
Abstract
Purpose
Isothiocyanates elicit anticancer effects by targeting cancer stem cells (CSCs). Here, we tested the antitumor activity of phenethyl-isothiocyanate (PEITC), either alone or in combination with trastuzumab, in HER2-positive tumor models.
Methods
We assessed the in vitro anticancer activity of PEITC, alone or combined with trastuzumab, in HER2-positive BT474, SKBR3, HCC1954 and SKOV3 cancer cells by measuring their sphere forming efficiency (SFE). The expression of the human/rodent CSC biomarkers aldehyde-dehydrogenase (ALDH) and CD29High/CD24+/Sca1Low was evaluated by cytofluorimetric analysis. The expression of wild type HER2 (WTHER2), its splice variant d16HER2 and NOTCH was analysed by quantitative RT-PCR and Western blotting. The in vivo activity of PEITC and trastuzumab was evaluated in mice orthotopically implanted with MI6 tumor cells transgenic for the human d16HER2 splice isoform. Magnetic resonance imaging/spectroscopy and immunohistochemistry were used to assess morpho-functional and metabolic profiles of treated versus untreated mice.
Results
We found that PEITC significantly impaired the SFE of HER2-positive human cancer cells by decreasing their ALDH-positive compartments. The anti-CSC activity of PEITC was demonstrated by a reduced expression/activation of established cancer-stemness biomarkers. Similar results were obtained with MI6 cells, where PEITC, alone or in combination with trastuzumab, significantly inhibited their SFE. We also found that PEITC hampered the in vivo growth of MI6 nodules by inducing hemorrhagic and necrotic intra-tumor areas and, in combination with trastuzumab, by significantly reducing spontaneous tumor development in d16HER2 transgenic mice.
Conclusions
Our results indicate that PEITC targets HER2-positive CSCs and that its combination with trastuzumab may pave the way for a novel therapeutic strategy for HER2-positive tumors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
HER2 overexpression or amplification occurs in subsets of different cancer types, including breast, colon, endometrial, ovarian, gastric/gastroesophageal junction, pancreas, head and neck and urothelial cancers [1]. HER2 overexpression, which is generally associated with a poor patient prognosis due to a high tumor aggressiveness and metastatic potential, predicts tumor sensitivity to anti-HER2 treatment in breast, gastric and gynecological cancers [1]. In the context of HER2-positive breast cancer (BC), the introduction of different effective anti-HER2 compounds, such as the humanized monoclonal antibodies trastuzumab (T) and pertuzumab, the tyrosine kinase inhibitor lapatinib and the drug-antibody conjugate T-emtansine (T-DM1), has remarkably improved the prognosis of patients with both limited-stage and advanced disease. However, not all patients with localized HER2-positive BC profit durably from currently available treatments and, in case of advanced disease, most initially responding patients finally develop resistance to these treatments and eventually die from it [2,3,4]. Even among HER2-enriched BC, as defined on basis of PAM50 gene expression signature, no more than 50% of the patients respond to T [5].
Increasing evidence indicates that various cancer types may be initiated and maintained by cancer stem cells (CSCs), a small but biologically relevant malignant cell subset with unlimited self-renewal capacity responsible for tumor growth, progression and metastasis. Due to these characteristics, CSCs are considered as the “roots” of cancer [6,7,8,9]. Specific gene signaling pathways, including the Wnt/β-catenin, Hedgehog and Notch pathways, contribute to the maintenance of CSCs, and inhibitors of these pathways are now entering clinical trials [10,11,12]. Furthermore, CSCs have been suggested to contribute to resistance to chemotherapy, radiation therapy, targeted therapy and immunotherapy, all features linked to a poor prognosis [11, 13,14,15]. In particular, emerging data suggest that the clinical efficacy of molecularly targeted therapies may be related to their ability to target CSCs [11]. Different studies performed in HER2-positive BC and ovarian cancer (OC) models report that HER2 may act as an important regulator of HER2-positive CSC properties through its functional interplay with NOTCH family members [10, 16,17,18,19]. Concordantly, anti-HER2 targeted therapies, such as T and lapatinib, have been found to be able to affect both breast and ovarian CSCs [10, 17, 18]. Previously, we reported that the d16HER2 splice variant, a highly oncogenic HER2 isoform characterized by the skipping of exon 16 [20], is expressed not only in human HER2-positive BCs [21, 22], but also in other HER2-positive cancer types [23]. In these cancers it promotes tumorigenesis [24], drives cancer aggressiveness [25], causes tumor addiction to HER2 signaling and its response to anti-HER2 treatments [23, 25, 26], regulates stemness [19] and drives dysregulated glycolitic metabolism [27]. However, in HER2-positive BCs not responding to T, CSCs transit into a therapy-resistant state as a consequence of their plasticity, thus becoming independent from HER2 signaling for their maintenance, expansion and activity. Instead, these CSCs may become dependent on other hitherto undefined, and potentially targetable genetic alterations or drivers [28].
Epidemiological and case-control studies indicate that consuming cruciferous vegetables may reduce the risk of cancer development. Isothiocyanates (ITCs), which are components of cruciferous vegetables, are bioactive products that have been found to exert both chemo-preventive and therapeutic activities against various human tumor types [29]. In particular, sulforaphane has been reported to suppress MCF-7 and SUM159 CSC activity, thus providing a rationale for future BC chemoprevention [30]. Also, β-phenethyl isothiocyanate (PEITC), one of the most studied ITCs, has been found to be able to target well-established hallmarks of cancer [31] by promoting cell cycle arrest, inducing apoptosis and inhibiting cell proliferation, angiogenesis, invasion and metastatic processes either in BC or OC [29, 32, 33]. Of note, PEITC has been found to induce apoptosis in MCF-7 and MDA-MB-231 BC cell models stably transfected with HER2, and to enhance the effects of doxorubicin by increasing the cleavage of caspase 3 [34]. However, the intrinsic effect of PEITC, alone and/or in combination with T, in CSC subsets from cellular models constitutively expressing HER2 remains to be addressed. In an attempt to increase the activity of T, which currently represents the gold standard therapy for early and advanced HER2-positive disease, our present study was designed to investigate the in vitro and in vivo antitumor activity of PEITC in HER2-positive human BC and OC cell models, as well as in orthotopic mouse models or mice transgenic for the highly oncogenic human d16HER2 splice variant [24, 25]. Our results indicate that PEITC may act as a potent anti-tumor agent that targets HER2-positive breast and ovarian CSCs as well as differentiated/bulk cells, and synergizes in vivo with T in d16HER2-positive BC pre-clinical models. These findings pave the way for the use of this ITC compound in combination with T to target HER2-positive CSCs, “the roots” of HER2-positive cancers.
2 Materials and methods
2.1 Cell lines and reagents
The d16HER2-positive transgenic mammary tumor cell line MI6 was generated in house from a spontaneous mammary lesion, as previously described [19, 25]. These cells were grown in complete MammoCult medium (StemCell Technologies, Vancouver, Canada) supplemented with 1% fetal bovine serum (FBS) (Sigma-Aldrich, St Louis, MO, USA) and penicillin–streptomycin (Sigma-Aldrich). The human HER2-positive BC cell lines BT474, SKBR3, HCC1954 and the Triple Negative BC (TNBC) cell line MDA-MB-231, as well as the human HER2-positive OC cell line SKOV3 were obtained from the ATCC (Rockville, MD, USA). The BT474 cells were grown in DMEM (Lonza, Basel, Switzerland) supplemented with 10% FBS (Gibco by Life Technologies, Waltham, MA, USA), whereas the SKBR3, HCC1954, MDA-MB-231 and SKOV3 cells were grown in RPMI-1640 medium with L-glutamine (Lonza) supplemented with 10% FBS. The human cell lines were purchased between 2000 and 2010 and authenticated yearly by short tandem repeat DNA fingerprinting using the AmpFISTR identifier PCR Amplification Kit (Thermo Fisher Scientific, Waltham, MA, USA; last verification, May 2017). All tumor cell lines were cultured at 37 °C in a humidified 5% CO2 atmosphere and routinely tested for mycoplasma contamination. PEITC was purchased from Sigma-Aldrich. The humanized monoclonal antibody T (Hoffmann-La Roche, Basel, Switzerland) was provided by Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy.
