 (2007)
J. Yoo, Y.H. Jeon, D.H. Lee, G.W. Kim, S.W. Lee, S.Y. Kim, J. Park, S.H. Kwon, HDAC6-selective inhibitors enhance anticancer effects of paclitaxel in ovarian cancer cells. Oncol. Lett. 21, 201 (2021)
T. Oba, M. Ono, H. Matoba, T. Uehara, Y. Hasegawa, K. Ito, HDAC6 inhibition enhances the anti-tumor effect of eribulin through tubulin acetylation in triple-negative breast cancer cells. Breast Cancer Res. Treat. 186, 37–51 (2021)
H.-J. Tu, Y.-J. Lin, M.-W. Chao, T.-Y. Sung, Y.-W. Wu, Y.-Y. Chen, M.-H. Lin, J.-P. Liou, S.-L. Pan, C.-R. Yang, The anticancer effects of MPT0G211, a novel HDAC6 inhibitor, combined with chemotherapeutic agents in human acute leukemia cells. Clin. Epigenetics 10, 162 (2018)
C. Corno, N. Arrighetti, E. Ciusani, E. Corna, N. Carenini, N. Zaffaroni, L. Gatti, P. Perego, Synergistic interaction of histone deacetylase 6- and MEK-inhibitors in castration-resistant prostate cancer cells. Front. Cell Dev. Biol. 8, 610 (2020)
M. Namdar, G. Perez, L. Ngo, P.A. Marks, Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. U.S.A. 107, 20003–20008 (2010)
L. Wang, S. Xiang, K.A. Williams, H. Dong, W. Bai, S.V. Nicosia, S. Khochbin, G. Bepler, X. Zhang, Depletion of HDAC6 enhances cisplatin-induced DNA damage and apoptosis in non-small cell lung cancer cells. PLoS ONE 7, e44265 (2012)
A. Bag, A. Schultz, S. Bhimani, O. Stringfield, W. Dominguez, Q. Mo, L. Cen, D. Adeegbe, Coupling the immunomodulatory properties of the HDAC6 inhibitor ACY241 with Oxaliplatin promotes robust anti-tumor response in non-small cell lung cancer. Oncoimmunology 11, 2042065 (2022)
Y. Ruan, L. Wang, Y. Lu, HDAC6 inhibitor, ACY1215 suppress the proliferation and induce apoptosis of gallbladder cancer cells and increased the chemotherapy effect of gemcitabine and oxaliplatin. Drug Dev. Res. 82, 598–604 (2021)
J. Sun, X. Qian, F. Zhang, X. Tang, C. Ju, R. Liu, R. Zhou, Z. Zhang, X.-B. Lv, C. Zhang, G. Huang, HDAC6 inhibitor WT161 induces apoptosis in retinoblastoma cells and synergistically interacts with cisplatin. Transl. Cancer Res. 8, 2759–2768 (2019)
X.-N. Wang, K.-Y. Wang, X.-S. Zhang, C. Yang, X.-Y. Li, 4-Hydroxybenzoic acid (4-HBA) enhances the sensitivity of human breast cancer cells to adriamycin as a specific HDAC6 inhibitor by promoting HIPK2/p53 pathway. Biochem. Biophys. Res. Commun. 504, 812–819 (2018)
H.-R. Won, H.-W. Ryu, D.-H. Shin, S.-K. Yeon, D.H. Lee, S.H. Kwon, A452, an HDAC6-selective inhibitor, synergistically enhances the anticancer activity of chemotherapeutic agents in colorectal cancer cells. Mol. Carcinog. 57, 1383–1395 (2018)
M. Bobrowicz, A. Slusarczyk, J. Domagala, M. Dwojak, D. Ignatova, Y.T. Chang, C. Iselin, N. Miazek-Zapala, K. Marhelava, E. Guenova, M. Winiarska, Selective inhibition of HDAC6 sensitizes cutaneous T-cell lymphoma to PI3K inhibitors. Oncol. Lett. 20, 533–540 (2020)
H. Losson, S.R. Gajulapalli, M. Lernoux, J.-Y. Lee, A. Mazumder, D. Gérard, C. Seidel, H. Hahn, C. Christov, M. Dicato, G. Kirsch, B.W. Han, M. Schnekenburger, M. Diederich, The HDAC6 inhibitor 7b induces BCR-ABL ubiquitination and downregulation and synergizes with imatinib to trigger apoptosis in chronic myeloid leukemia. Pharmacol. Res. 160, 105058 (2020)
Y. Qin, Y. Liang, G. Jiang, Y. Peng, W. Feng, ACY-1215 suppresses the proliferation and induces apoptosis of chronic myeloid leukemia cells via the ROS/PTEN/Akt pathway. Cell Stress Chaperones 27, 383–396 (2022)
D.H. Lee, G.W. Kim, S.H. Kwon, The HDAC6-selective inhibitor is effective against non-Hodgkin lymphoma and synergizes with ibrutinib in follicular lymphoma. Mol. Carcinog. 58, 944–956 (2019)
S.-J. Park, S.H. Joo, N. Lee, W.-J. Jang, J.H. Seo, C.-H. Jeong, ACY-241, an HDAC6 inhibitor, overcomes erlotinib resistance in human pancreatic cancer cells by inducing autophagy. Arch. Pharm. Res. 44, 1062–1075 (2021)
U. Peng, Z. Wang, S. Pei, Y. Ou, P. Hu, W. Liu, J. Song, ACY-1215 accelerates vemurafenib induced cell death of BRAF-mutant melanoma cells via induction of ER stress and inhibition of ERK activation. Oncol. Rep. 37, 1270–1276 (2017)
