
Abstract
A novel xylanase gene (denominated xynDRTY1) was identified from Tengchong hot spring by a metagenomic approach. Its amino acid sequence was 73.43% identical to a hypothetical protein from Bryobacterales bacterium. The codon-optimized XynDRTY1 gene was synthesized and overexpressed in Escherichia coli. The XynDRTY1 was purified by using Ni–NTA affinity chromatography. It exhibited activity with natural glycosides, such as beechwood xylan (21.2 ± 3 U/mg) and oat spelt xylan (8.2 ± 0.3 U/mg). Its optimum pH was determined to be 6.0 and optimum temperature of 65 ℃, along with its stability over 140% and 110% relative enzyme activity after incubation at 60 ℃ for 20 min and 120 min, respectively. Based on these findings, we believe that XynDRTY1, as thermostable xylanase, may prove useful for biotechnological applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Xylan, as the main constituent of hemicellulose, consists of xylose monomers connected by beta-1,4-glycoside linkages. Its contents are different in various plants, such as 30–35% in hardwoods, 15–30% in graminaceous plants, and 7–12% in gymnosperms [1]. At present, xylan is widely used in the production of biofuels and bio-based chemicals [2]. Xylanase can hydrolyze xylan into xylo-oligosaccharides for application in various industries such as pharmaceuticals, animal feed, paper, biofuels, and waste treatment [3]. In many application processes, the thermal stability of xylanase is required. The use of thermophilic xylanase is conducive to reducing costs and improving efficiency.
However, existing xylanases, which are mainly derived from fungi, exhibited low thermal stability, like xylanases from Trichoderma reesei lost activity when the temperature was over 55 ℃ [4]. In a typical high-temperature environment [5], hot springs are an important source of various thermophilic xylanases [6]. While less than 1% of identified prokaryotes can be cultured in the existing laboratory pure culture techniques [7, 8]. At present, with the development of metagenomic sequencing technology, a large number of novel glycoside hydrolase sequences have been obtained, but their potential functions have not been fully verified [9].
In the present study, a novel xylanase gene (xynDRTY1) was isolated from metagenomic data of Diretiyan hot spring in the Rehai area of Tengchong County, south-west of China. This gene sequence was synthesized artificially, and the recombinant vector was constructed and imported into Escherichia coli. The activity of recombinant enzymes was determined by allogenic expression and protein purification. The results show that it is thermophilic xylanase and has potential in food, feed, papermaking, and lignocellulosic ethanol.
2 Materials and methods
2.1 Sample collection and metagenomic sequencing
Sandy soil sample was collected from Diretiyan hot spring in the Rehai area of Tengchong County, Yunnan Province, south-west of China. Coordinates are latitude 24.95002°N and longitude 98.43729°E. The surface temperature of geothermal water was around 72 ℃ with pH 2.5. Samples were quickly frozen on dry ice for laboratory DNA isolation and metagenome sequencing. These activities did not require specific permissions. DNA isolation was performed with the power soil Kit ( MOBIO DNeasy PowerSoil Kit, USA) according to the operation manual. HiSeq 2500 instrument was used for metagenome sequencing at the GENWIZ in Suzhou. De novo assembly was performed with the Velvet assembly program version 1.2.08 [10]. The IMG server (https://img.jgi.doe.gov/cgi-bin/mer/main.cgi) was used for investigating the sequences. To further analyze the possible functions of individual genes and ORFs, the COG [11], the KEGG [12], and the Pfam [13] databases were employed.