2.2 SRB cell viability/proliferation assay
All cell lines were seeded in 96-well plates at a density of 3,000–5,000 cells per well (according to the cell line) either at 24 h (for BT474, SKBR3, HCC1954, SKOV3 and MDA-MB-231 cells) or at 72 h (for MI6 cells) before PEITC treatment to allow attachment to plastic. Next, the cells were treated with increasing serial concentrations of PEITC starting from 3 to 48 μM during different time periods (24 h, 48 h and 72 h). Cell viability was assessed using a sulforhodamine B (SRB) assay as reported before [35]. Briefly, after each incubation period, cell monolayers were fixed with 10% trichloroacetic acid and stained with sulforhodamine B for 30 min. The excess dye was subsequently removed by repeated washing with 1% acetic acid, after which the protein-bound dye was dissolved in 10 mM Tris solution for OD determination at 564 nm. The number of living cells is proportional to the solubilized dye. Cell proliferation inhibition (%) was calculated as {(untreated - treated)/(untreated)}X100 and plotted using GraphPad Prism 5.02.
2.3 Flow cytometry (FACS)
MI6 cells were analyzed by multiparametric FACS for the expression of molecular stemness markers by incubating the cells for 30 min at 0 °C with the fluorochrome-labeled antibodies anti-mouse CD24 PerCP-Cyanine5.5 (1:150), anti-mouse/rat CD29 (Integrin beta 1) PE-Cyanine7 (1:150), and anti-mouse Ly-6A/E (Sca-1) FITC (1:100) (all from eBioscience, San Diego, CA, USA). This incubation was followed by 2 washes with PBS 1X-2% FBS after which the cells were analyzed using a BD LSR II Fortessa cytometer (BD Biosciences, Franklin Lakes, NJ, USA). In all experiments, the data were processed using the FlowJo software package.
2.4 ALDH activity assay
Aldehyde-dehydrogenase (ALDH) activity was measured using an ALDEFLUOR assay kit (StemCell Technologies) in BT474, SKBR3, HCC1954 and SKOV3 cells as per manufacturer’s instructions. Briefly, following treatment with PEITC for 72 h, 1 × 106 cells were suspended in ALDEFLUOR assay buffer containing the ALDH substrate bodipy-aminoacetaldehyde (BAAA), and incubated for 1 h at 37 °C. A specific inhibitor of ALDH, diethylaminobenzaldehyde (DEAB), was used to distinguish ALDH-positive and -negative cell subsets.
2.5 Sphere formation efficiency (SFE) assay
Dissociated MI6, BT474, SKBR3, HCC1954 and SKOV3 cells were seeded in 6-well ultra-low attachment plates (Corning, Corning, NY, USA) at densities of 2,500–3,000 cells per well for 7 days in serum-free Mammocult medium (StemCell Technologies) as previously reported [19]. Among the cell lines tested, we found that SKBR3 formed spheres with a “grape-like” morphology as documented by Shaw et al. [36]. Briefly, MI6, BT474, SKBR3, HCC1954 and SKOV3 cells were treated at day 0 with 12 μM PEITC or with 0.1% of the diluent DMSO. MI6 cells were also incubated with T and the combination PEITC+T for 7 days. The spheres were counted microscopically at day 7 and representative images were acquired using an EVOS XL Core Cell Imaging System (Thermo Fisher Scientific) (Magnification 10x). The SFE was calculated as the number of spheres/number of single cells that were initially seeded. The SFE inhibition percentage, i.e., the capability to form spheres under 3D culture conditions, was calculated as {(untreated SFE-treated SFE)/(untreated SFE)}X100, as reported before [19].
2.6 Self renewal assay
Dissociated BT474, SKBR3, HCC1954 and SKOV3 cells were seeded and grown in 6-well, ultra-low attachment plates at a density of 2,500–3,000 cells per well in the presence of DMSO or PEITC (12 μM) for 7 days. After dissociation of primary spheres, the cells were re-plated at the same density without any treatment. The self-renewal capacities were calculated as follows: secondary SFE/primary SFE, and the results were compared between cells treated with DMSO or PEITC.
2.7 Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from BT474, SKBR3, HCC1954 and SKOV3 cells using Qiazol (Qiagen, Hilden, Germany) according to the manufacturer’s instructions, and as reported before [25]. cDNA was reverse-transcribed from 1 μg total RNA using a High-Capacity RNA-to-cDNA™ Kit (Applied Biosystems, Foster City, CA, USA) and subjected to qRT-PCR using an Applied Biosystems SYBR® Green dye-based PCR assay in conjunction with an ABI Prism 7900HT sequence detection system (Applied Biosystems).The primers used to measure gene expression levels are listed in supplementary Table 1. All primer sets were used under the same qRT-PCR cycling conditions with similar efficiencies to obtain simultaneous amplification in the same run. The data were normalized to GAPDH using the comparative 2-ΔCt method. The mRNA levels of each analyzed gene were calculated using the comparative Ct method and presented as relative expression.
2.8 Western blot analysis
Human and murine protein extracts from tumor cells were solubilized for 40 min at 0 °C using a lysis buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 2 mM Na-orthovanadate and a protease inhibitor cocktail (Complete Mini, Roche, Basel, Switzerland), as previously reported [25]. Briefly, the samples were processed under reducing and non-reducing conditions, heated for 5 min at 95 °C, and resolved by electrophoresis on pre-cast 4–12% and 3–8% Bis-Tris gels (Invitrogen, Carlsbad CA, USA). Separated proteins were electrophoretically transferred onto nitrocellulose filters, stained with Red Ponceau to check protein loading, washed extensively with TBS + 0.5% Tween-20 and saturated for 1 h at room temperature with 5% low-fat milk in TBS + 0.1% Tween-20 before the addition of primary antibodies in 3% low-fat milk containing TBS + 0.1% Tween-20 for 1 h at room temperature or overnight at 4 °C with gentle shaking. The following primary mouse mAb was used: Ab3 c-erbB-2/HER2/neu (1:300, Calbiochem, Darmstadt, Germany) directed against the human HER2 intracellular domain and the following primary rabbit polyclonal antibodies were used: anti-phospho-HER2 p-Neu (Tyr1248) (1:2000, Santa Cruz Biotechnology, Dallas, TX, USA), anti-Cleaved NOTCH1 (Val1744) (1:1000, Cell Signaling Technology, Leiden, Netherland) and anti-β-Actin-peroxidase (AC-15 clone) (1:30000, Sigma-Aldrich). After incubation, the filters were washed extensively with TBS + 0.5% Tween-20 and subsequently incubated with HRP-conjugated goat anti-mouse IgG (1:5000) or donkey anti-rabbit IgG (1:10000) (Amersham GE Healthcare, Little Chalfont, UK) for 1 h at room temperature. The resulting signals were detected using enhanced chemiluminescence (Pierce™ ECL Western Blotting Substrate, Thermo Fisher Scientific). Phosphorylated and basal protein expression levels were assessed relative to those of housekeeping proteins in the same sample. Phosphorylated protein expression levels were normalized relative to basal protein expression levels using Quantity One (Bio-Rad, Hercules, CA, USA).
2.9 High-resolution magnetic resonance spectroscopy (MRS)
Tissue extracts were prepared in EtOH:H2O (70:30, v/v) as reported before [37]. To this end, samples were ultra-sonicated at 20 kHz with an exponential probe (8 mm peak-to-peak) in a MSE ultrasonic disintegrator Mk2 (Crawley, Sussex, UK) and centrifuged at 14000×g for 30 min. The resulting supernatants were lyophilized twice in a RVT 4104 Savant lyophilizer (Mildford, Main, USA). For MRS analyses, the residues were resuspended in 0.7 ml D2O (Sigma-Aldrich) containing 0.1 mM 3(trimethylsilyl)-propionic-2,2,3,3-d4 acid sodium salt as an internal standard (Merck & Co, Montreal, Canada). High-resolution MRS analyses (25 °C) were performed at 9.4 T (Bruker AVANCE spectrometer, Karlsruhe, Germany). Spectra of tissue extracts were obtained using RF pulses for excitation, water signal pre-saturation, data processing and data analysis as reported before [38]. Relative metabolite quantifications are presented as percentage of an individual metabolite among all metabolites.