G. Zhang, Y.H. Gan, Synergistic antitumor effects of the combined treatment with an HDAC6 inhibitor and a COX-2 inhibitor through activation of PTEN. Oncol. Rep. 38, 2657–2666 (2017)
M. Cosenza, M. Civallero, L. Marcheselli, S. Sacchi, S. Pozzi, Ricolinostat, a selective HDAC6 inhibitor, shows anti-lymphoma cell activity alone and in combination with bendamustine. Apoptosis 25, 370–387 (2020)
J.S. Carew, C.M. Espitia, W. Zhao, V. Visconte, F. Anwer, K.R. Kelly, S.T. Nawrocki, Rational cotargeting of HDAC6 and BET proteins yields synergistic antimyeloma activity. Blood Adv. 3, 1318–1329 (2019)
B.J. North, I. Almeciga-Pinto, D. Tamang, M. Yang, S.S. Jones, S.N. Quayle, Enhancement of pomalidomide anti-tumor response with ACY-241, a selective HDAC6 inhibitor. PLoS ONE 12, e0173507 (2017)
K. Miyake, N. Takano, H. Kazama, H. Kikuchi, M. Hiramoto, K. Tsukahara, K. Miyazawa, Ricolinostat enhances adavosertib-induced mitotic catastrophe in TP53-mutated head and neck squamous cell carcinoma cells. Int. J. Oncol. 60, 54 (2022)
D.O. Adeegbe, Y. Liu, P.H. Lizotte, Y. Kamihara, A.R. Aref, C. Almonte, R. Dries, Y. Li, S. Liu, X. Wang, T. Warner-Hatten, J. Castrillon, G.C. Yuan, N. Poudel-Neupane, H. Zhang, J.L. Guerriero, S. Han, M.M. Awad, D.A. Barbie, J. Ritz, S.S. Jones, P.S. Hammerman, J. Bradner, S.N. Quayle, K.K. Wong, Synergistic immunostimulatory effects and therapeutic benefit of combined histone deacetylase and bromodomain inhibition in non-small cell lung cancer. Cancer Discov. 7, 852–867 (2017)
H.Y. Cho, S.W. Lee, Y.H. Jeon, D.H. Lee, G.W. Kim, J. Yoo, S.Y. Kim, S.H. Kwon, Combination of ACY-241 and JQ1 synergistically suppresses metastasis of HNSCC via regulation of MMP-2 and MMP-9. Int. J. Mol. Sci. 21, 6873 (2020)
Y. Liu, Y. Li, S. Liu, D.O. Adeegbe, C.L. Christensen, M.M. Quinn, R. Dries, S. Han, K. Buczkowski, X. Wang, T. Chen, P. Gao, H. Zhang, F. Li, P.S. Hammerman, J.E. Bradner, S.N. Quayle, K.-K. Wong, NK cells mediate synergistic antitumor effects of combined inhibition of HDAC6 and BET in a SCLC preclinical model. Cancer Res. 78, 3709–3717 (2018)
S. Moufarrij, A. Srivastava, S. Gomez, M. Hadley, E. Palmer, P.T. Austin, S. Chisholm, N. Diab, K. Roche, A. Yu, J. Li, W. Zhu, M. Lopez-Acevedo, A. Villagra, K.B. Chiappinelli, Combining DNMT and HDAC6 inhibitors increases anti-tumor immune signaling and decreases tumor burden in ovarian cancer. Sci. Rep. 10, 3470 (2020)
T. Fukumoto, N. Fatkhutdinov, J.A. Zundell, E.N. Tcyganov, T. Nacarelli, S. Karakashev, S. Wu, Q. Liu, D.I. Gabrilovich, R. Zhang, HDAC6 inhibition synergizes with anti-PD-L1 therapy in ARID1A-inactivated ovarian cancer. Cancer Res. 79, 5482–5489 (2019)
G.W. Kim, J. Yoo, H.-R. Won, S.-K. Yeon, S.W. Lee, D.H. Lee, Y.H. Jeon, S.H. Kwon, HDAC6-selective inhibitor synergistically enhances the anticancer activity of immunomodulatory drugs in IMiDs-resistant multiple myeloma. Leuk. Res. 95, 106398 (2020)
Funding
Sumeet Kaur would like to thank the University Grants Commission (UGC), New Delhi, India, for awarding her a UGC fellowship. Prerna Rajoria would like to thank the Department of Biotechnology (DBT), Ministry of Science & Technology, Government of India for awarding her a DBT-JRF fellowship (Grant Number: DBT/2021–22/CBR/1593).
Author information
Authors and Affiliations
Contributions
Madhu Chopra conceptualized the article and critically revised the work. Sumeet Kaur conceptualized the article, performed the literature search, and drafted and critically revised the work. Prerna Rajoria performed the literature search and prepared the figures.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No ethical approval and consent to participate was required for this study.
Consent for publication
There is no conflict of interest (either financial or personal). All authors and acknowledged contributors have read and approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kaur, S., Rajoria, P. & Chopra, M. HDAC6: A unique HDAC family member as a cancer target. Cell Oncol. 45, 779–829 (2022). https://doi.org/10.1007/s13402-022-00704-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-022-00704-6