2.2 Prediction of xylanase sequence, gene synthesis, and sequence analysis
Based on the functional prediction, a xylanase gene sequence (named xyndrty1) was isolated from the metagenomic database. The nucleotide sequence of the xylanase gene was submitted to GenBank under accession number MW131968. The xynDRTY1 gene was codon-optimized and synthesized according to E. coli base preference (Supplementary Fig. S1) and cloned to the pUC18 vector. BLASTx and BLASTp programs (http://blast.ncbi.nlm.nih.gov/Blast.cgi) were used to align DNA and protein sequences of xyndrty1, respectively. Signal peptides were predicted using SignalP (http://www.cbs.dtu.dk/services/SignalP/). The primary structures of the amino acid sequences were deduced and analyzed using EXPASY tools (http://web.expasy.org/protparam/). Multiple alignments with the protein sequence of the closely related (retrieved from the NCBI database) were conducted using Clustal X [14]. Phylogenetic analyses were performed using the MEGA 7 software package [15]. Phylogenetic trees were constructed using the maximum likelihood (ML) method with a Poisson correction model. The sequence of XynDRTY1 was compared among the protein structure data of the protein data bank (http://www.rcsb.org/). A structural model of XynDRTY1 was generated with the MODELLER package [16] using endo-β-1, 4-xylanase (PDB ID, 1VBU; sequence identity, 29.13%) from Thermotoga maritima as the template. Multiple sequence alignment was performed by Clustal W and Clustal X version 2.0 [17], and the figure was produced by using Espript 3 [18] (http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi).
2.3 Cloning, expression, and purification of xyndrty1
The full-length xylanase gene was moved out from the pUC18 vector by PCR using the following primers: xynDRTY1-F (CATCATCATCATCATCATGAA GTCGCGAAATGCAGCGATATTG) and xynDRTY1-R (GTGCTCGAGTGCGGCCGCAAGTTAGTCCAGACGAATACGAACA). The PCR was performed by TransStarFastPfu Fly DNA Polymerase (TransGen Biotech, China). Underlined sequences represent the homologous recombinant fragment with the pET28a vector (Novagen, USA), which had been previously digested using BamH I and Hind III. The PCR program consisted of denaturation at 95 ℃ for 3 min, followed by 30 cycles at 98 ℃ for 20 s, 65 ℃ for 30 s, and 72 ℃ for 45 s, and then a final incubation at 72 ℃ for 5 min for the final extension. The PCR product was inserted into the pET28a using the pEASY-Uni Seamless Cloning and Assembly Kit (TransGen Biotech, China) to yield the expression plasmid pET28a-xynDRTY1. Escherichia coli DH5α and E. coli BL21 (DE3) for xylanase gene clone and expression, respectively. E. coli strains were grown on LB medium with 50 μg/mL Kanamycin. DNA isolation and purification kits were purchased from Sangon (China).
The recombinant plasmid was transformed into E. coli BL21 (DE3) for xynDRTY1 expression. Transformants were cultured in 200 mL of LB broth containing 50 µg/mL kanamycin at 37 ℃ with shaking at 200 rpm. To induce expression of the recombinant xylanase, 0.2 mL of 100 mM IPTG (isopropyl β-D-1-thiogalactopyranoside) was added to the cell suspension when absorbance (600 nm) reached 0.7. Afterward, the suspension was incubated at 25 ℃ with shaking at 200 rpm for 4 h. Induced cells were harvested by centrifugation at 10,000 × g, 4 ℃ for 15 min, and cell lysis was done by ultrasonication. After centrifugation, cell-free extracts were purified using a Ni-chelating affinity column (Histrap, TransGen Biotech, China) according to the method previously reported by Yin et al. (2017) [19]. Purified xylanase was loaded at 10% SDS-PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis). Protein bands were stained by Coomassie brilliant blue dye R-250. Protein concentrations were determined with Bradford Protein Assay Kit (Order NO. C503031, Sangon Biotech, China) using bovine serum albumin as the standard.
2.4 Xylanase assay
The recombinant XynDRTY1 activity was assayed by spectrophotometry at 540 nm using beechwood xylan (Sigma, USA) as a substrate described by Yin et al. (2016) [20]. Reducing sugars were determined by the DNS (3, 5-dinitrosalicylic acid) method [21] with xylose as standard. One unit (U) of XynDRTY1 activity was identified as the amount of enzyme releasing 1 µmol reducing sugar per min.