2.10 In vivo magnetic resonance imaging (MRI)/MRS
In vivo MRI/MRS analyses were performed in d16HER2 mammary tumors between 68 and 88 days post injection using an Agilent Inova MRI/MRS system (4.7 T) equipped with a volume coil for homogeneous transmission in combination with a receiver surface coil (RAPID Biomedical, Rimpar, Germany). MRI evaluation was performed by T1-weighted (TR/TE = 400/7 ms), T2-weighted (TR/TE = 3000/70 ms) and diffusion-weighted (TR/TE = 2500/50 ms, ranging between 0 and 1100 s/mm2) multi-slice spin echo images. Quantitative parameters related to water tissue diffusion (apparent diffusion coefficient (ADC) maps) were derived from diffusion weighted imaging (DWI). Histogram analyses of ADC values were also performed in order to study the biological heterogeneity of tumors by classifying domains of different diffusivity, which may have both prognostic and predictive implications. ADC mean, ADC median, kurtosis and skewness were determined from ADC histograms [Canese et al., unpublished data]. Kurtosis indicates how tall and sharp the peak is relative to a standard bell curve, i.e., how homogeneous the analyzed tumor is. Skewness provides a measure of the amount of departure from horizontal symmetry, which indicates the presence of areas with a slightly different ADC within a given tumor.
2.11 Transgenic and orthotopic mouse models
A breeding colony of d16HER2 transgenic mice, a mouse model that expresses the d16HER2 splice variant under the control of a MMTV promoter, was generated as reported before [24] and bred in the Animal Facility of Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy. All animal care and experimental procedures were approved by the Ethics Committee for Animal Experimentation of the Institute according to Italian law and the Italian Ministry of Health (Authorization code: INT 02/2014). DNA extracted from tail biopsies was used for routine genotyping by PCR analysis, as reported before [25]. Transgenic mice were inspected twice weekly by palpation and progressively growing masses ≥ 50 mm3 were scored as tumors. Susceptibility to PEITC, T and their combination was assessed in d16HER2-transgenic mice that were randomized at 8 weeks of age into four groups to be subjected to biweekly intra-peritoneal injections of 1 mg/kg of T (n = 6) and DMSO as a negative control (n = 8), as reported before [25]. PEITC (6 μmol/mouse) alone (n = 7) or in combination with T (1 mg/Kg/mouse) (n = 7) was orally (per os) administered every day for five days per week except for T (see above). Mice were sacrificed when the total tumor volumes reached 2,000 mm3. The anti-tumor activity of PEITC (6 μmol/mouse) alone (n = 11) was tested in syngeneic FVB mice grafted with 1 × 106 MI6 cells into the mammary fat pad (m.f.p.). When the tumors became palpable (~50 mm3) they were randomized into two groups to receive 6 μmol/mouse per os of PEITC every day (n = 11) and DMSO as a negative control (n = 8). FVB female mice (6–8 weeks old; body weight 20–25 g) were purchased from Charles River Laboratories (Charles River Laboratories, Wilhington, MA, USA). Tumors were calibrated twice weekly and tumor volumes were calculated as 0,5 x d12 x d2, where d1 and d2 are the smallest and largest diameters, respectively. Mice were sacrificed when the tumor volumes of the untreated group reached ~ 1,000 mm3 and of the PEITC-treated group ~ 250 mm3.
2.12 Immunohistochemistry (IHC)
MI6 lesions implanted into the m.f.p. of FVB mice were harvested at the end of the in vivo experiment and fixed in 10% neutral buffered formalin, paraffin embedded, sectioned and stained with hematoxylin and eosin (Bio-Optica, Milan, Italy) for histological examination. For IHC, the sections were deparaffinized, serially rehydrated and, after appropriate heat-induced antigen retrieval using sodium citrate buffer, pH 6.0, for 6 min in a pressure cooker, stained with an anti-active caspase 3 antibody (AF 835, R&D), followed by a Dako Envision + System-HRP Labelled Polymer Anti-Rabbit secondary antibody (K4003, Dako, Jena, Germany). Immunoreactive antigen was detected using a DAB Chromogen System (Dako) and, after chromogen incubation, the sections were counterstained with hematoxylin (BioOptica). Images were acquired using a Leica DMRD optical microscope (Leica, Wetzlar, Germany) at 100-200x magnification. ImageJ software (version 1.52 J) was used to evaluate the spheroids areas.
2.13 Statistical analysis
Statistical analyses were performed using GraphPad software Prism 5.02. Statistical significance was determined by two-tailed unpaired or paired Student’s t test. When p < 0.05, the difference between the compared groups was considered to be statistically significant. Data are presented as mean ± SD or SEM (n ≥ 3).
3 Results
3.1 PEITC targets HER2-positive CSCs
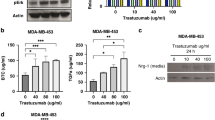
We incubated HER2-positive BT474, SKBR3, HCC1954 and SKOV3 cells, and TNBC MDA-MB-231 cells with serial dilutions of PEITC (from 48 to 3 μM) for 24, 48 and 72 h to test their susceptibility to PEITC (Supplementary Fig. 1). By doing so, we observed about 50% cell growth inhibition in all HER2-positive cell lines tested, regardless of their histotype, after incubation (IC50) with 12 μM PEITC for 48 and 72 h. Based on these findings, the 12 μM (IC50) concentration was used in all in vitro assays shown in Figs. 1 and 2. Of note, in the human TNBC cell line MDA-MB-231, which was included as an internal positive control, PEITC reached an IC50 at 8 and 6 μM after 48 and 72 h treatment, respectively (Supplementary Fig. 1). To investigate whether PEITC can target HER2-positive CSCs, we tested the % of cells expressing ALDH, a canonical CSC marker [39], following 72 h of treatment with PEITC (Fig. 1a; Supplementary Fig. 2). We found that PEITC significantly reduced the number of ALDH-positive cells by 50–70% versus control treatment (DMSO), with the percentage of inhibition varying across the different cell lines tested (Fig. 1a; Supplementary Fig. 2). To confirm the anti-CSC activity of PEITC, we evaluated the inhibition of SFE (%) of all cell lines after incubation in the presence or absence of PEITC for 7 days. We found that PEITC significantly reduced the % of generated spheres by 50% to 85%, depending to the cell line tested (Fig. 1b). Furthermore, we found that the self-renewal capacities of the BT474, SKBR3, HCC1954 and SKOV3 cell lines were significantly decreased upon PEITC treatment (12 μM) for 7 days (Fig. 1c). Additionally, we found that not only the number of spheres and their self-renewal capacity, but also the sizes of the spheres consistently decreased upon treatment with PEITC (Fig. 1d) as denoted by the calculated sphere areas (Fig. 1e). Together these data indicate that PEITC exerts a significant activity against HER2-positive CSCs.

Effect of PEITC on HER2-positive CSCs. a % of ALDH-positive BT474, SKBR3, HCC1954 and SKOV3 cells upon treatment with PEITC (12 μM) for 72 h. The values were normalized to the percentage of ALDH-positive cells in DMSO treated cells. The results are presented as mean ± SEM (n = 3). Significance was calculated using a two-tailed unpaired t-test. b Inhibition of SFE (%), normalized to internal controls, in BT474, SKBR3, HCC1954 and SKOV3 cells incubated for 7 days with PEITC (12 μM). The results are presented as mean ± SEM (n = 3). Significance was calculated using a two-tailed unpaired t-test. c Self renewal in BT474, SKBR3, HCC1954 and SKOV3 cells incubated with PEITC (12 μM) or DMSO. The results are presented as mean ± SEM (n = 3). Significance was calculated using a two-tailed unpaired t-test. d Representative images of spheres formed after 7 days in presence or not of PEITC (12 μM) for 7 days or DMSO under 3D conditions. Magnification 10x. e Normalized sphere areas of BT474, SKBR3, HCC1954 and SKOV3 cells. Significance was calculated using a two-tailed unpaired t-test

Dysregulation of HER2 and NOTCH1 expression/activation in human HER2-positive cells after PEITC treatment. a Wild type (WT) HER2 and b mutant d16HER2 relative expression levels evaluated by qRT–PCR analyses of BT474, SKBR3, HCC1954 and SKOV3 cells after treatment or not with PEITC (12 μM) for 48 and 72 h. The results are presented as the mean ± SEM (n = 4). Significance was calculated using a two-tailed paired t-test. c Western blot analyses of BT474, SKBR3, HCC1954 and SKOV3 protein extracts separated by 4–12% gradient SDS-PAGE under reducing conditions after treatment with PEITC (12 μM) (lanes P) or DMSO (lanes D) for 72 h to evaluate basal HER2 (HER2M), activated HER2 (pHER2M) and cleaved NOTCH1 proteins. ACTIN was used to normalize protein loading. Autoradiographs were acquired at different exposure times to obtain optimal image resolution. d Hes1 relative expression levels assessed by qRT-PCR analyses of BT474, SKBR3, HCC1954 and SKOV3 cells after treatment or not with PEITC (12 μM) for 72 h. The results are presented as mean ± SEM (n = 5). Significance was calculated using a two-tailed paired t-test
3.2 PEITC affects interplay between HER2 and the NOTCH signaling pathway
Since it was previously found that the d16HER2 variant facilitates HER2-driven tumor growth and stemness more efficiently than wild type HER2 (WTHER2) [19, 25], we set out to evaluate the expression of both WTHER2 and d16HER2 by qRT-PCR analysis in all target cells upon treatment with PEITC for 48 and 72 h. We found significantly decreased WTHER2 (Fig. 2a) and d16HER2 (Fig. 2b) transcript levels in all treated target cells compared to control treatment, thus supporting the notion that PEITC targets HER2-positive CSCs. To test whether the decrease in HER2 mRNA levels corresponds to reduced activation of the HER2 pathway, we evaluated by Western blotting total and phosphorylated (i.e., activated) levels of HER2 monomer (HER2M and pHER2M, respectively) at baseline (untreated, lane D) and after treatment with PEITC (lane P) for 72 h (Fig. 2c). Although PEITC treatment did not significantly modify total HER2 levels in BT474 and SKBR3 cells, it clearly reduced HER2 levels in HCC1954 and SKOV3 cells (P versus D). Of note, we found that PEITC treatment down-regulated the pHER2M level in all cell lines tested, albeit to a different extent. Consistent with data published by us and others suggesting the existence of a close interplay between HER2-activation and the NOTCH signaling pathway [17, 19, 40], we also found a decreased expression of activated NOTCH1 (cleaved NOTCH1) in all cell lines tested after 72 h of treatment with PEITC (Fig. 2c). To confirm that NOTCH1 signaling is affected by PEITC, we evaluated by qRT-PCR the expression of the Hes1 gene, one of its downstream targets that plays a critical role in self-renewal [41]. Consistent with a reduction in cleaved NOTCH1, we found a significant decrease in Hes1 mRNA levels in all cell lines tested (Fig. 2d). Overall, these results suggest that PEITC affects a CSC-related interplay between HER2 and the NOTCH signaling pathway.
3.3 PEITC affects d16HER2-driven signaling, stemness, in vivo tumor growth and metabolism
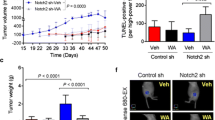
To further evaluate whether PEITC exerts an inhibitory activity only against WTHER2, or also against the d16HER2 splice isoform, we used a d16HER2-positive BC model that was previously characterized in our laboratory [19, 25]. In particular, we tested PEITC activity against both in vitro and in vivo transgenic d16HER2-positive models. MI6 cells, which express only the human d16HER2-positive splice isoform, were found to be less susceptible to PEITC than human HER2-positive cells, with an IC50 of ~48 μM and ~12 μM after 48 h and 72 h, respectively (Supplementary Fig. 3). To assess whether PEITC affects the expression (d16HER2D) and/or activation (pd16HER2D) of d16HER2 homodimers (D), we performed Western blot analyses under non-reducing conditions using MI6 cell extracts obtained after treating the cells with PEITC at 12 μM for 72 h. We found that PEITC (lane P) reduced the levels of pd16HER2D and pd16HER2M, whereas the basal d16HER2D and d16HER2M levels were only slightly affected versus control cell extracts (Fig. 3a, lane D). These data suggest that PEITC exerts inhibitory effects also against the d16HER2 variant that is constitutively expressed in all clinical and pre-clinical HER2-positive models, even if at variable levels [19, 22, 23, 25]. To investigate the capacity of PEITC to impair the generation of d16HER2-driven spheres, we performed SFE bioassays by treating MI6 cells with 12 μM PEITC for 7 days. We found that PEITC significantly inhibited the development of spheres by about 50% compared to control (DMSO) cells (Fig. 3b) (p < 0.0001). We next set out to test the in vivo therapeutic activity of PEITC upon injection of MI6 cells (1 × 106 per animal) into the m.f.p. of parental FVB female mice (Fig. 3c). PEITC (6 μmol/mouse) was administered daily (5 days/week; n = 11) starting when the mammary tumors became palpable until the mice were sacrificed. After more than 70 days following the initiation of treatment, we found that PEITC remarkably slowed down d16HER2-driven tumor growth compared to control treatment (i.e., diluent DMSO; n = 8) (p < 0.0001) (Fig. 3c). Hematoxylin-eosin staining of the tumor samples revealed areas of necrosis in PEITC-treated mice (Fig. 3d). In addition, in vivo T2-weighted MRI (Fig. 3e) revealed intra-tumor hemorrhagic (hypo-intense areas) and necrosis (hyper-intense areas) following PEITC treatment in comparison with the control group, thus suggesting that PEITC impairs tumor growth through a pro-apoptotic mechanism. In keeping with this hypothesis, we also observed an increase in the ADC parameter measured by DWI (related to extracellular water diffusion), which corresponds to a shift towards the right (higher ADC values) of the main peak in the ADC distribution (Fig. 3f). This suggests the presence of cellular death also in regions that appear as normal in conventional T2-weighted MRI. This shift was attributed to the presence of increased, diffused extracellular volumes, possibly due to necrosis apoptosis within the tumors, thus confirming the anti-tumor activity of PEITC [42].