2.5 Biochemical characterization
Optimum pH was determined by incubating the purified XynDRTY1 in various buffers ranging from pH 3.0 to 9.0 (citrate buffer, pH 3.0–8.0; borate buffer, pH 7.6–9.0). Optimum temperature was determined by measuring xylanase activity at various temperatures (30–80 ℃) at optimum pH. To assess thermostability and pH stability, the residual xylanase activity was measured after incubating the purified XynDRTY1 at different temperatures (60 ℃, 65 ℃, and 70 ℃) for different times (0 min, 20 min, 40 min, 60 min, 80 min, 100 min, and 120 min) and pH 3.0 to 9.0 for different times (12 h and 24 h), respectively.
To evaluate the influences of metal ions and chemical reagents on XynDRTY1 activity, 10 mM of various metal ions such as KCl, MgSO4, FeSO4, FeCl3, CaCl2, NiSO4, CoCl2, BaCl2, MnCl2, AgNO3, Pb(NO3)2, CuSO4, ZnSO4, and AlCl3, 1% of detergents, SDS, Tween 20, Tween 60, and Tween 80, 1% of enzyme inhibitor EDTA (ethylene diamine tetraacetic acid), DTT (Deloitte Touche Tohmatsu) and PMFS (phenylmethylsulfonyl fluoride), 10% of ionic liquid (1-allyl-3-methylimidazolium chloride), and alcohols (methyl alcohol, ethyl alcohol, and isopropyl alcohol), were added individually to the reaction system. Control conditions were tested using the same process described above without any additives to the reaction mixture.
To investigate the substrate specificity of XynDRTY1, beechwood xylan, oat xylan, beta-(1,3;1,4)-glucan, avicel, CMC (carboxyl methyl cellulose), soluble starch, and pNPX (p-nitrophenyl β-D-xylopyranoside) were used as substrates (1%, w/v) to measure enzymatic activity. The kinetic constants of XynDRTY1 were determined using different concentrations of beechwood xylan (0.1 to 20 mg/ml) at optimum pH and temperature for 5 min. The Km (Michaelis–Menten constant) and Vmax (maximum velocity of the reaction) were calculated by the Lineweaver–Burk plot.
2.6 TLC analysis
The reaction mixture, which consisted of 1% beechwood xylan and 10 μg purified enzyme, was incubated at 65 ℃ for 2 h, respectively. The hydrolytic products of beechwood xylan were characterized by TLC (thin-layer chromatography) with silica gel 60 plate (Merck, Darmstadt, Germany). The solvent was 1-butanol/acetic acid/water (2:1:1, v/v/v). Sugars were detected by treating at 120 ℃ for 10 min after spraying the plates with freshly prepared 5% (v/v) H2SO4 in ethanol. Xylose (X1), xylobiose (X2), xylotriose (X3), and xylotetraose (X4) were used as sugar standards.
2.7 Statistical analysis
Unless otherwise stated, all assays were carried out in triplicate, and the average was used in all analyses. The results were analyzed by SPSS 20.0 and expressed as means ± SEM. Statistical analyses were performed by using one-way ANOVA, followed by Tukey’s test for comparison of multiple treatment groups. In all comparisons, p values < 0.05 were considered statistically significant.