Effect of PEITC on d16HER2-driven signaling, stemness and in vivo tumor growth. a Western blot analysis of MI6 protein extracts separated by 3–8% gradient SDS-PAGE under non-reducing conditions after treatment (lanes P) or not (lanes D) with PEITC (12 μM) for 72 h to evaluate basal (HER2) and activated (pHER2) HER2 monomers (M) and homodimers (D). ACTIN was used to normalize protein loading. Autoradiographs were acquired at different exposure times to obtain optimal image resolution. b SFE (%), normalized to internal controls (DMSO), in MI6 cells incubated for 7 days with PEITC (12 μM). The results are presented as mean ± SEM (n = 3). Significance was calculated using a two-tailed unpaired t-test. c PEITC-mediated anti-tumor growth activity in FVB mice implanted with MI6 cells into the m.f.p. and treated (n = 11) or not (n = 8) with PEITC (6 μmol/mouse). Data are presented as mean ± SEM. Significance was calculated using a unpaired t-test. d Representative hematoxylin and eosin staining of MI6 nodules from mice treated with DMSO or PEITC. Tumor areas with necrotic phenotypes are marked by a black line, magnification 20X. e Representative examples of a coronal slice of T2-weighted MRI (left) and ADC map (right) of PEITC-treated and DMSO-treated xenografts acquired during treatment. Hypo-intense areas indicate hemorrhage while hyper-intense areas are mainly due to necrosis. f Comparison of the distribution of ADC values in PEITC-treated versus DMSO-treated xenografts
3.4 PEITC potentiates T activity in d16HER2-positive pre-clinical transgenic models
Aiming to test whether or not PEITC exhibits additive anticancer activity when combined with T, the standard-of-care treatment for HER2-positive BC in all disease stages, we treated MI6 cells with PEITC, T, or with a PEITC+T combination, and tested their SFE% (Fig. 4a). In these experiments, PEITC was used at a lower concentration (6 μM) than in previous in vitro assays, thus increasing the chance of observing a potential additive or synergistic effect between the two treatment modalities. At the drug concentrations used in this experiment, we found that PEITC reduced the SFE% significantly more efficiently than T when both compounds were used as monotherapy. Remarkably, the PEITC+T combination readily inhibited sphere formation (p < 0.0001) (Fig. 4a), as well as sphere dimensions compared to DMSO (Supplementary Fig. 4). To investigate whether the PEITC+T combination synergistically prevents d16HER2-driven CSC generation, we evaluated the number and proportion of CD29high/CD24+/SCA-1low MI6 cells, a subset enriched in mammary stem cells [43], following treatment with PEITC, T or the PEITC+T combination for 72 h (Fig. 4b). In keeping with the SFE% data (Fig. 4a), we found that the combination strategy significantly reduced the percentage of CD29high/CD24+/SCA-1low cells (p = 0.0308), as revealed by multiparametric FACS analysis (Fig. 4b and Supplementary Fig. 5). To confirm the relevance of these findings in an in vivo setting, we evaluated the effect of PEITC on the occurrence of d16HER2-positive mammary lesions in transgenic animals that develop spontaneous palpable tumors with a rapid onset and an average latency of 15.11 weeks of age [24]. At the 8th week of age, i.e., when non palpable mammary lesions are already developing [24], the mice were divided into four treatment groups: control (DMSO), PEITC, T or PEITC+T. As expected, all mice in the control treatment arm (n = 8) developed tumors between the 10th and 20th weeks of age (Fig. 4c). In addition, we found that PEITC alone (n = 7) delayed tumor appearance by a median of 5 weeks (p = 0,0657), while T alone (n = 6) was more effective than PEITC in delaying the formation of detectable nodules (p = 0,0024). However all animals treated with PEITC or T alone finally developed mammary tumors within 31 weeks of age. Notably, the PEITC+T combination significantly delayed the onset of spontaneous lesions compared to both the control group and the single treatment groups (p < 0,0001) (Supplementary Table 2). Of note, we found that ~40% of mice receiving the combination strategy was cancer-free at the end of the experiment (~50 weeks of age) (Fig. 4c). IHC analyses performed in tumor samples arising in transgenic mice treated with or without PEITC (6 μmol/mouse), T (1 mg/kg/mouse) and the combination PEITC+T revealed numerous and large zones of extensive cell damage with positivity for the active (cleaved) form of caspase 3 mainly in the PEITC+T group compared to the PEITC and T alone groups. Only sporadic positive cells were noted in samples derived from control mice (Fig. 4d). Furthermore, we found that the metabolic profiles of d16HER2-positive tumors excised at the end of the in vivo treatments showed significantly increased lactate levels when treated with a PEITC+T combination (p = 0.003), but not statistically significant for the individual (PEITC and T) treatments, compared to DMSO alone (Fig. 4e; p = 0.06). This suggests that the treatments cause mitochondrial dysfunction that may be associated with apoptosis [44]. In fact, loss of mitochondrial function during apoptosis may lead to compensatory glycolysis and the accumulation of lactate [45]. Together, the in vivo data presented here indicate that single-agent PEITC has a relatively weak antitumor/tumor preventing activity, but that its combination with T shows synergistic effects in terms of delaying, or even preventing, the formation of HER2-positive BC.

Therapeutic effects of PEITC combined with T in in vitro and in vivo transgenic d16HER2-positive pre-clinical models. a Inhibition of SFE (%), normalized to internal controls, in MI6 cells treated with T (50 μg/ml), PEITC (6 μM) and their combination. The results are presented as mean ± SEM (n = 3). Significance was calculated using a two-tailed unpaired t-test. b Frequency of CD29High/CD24+ SCA1Low cells in MI6 cells upon treatment with DMSO, T, PEITC and their combination evaluated by multiparametric FACS analysis The results are presented as mean ± SEM (n = 2). Significance was calculated using a two-tailed unpaired t-test. c d16HER2 transgenic tumor-free mice (%) after treatment with PEITC (n = 7) (6 μmol/animal) and T (n = 6) alone (1 mg/kg/animal), their combination (n = 7) or left untreated (DMSO) (n = 8). Treatments started at the 8th week of age. Mice were injected with PEITC (per os) every day and with T (i.p.) twice a week. Differences were assessed by Log-rank test. d Representative IHC staining of cleaved Caspase 3 (brown cells) of d16HER2-positive lesions from mice treated with DMSO, PEITC, T and PEITC+T combination. Magnification 40x and resolution 600 dpi. e Lactate levels detected by high resolution MRS (9.4 T) in d16HER2-positive mammary tumor lysates after treatment with DMSO (n = 5), PEITC (n = 6), T (n = 6) and PEITC+T combination (n = 4). The results are presented as mean ± SEM. Significance was calculated using a two-tailed unpaired t-test
4 Discussion
HER2-positive cancers are characterized by a high clinical aggressiveness, a high metastatic potential and a poor prognosis in the absence of anti-HER2 treatment. In particular, HER2-positive BC was associated with a notorious low patient survival before the advent of anti-HER2 treatment options [3]. Although T, pertuzumab, lapatinib and T-DM1 have considerably improved HER2-positive BC patient prognosis in recent years, there is still a certain percentage of patients in which the disease recurs after surgery and adjuvant treatments. In addition, almost all patients with metastatic HER2-positive BC still die of the disease.
Emerging data indicate that the clinical benefit provided by anti-HER2 therapies is mostly related to their ability to efficiently target CSCs. Improving the targeting of CSCs is, therefore, considered to be a high priority goal to achieve long-lasting tumor control and/or eradication and, eventually, cure [6]. Therefore, the development of anti-tumor strategies that are able to concomitantly target CSCs, the “roots” of cancer, is of pivotal importance to improve the outcome of patients with HER2-positive cancer [46].
Several epidemiological and case-control studies suggest that a high intake of cruciferous vegetables may reduce the risk of cancer development, presumably due to the presence of ITCs [29, 47,48,49]. Several preclinical studies have also shown broad anticancer effects of PEITC, thus suggesting that it may exert an activity directed against various tumor cells [50, 51]. In addition, Li and coworkers have shown that sulforaphane targets TNBC BCSCs [30] and Gupta and colleagues have reported that PEITC exerts anti-tumor activity against luminal and TNBC cells engineered to overexpress HER2 [34]. However, no data are available on cell lines constitutively overexpressing HER2 and/or its d16HER2 splice variant. Our in vitro and in vivo data provide the first evidence that PEITC may efficiently target HER2-positive CSCs. We found that PEITC consistently reduced SFE% and sphere dimensions, thus indicating cytotoxic and cytostatic effects against CSCs. These findings are consistent with previous results obtained by others in TNBC [30] and colorectal cancer [52], which supports the use of sulforaphane for eradicating CSCs. We also found that PEITC negatively modulates the activation of WTHER2 and d16HER2, two key regulators of HER2-positive breast CSC properties [16,17,18,19], thus suggesting a potential anti-CSC activity mediated by inhibition of oncogenic signaling downstream of both HER2 isoforms.
Consistent with literature data indicating a direct bidirectional functional crosstalk between HER2 and CSC-related NOTCH signaling [17, 19, 40], we found that PEITC reduces NOTCH1 activation by acting as a negative modulator of crosstalk between HER2 and the NOTCH pathway, known to be critical for the self-renewal of CSCs [53]. The observed decrease in expression of the nuclear target Hes1, a well-established key regulator of CSC self-renewal, tumor progression and metastasis downstream of NOTCH [41], indeed indicates that PEITC may inhibit BCSCs and down-regulate the NOTCH self-renewal pathway.
In the in vivo setting, we observed by in vivo MRI a long-term efficacy of PEITC as a single agent against MI6 cells orthotopically implanted in syngeneic animals, both in terms of tumor size and composition. This observation confirms previous findings of a potential role of this compound in eradicating HER2-positive CSC clones, which may be responsible for tumor resistance to pharmacological treatment. This interpretation is supported by our finding that PEITC, and in particular the PEITC+T combination, impaired the SFE% of MI6 cells and reduced the number and frequency of mammary tumor cells with a CD29high/CD24+/SCA-1low immunophenotype, a cell population known to be enriched for murine mammary stem cells [43].
Finally, we observed a potent therapeutic potential of PEITC in d16HER2 transgenic mice, a strain reported to develop highly aggressive asynchronous spontaneous palpable mammary lesions after the 8th week of age [24, 25]. We found that in this model the PEITC+T combination prevented tumor formation in ~40% of the treated mice, while tumor formation invariably occurred in the control group or in mice receiving single treatments. We speculate that PEITC and T exert synergistic anticancer effects by targeting different populations of cancer cells, i.e., the bulk of proliferating cancer cells and CSCs, respectively. Collectively, these findings support not only the chemo-preventive/chemotherapeutic effects exerted by PEITC [29, 47,48,49] even in the highly oncogenic d16HER2-positive pre-clinical model, but also its effectiveness in differentiated HER2-positive tumor cells.
We also observed different in vivo anti-cancer activities of single-agent PEITC according to the treatment setting, i.e., in the orthotopic model, PEITC considerably delayed the growth of already established tumors, while only a mild delay in the formation of palpable tumors was observed in the transgenic d16HER2 model. Different hypotheses may explain these discordant results in distinct in vivo settings: 1) differences in tumor biology and drug pharmacokinetics in tumors growing subcutaneously or arising spontaneously in mammary tissue; 2) basic biological differences in terms of HER2 addiction in the two settings, which may require the use of T in combination with PEITC in the transgenic mouse model; 3) while in the orthotopic setting the inoculated tumor cells are derived from the same cell clone, the tumors arising in the transgenic mouse model may be more heterogeneous, genetically and/or epigenetically [25]. Increasing evidence underscores the coexistence of genetically distinct intra-tumor CSC clones, which may result in therapy resistance [54] as well as CSC plasticity during therapy [28].
In conclusion, we found that PEITC acts synergistically with T to reduce HER2 activation, sphere formation and in vivo growth of d16HER2-positive mammary cancer cells, thereby contributing to our understanding of anti-cancer activities of ITCs. Our data reinforce previously published findings on the efficacy of ITCs in enhancing the activity of different cytotoxic agents [55] and to exert additive effects in combination with TRAIL in a pancreatic xenograft model reducing the proportion of CSCs [56]. Our data also indicate that PEITC in combination with T may represent a promising approach to target HER2-positive BC cells and eradicate HER2-positive CSCs. In recent years, chemotherapy-free strategies to treat HER2-positive BC have shown antitumor efficacies and a good tolerability, in particular for the combination of endocrine treatment and single or dual HER2 blockade in the subgroup of patients with hormone receptor- and HER2-positive disease [57, 58]. Additionally, we show that PEITC effectively targets in vitro the stemness compartment of HER2-positive OC cells, thus supporting its putative applicability also in other HER2-driven cancer types. Of note, PEITC is a natural compound with minimal or no toxicity that can be produced at a low cost [32]. Several studies have shown anti-cancer effects of PEITC in various cancer types [32], which may be elicited either by the generation of reactive oxygen species or by the induction of cell cycle arrest and, ultimately apoptosis [29]. Within this perspective, phase I trials to determine the maximum tolerated doses of PEITC that are active against human HER2-positive BCs are warranted.
References
S. Parakh, H.K. Gan, A.C. Parslow, I.J.G. Burvenich, A.W. Burgess, A.M. Scott, Evolution of anti-HER2 therapies for cancer treatment. Cancer Treat. Rev. 59, 1–21 (2017). https://doi.org/10.1016/j.ctrv.2017.06.005
B.N. Rexer, C.L. Arteaga, Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: Mechanisms and clinical implications. Crit. Rev. Oncog. 17, 1–16 (2012)
S. Loibl, L. Gianni, HER2-positive breast cancer. Lancet 389, 2415–2429 (2017). https://doi.org/10.1016/S0140-6736(16)32417-5
P. Samadi, S. Saki, F.K. Dermani, M. Pourjafar, M. Saidijam, Emerging ways to treat breast cancer: Will promises be met? Cell. Oncol. (Dordr). 41(605–621), 605–621 (2018). https://doi.org/10.1007/s13402-018-0409-1
T. Triulzi, G.V. Bianchi, E. Tagliabue, Predictive biomarkers in the treatment of HER2-positive breast cancer: An ongoing challenge. Future Oncol. 12, 1413–1428 (2016). https://doi.org/10.2217/fon-2015-0025
C.T. Jordan, M.L. Guzman, M. Noble, Cancer stem cells. N. Engl. J. Med. 355, 1253–1261 (2006). https://doi.org/10.1056/NEJMra061808
H. Korkaya, S. Liu, M.S. Wicha, Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Invest. 121, 3804–3809 (2011). https://doi.org/10.1172/JCI57099
H. Korkaya, M.S. Wicha, Breast cancer stem cells: we've got them surrounded. Clin. Cancer Res. 19, 511–513 (2013). https://doi.org/10.1158/1078-0432.CCR-12-3450
R. Castro-Oropeza, J. Melendez-Zajgla, V. Maldonado, K. Vazquez-Santillan, The emerging role of lncRNAs in the regulation of cancer stem cells. Cell. Oncol. (Dordr) 41(585–603), 585–603 (2018). https://doi.org/10.1007/s13402-018-0406-4
H. Korkaya, M.S. Wicha, HER2 and breast cancer stem cells: more than meets the eye. Cancer Res. 73, 3489–3493 (2013). https://doi.org/10.1158/0008-5472.CAN-13-0260
S. Annett, T. Robson, Targeting cancer stem cells in the clinic: Current status and perspectives. Pharmacol. Ther. 187, 13–30 (2018). https://doi.org/10.1016/j.pharmthera.2018.02.001
S.G. Pohl, N. Brook, M. Agostino, F. Arfuso, A.P. Kumar, A. Dharmarajan, Wnt signaling in triple-negative breast cancer. Oncogenesis 6, e310 (2017). https://doi.org/10.1038/oncsis.2017.14
N. Shafee, C.R. Smith, S. Wei, Y. Kim, G.B. Mills, G.N. Hortobagyi, E.J. Stanbridge, E.Y. Lee, Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res. 68, 3243–3250 (2008). https://doi.org/10.1158/0008-5472.CAN-07-5480
D. Hambardzumyan, M. Squatrito, E.C. Holland, Radiation resistance and stem-like cells in brain tumors. Cancer Cell 10, 454–456 (2006). https://doi.org/10.1016/j.ccr.2006.11.008
L. Castagnoli, V. Cancila, S.L. Romero-Cordoba, S. Faraci, G. Talarico, B. Belmonte, M.V. Iorio, T. Volpari, C. Chiodoni, A. Hidalgo-Miranda, E. Tagliabue, C. Tripodo, S. Sangaletti, M. Di Nicola, S.M. Pupa, WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 38, 4047–4060 (2019). https://doi.org/10.1038/s41388-019-0700-2
H. Korkaya, A. Paulson, F. Iovino, M.S. Wicha, HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 27, 6120–6130 (2008). https://doi.org/10.1038/onc.2008.207
A. Magnifico, L. Albano, S. Campaner, D. Delia, F. Castiglioni, P. Gasparini, G. Sozzi, E. Fontanella, S. Ménard, E. Tagliabue, Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to Trastuzumab. Clin. Cancer Res. 15, 2010–2021 (2009). https://doi.org/10.1158/1078-0432.CCR-08-1327
G. Farnie, R.L. Johnson, K.E. Williams, R.B. Clarke, N.J. Bundred, Lapatinib inhibits stem/progenitor proliferation in preclinical in vitro models of ductal carcinoma in situ (DCIS). Cell Cycle 13, 418–425 (2014). https://doi.org/10.4161/cc.27201
L. Castagnoli, G.C. Ghedini, A. Koschorke, T. Triulzi, M. Dugo, P. Gasparini, P. Casalini, A. Palladini, M. Iezzi, A. Lamolinara, P.L. Lollini, P. Nanni, C. Chiodoni, E. Tagliabue, S.M. Pupa, Pathobiological implications of the d16HER2 splice variant for stemness and aggressiveness of HER2-positive breast cancer. Oncogene 36, 1721–1732 (2017). https://doi.org/10.1038/onc.2016.338
K.Y. Kwong, M.C. Hung, A novel splice variant of HER2 with increased transformation activity. Mol. Carcinog. 23, 62–68 (1998)
F. Castiglioni, E. Tagliabue, M. Campiglio, S.M. Pupa, A. Balsari, S. Ménard, Role of exon-16-deleted HER2 in breast carcinomas. Endocr. Relat. Cancer 13, 221–232 (2006). https://doi.org/10.1677/ERC.1.01047
D. Mitra, M.J. Brumlik, S.U. Okamgba, Y. Zhu, T.T. Duplessis, J.G. Parvani, S.M. Lesko, E. Brogi, F.E. Jones, An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol. Cancer Ther. 8, 2152–2162 (2009). https://doi.org/10.1158/1535-7163.MCT-09-0295
C.C. Volpi, F. Pietrantonio, A. Gloghini, G. Fuca, S. Giordano, S. Corso, G. Pruneri, M. Antista, C. Cremolini, E. Fasano, S. Saggio, S. Faraci, M. Di Bartolomeo, F. De Braud, M. Di Nicola, E. Tagliabue, S.M. Pupa, L. Castagnoli, The landscape of d16HER2 splice variant expression across HER2-positive cancers. Sci. Rep. 9, 3545 (2019). https://doi.org/10.1038/s41598-019-40310-5
C. Marchini, F. Gabrielli, M. Iezzi, S. Zanobi, M. Montani, L. Pietrella, C. Kalogris, A. Rossini, V. Ciravolo, L. Castagnoli, E. Tagliabue, S.M. Pupa, P. Musiani, P. Monaci, S. Ménard, A. Amici, The human splice variant delta16HER2 induces rapid tumor onset in a reporter transgenic mouse. PLoS One 6, e18727 (2011). https://doi.org/10.1371/journal.pone.0018727
L. Castagnoli, M. Iezzi, G.C. Ghedini, V. Ciravolo, G. Marzano, A. Lamolinara, R. Zappasodi, P. Gasparini, M. Campiglio, A. Amici, C. Chiodoni, A. Palladini, P. Lollini, T. Triulzi, S. Ménard, P. Nanni, E. Tagliabue, S.M. Pupa, Activated d16HER2 homodimers and Src kinase mediate optimal efficacy for trastuzumab. Cancer Res. 74, 6248–6259 (2014). https://doi.org/10.1158/0008-5472.CAN-14-0983
A. Alajati, N. Sausgruber, N. Aceto, S. Duss, S. Sarret, H. Voshol, D. Bonenfant, M. tires-Alj, Mammary tumor formation and metastasis evoked by a HER2 splice variant. Cancer Res. 73, 5320–5327 (2013). https://doi.org/10.1158/0008-5472.CAN-12-3186
L. Castagnoli, E. Iorio, M. Dugo, A. Koschorke, S. Faraci, R. Canese, P. Casalini, P. Nanni, C. Vernieri, M. Di Nicola, D. Morelli, E. Tagliabue, S.M. Pupa, Intra-tumor lactate levels reflect HER2 addiction status in HER2-positive breast cancer. J. Cell. Physiol. 234, 1768–1779 (2019). https://doi.org/10.1002/jcp.27049
M.R. Doherty, J.M. Smigiel, D.J. Junk, M.W. Jackson, Cancer stem cell plasticity drives therapeutic resistance. Cancers (Basel) 8 (2016). https://doi.org/10.3390/cancers8010008
P. Gupta, B. Kim, S.H. Kim, S.K. Srivastava, Molecular targets of isothiocyanates in cancer: Recent advances. Mol. Nutr. Food Res. 58, 1685–1707 (2014). https://doi.org/10.1002/mnfr.201300684
Y. Li, T. Zhang, H. Korkaya, S. Liu, H.F. Lee, B. Newman, Y. Yu, S.G. Clouthier, S.J. Schwartz, M.S. Wicha, D. Sun, Sulforaphane, a dietary component of broccoli/broccoli sprouts, inhibits breast cancer stem cells. Clin. Cancer Res. 16, 2580–2590 (2010). https://doi.org/10.1158/1078-0432.CCR-09-2937
D. Hanahan, R.A. Weinberg, Hallmarks of cancer: The next generation. Cel. 144, 646–674 (2011). https://doi.org/10.1016/j.cell.2011.02.013
P. Gupta, S.E. Wright, S.H. Kim, S.K. Srivastava, Phenethyl isothiocyanate: A comprehensive review of anti-cancer mechanisms. Biochim. Biophys. Acta 1846, 405–424 (2014). https://doi.org/10.1016/j.bbcan.2014.08.003
K.S. Satyan, N. Swamy, D.S. Dizon, R. Singh, C.O. Granai, L. Brard, Phenethyl isothiocyanate (PEITC) inhibits growth of ovarian cancer cells by inducing apoptosis: Role of caspase and MAPK activation. Gynecol. Oncol. 103, 261–270 (2006). https://doi.org/10.1016/j.ygyno.2006.03.002
P. Gupta, S.K. Srivastava, Antitumor activity of phenethyl isothiocyanate in HER2-positive breast cancer models. BMC Med. 10, 80 (2012). https://doi.org/10.1186/1741-7015-10-80
G.C. Ghedini, V. Ciravolo, M. Tortoreto, S. Giuffre, F. Bianchi, M. Campiglio, M. Mortarino, M. Figini, A. Coliva, M.L. Carcangiu, M. Zambetti, T. Piazza, S. Ferrini, S. Menard, E. Tagliabue, S.M. Pupa, Shed HER2 extracellular domain in HER2-mediated tumor growth and in trastuzumab susceptibility. J. Cell. Physiol. 225, 256–265 (2010). https://doi.org/10.1002/jcp.22257
F.L. Shaw, H. Harrison, K. Spence, M.P. Ablett, B.M. Simoes, G. Farnie, R.B. Clarke, A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J. Mammary Gland Biol. Neoplasia 17, 111–117 (2012). https://doi.org/10.1002/jcp.22257
F. Granata, E. Iorio, G. Carpinelli, M. Giannini, F. Podo, Phosphocholine and phosphoethanolamine during chick embryo myogenesis: A (31)P-NMR study. Biochim. Biophys. Acta 1483, 334–342 (2000)
M.E. Pisanu, A. Ricci, L. Paris, E. Surrentino, L. Liliac, M. Bagnoli, S. Canevari, D. Mezzanzanica, F. Podo, E. Iorio, R. Canese, Monitoring response to cytostatic cisplatin in a HER2(+) ovary cancer model by MRI and in vitro and in vivo MR spectroscopy. Br. J. Cancer 110, 625–635 (2014). https://doi.org/10.1038/bjc.2013.758
C. Ginestier, M.H. Hur, E. Charafe-Jauffret, F. Monville, J. Dutcher, M. Brown, J. Jacquemier, P. Viens, C.G. Kleer, S. Liu, A. Schott, D. Hayes, D. Birnbaum, M.S. Wicha, G. Dontu, ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1, 555–567 (2007). https://doi.org/10.1016/j.stem.2007.08.014
A.T. Baker, A. Zlobin, C. Osipo, Notch-EGFR/HER2 bidirectional crosstalk in breast Cancer. Front. Oncol. 4, 360 (2014). https://doi.org/10.3389/fonc.2014.00360
Z.H. Liu, X.M. Dai, B. Du, Hes1: A key role in stemness, metastasis and multidrug resistance. Cancer Biol. Ther. 16, 353–359 (2015). https://doi.org/10.1080/15384047.2015.1016662
H.C. Thoeny, B.D. Ross, Predicting and monitoring cancer treatment response with diffusion-weighted MRI. J. Magn. Reson. Imaging 32, 2–16 (2010). https://doi.org/10.1002/jmri.22167
M. Shackleton, F. Vaillant, K.J. Simpson, J. Stingl, G.K. Smyth, M.L. sselin-Labat, L. Wu, G.J. Lindeman, J.E. Visvader, Generation of a functional mammary gland from a single stem cell. Nature 439, 84–88 (2006). https://doi.org/10.1038/nature04372
E. Iorio, M. Di Vito, F. Spadaro, C. Ramoni, E. Lococo, R. Carnevale, L. Lenti, R. Strom, F. Podo, Triacsin C inhibits the formation of 1H NMR-visible mobile lipids and lipid bodies in HuT 78 apoptotic cells. Biochim. Biophys. Acta 1634, 1–14 (2003)
M. Tiefenthaler, A. Amberger, N. Bacher, B.L. Hartmann, R. Margreiter, R. Kofler, G. Konwalinka, Increased lactate production follows loss of mitochondrial membrane potential during apoptosis of human leukaemia cells. Br. J. Haematol. 114, 574–580 (2001)
G. Xu, J. Shen, Y. Ou, X, M. Sasahara, X. Su, Cancer stem cells: The 'heartbeat' of gastric cancer. J. Gastroenterol. 48, 781–797 (2013). https://doi.org/10.1007/s00535-012-0712-y
S. Palmer, Diet, nutrition, and cancer. Prog. Food Nutr. Sci. 9, 283–341 (1985)
D.T. Verhoeven, R.A. Goldbohm, P.G.H. van Verhagen, P.A. Van den Brandt, Epidemiological studies on brassica vegetables and cancer risk. Cancer Epidemiol. Biomark. Prev. 5, 733–748 (1996)
J.V. Higdon, B. Delage, D.E. Williams, R.H. Dashwood, Cruciferous vegetables and human cancer risk: Epidemiologic evidence and mechanistic basis. Pharmacol. Res. 55, 224–236 (2007). https://doi.org/10.1016/j.phrs.2007.01.009
C.B. Ambrosone, S.E. McCann, J.L. Freudenheim, J.R. Marshall, Y. Zhang, P.G. Shields, Breast cancer risk in premenopausal women is inversely associated with consumption of broccoli, a source of isothiocyanates, but is not modified by GST genotype. J. Nutr. 134, 1134–1138 (2004)
D.A. Boggs, J.R. Palmer, L.A. Wise, D. Spiegelman, M.J. Stampfer, L.L. ms-Campbell, L. Rosenberg, Fruit and vegetable intake in relation to risk of breast cancer in the Black Women's Health Study. Am. J. Epidemiol. 172, 1268–1279 (2010). https://doi.org/10.1093/aje/kwq293
Y. Chen, Y. Li, X.Q. Wang, Y. Meng, Q. Zhang, J.Y. Zhu, J.Q. Chen, W.S. Cao, X.Q. Wang, C.F. Xie, X.T. Li, S.S. Geng, J.S. Wu, C.Y. Zhong, H.Y. Han, Phenethyl isothiocyanate inhibits colorectal cancer stem cells by suppressing Wnt/beta-catenin pathway. Phytother. Res. 32, 2447–2455 (2018). https://doi.org/10.1002/ptr.6183
G. Dontu, K.W. Jackson, E. McNicholas, M.J. Kawamura, W.M. Abdallah, M.S. Wicha, Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 6, R605–R615 (2004)
A. Marusyk, K. Polyak, Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 1805, 105–117 (2010). https://doi.org/10.1016/j.bbcan.2009.11.002
J.P. Burnett, G. Lim, Y. Li, R.B. Shah, R. Lim, H.J. Paholak, S.P. McDermott, L. Sun, Y. Tsume, S. Bai, M.S. Wicha, D. Sun, T. Zhang, Sulforaphane enhances the anticancer activity of taxanes against triple negative breast cancer by killing cancer stem cells. Cancer Lett. 394, 52–64 (2017). https://doi.org/10.1016/j.canlet.2017.02.023
G. Kallifatidis, V. Rausch, B. Baumann, A. Apel, B.M. Beckermann, A. Groth, J. Mattern, Z. Li, A. Kolb, G. Moldenhauer, P. Altevogt, T. Wirth, J. Werner, P. Schemmer, M.W. Buchler, A.V. Salnikov, I. Herr, Sulforaphane targets pancreatic tumour-initiating cells by NF-kappaB-induced antiapoptotic signalling. Gut 58, 949–963 (2009). https://doi.org/10.1136/gut.2008.149039
Arpino, G., Ferrero, J. M., de la Haba-Rodriguez J., Easton, V., Schuhmacher, C., Restuccia, E. et al. Primary analysis of PERTAIN: a randomized, two-arm, open-label, multicenter phase II trial assessing the efficacy and safety of pertuzumab given in combination with trastuzumab plus an aromatase inhibitor in first-line patients with HER2-positive and hormone receptor-positive metastatic or locally advanced breast cancer. San Antonio Breast Cancer Symposium; December 6–10, 2016 San Antonio, Texas. 2017
S.R.D. Johnston, R. Hegg, S.A. Im, I.H. Park, O. Burdaeva, G. Kurteva, M.F. Press, S. Tjulandin, H. Iwata, S.D. Simon, S. Kenny, S. Sarp, M.A. Izquierdo, L.S. Williams, W.J. Gradishar, Phase III, randomized study of dual human epidermal growth factor receptor 2 (HER2) blockade with Lapatinib plus Trastuzumab in combination with an aromatase inhibitor in postmenopausal women with HER2-Positive, hormone receptor-positive metastatic Breast Cancer: ALTERNATIVE. J. Clin. Oncol. 36, 741–748 (2018). https://doi.org/10.1200/JCO.2017.74.7824
Acknowledgments
The authors thank Mrs. C. Ghirelli for technical assistance, the Flow Cytometry and Cell Sorting Facility for their support in the cytofluorimetric analyses, Dr. P. Casalini for her support in the ImageJ analyses, Mrs. L. Mameli, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, for secretarial assistance and Dr. M. Borghi, Istituto Superiore di Sanità, Rome, for her contribution to the in vivo MRI study. This work was funded by grants from the Associazione Italiana Ricerca Cancro (AIRC; call 2010) 10352 to S.M. Pupa and Ministero Italiano della Salute RF-2009-1532281 to S.M. Pupa.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 16 kb)
ESM 2
(DOCX 12 kb)
Supplementary Figure 1
Susceptibility of human HER2-positive BC cell lines to PEITC treatment. Cell growth of BT474, SKBR3, HCC1954, MDAMB231 and SKOV3 cell lines cultured under 2D conditions evaluated by SRB assay. MDAMB231 cell line was used as positive control for in vitro PEITC activity. Cells (3,000/well) were cultured and treated per 24, 48 and 72 h in presence of increasing concentrations of PEITC diluted in DMSO. (PDF 26 kb)
Supplementary Figure 2
Representative plots of ALDH-positive cells in BT474, SKBR3, HCC1954 and SKOV3 targets upon treatment with PEITC (12 μM) for 72 h. (PDF 231 kb)
Supplementary Figure 3
Susceptibility of MI6 cells to PEITC treatment. Cell growth of MI6 cell line cultured under 2D conditions evaluated by SRB assay. Cells (5,000/well) were cultured and treated per 24, 48 and 72 h in presence of increasing concentrations of PEITC diluted in DMSO. (PDF 102 kb)
Supplementary Figure 4
Representative images of morphology and size of MI6 mammospheres formed after 7 days in presence or not of T, PEITC and PEITC-T combination. Magnification 10X. (PDF 99 kb)
Supplementary Figure 5
Multiparametric FACS analysis of the CD29High/CD24+ SCA1Low stem cell subset in MI6 treated with DMSO, T, PEITC, and their combination (PEITC+T). (PDF 117 kb)
Rights and permissions
About this article

Cite this article
Koschorke, A., Faraci, S., Giani, D. et al. Phenethyl isothiocyanate hampers growth and progression of HER2-positive breast and ovarian carcinoma by targeting their stem cell compartment. Cell Oncol. 42, 815–828 (2019). https://doi.org/10.1007/s13402-019-00464-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-019-00464-w