3 Results and discussion
3.1 Cloning and molecular analysis of the XynDRTY1 gene
DNA samples obtained from the sandy soil of Diretiyan hot spring (72 ℃, pH 2.5) were subjected to sequencing, which generated a total of 5.2 Gbp with 30,917 contigs of > 500 bp in length. Similarity search for beta-xylanase in the contigs revealed a new candidate xylanase gene sequence, which denominated xynDRTY1. Nucleotide sequence analysis of the complete XynDRTY1 gene revealed 1221 bp ORF encoding a xylanase protein of 406 amino acid residues. A signal peptide sequence of 31 amino acids was found at the N-terminal end by using SignalP 5.0 server, which indicated that xylanase is a secreted protein. The deduced protein without signal peptide consisted of 375 amino acids with a theoretical calculated molecular size of 43.85 kDa and theoretical pI of 6.48. The amino acid sequence of XynDRTY1 showed 75.40%, 63.32%, and 63.04% identity to hypothetical protein (GenBank: HFE91178.1) from Bryobacterales bacterium, endo-1,4-beta-xylanase (GenBank: MBI5280435.1) from Candidatus Solibacter usitatus, and endo-1,4-beta-xylanase (GenBank: MBI5083254.1) from Acidobacteria bacterium.
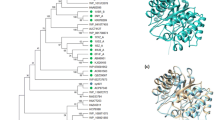
As observed in Fig. 1, XynDRTY1 has a catalytic domain, which was similar to the GH10 family domain of endo-β-1,4-xylanase from Cellvibrio japonicus. Like other GH10 xylanases, its tertiary structure shows a (β/α)8 or TIM-barrel folding [22, 23]. A phylogenetic analysis of protein sequences revealed XynDRTY1 clustered with hypothetical protein (GenBank: HFE91178.1) from the Bryobacterales bacterium. This indicated that the hypothetical protein (GenBank: HFE91178.1) was a potential endo-1,4-beta-xylanase. Multiple sequence alignments of XynDRTY1 with the closest structure-resolved xylanase were performed (Supplementary Fig. S2). Two putative catalytic residues (E188 and E291) were found in XynDRTY1 [22].

Sequence analysis of GH10 xylanase XynDRTY1. A, The structural domain and tertiary structure of XynDRTY1. B, Phylogenetic dendrogram obtained by maximum likelihood analysis based on amino acid sequences showing the phylogenetic position of XynDRTY1 with related xylanase. Bootstrap values (expressed as a percentage of 1000 replications) are given at nodes
3.2 Heterologous expression and purification of XynDRTY1
Xylanase gene without signal sequence was successfully cloned into pET28a C-His as a fusion protein with His-tag which was further confirmed by sequencing. The recombinant xylanase (XynDRTY1) was purified by Ni2+-NTA resin affinity chromatography. The purified protein showed a single band of ~ 44 kDa against the protein marker on 10% SDS-PAGE (Fig. 2).

SDS-PAGE analysis of the recombinant XynDRTY1 produced by E. coli BL21. Lane 1, protein molecular weight marker, mass indicated on the left; lane2, total protein in IPTG-induced E. coli BL21/pET28a-xynDRTY1; lane 3, purified XynDRTY1
3.3 Effect of temperature and pH on XynDRTY1
The optimal reaction temperature for XynDRTY1 activity was 65 ℃, and over 50% of the maximal activity was observed at 55 to 70 ℃ (Fig. 3A). Optimum pH for XynDRTY1 activity was pH 6.0, and over 70% of the maximal activities were kept between pH 4.6 and 7.0 (Fig. 3B). Thermostability analysis showed that XynDRTY1 retained 100% activity after heat treatment at 60 ℃ for 2 h, and its half-lives at 65 ℃ and 70 ℃ were about 38 min and 5 min (Fig. 3C). Interestingly, its activity was increased to 140% after incubation at 60 ℃ for 20 min. The pH stability analysis revealed that it retained more than 60% of its initial activity at pH 3–9 and more than 80% at pH 6–7 after incubation at 25 ℃ for 12 h and 24 h (Fig. 3D). Thermostable xylanases have advantages since the various industrial processes need to go through high-temperature processes [24]. However, most of the reported GH 10 xylanases cannot tolerate a temperature of over 50 ℃ [25]. For example, Xyn10A (GenBank NO., AGA16736) from Bacillus sp. SN5 lost more than 90% enzyme activity after incubation at 50 ℃ for 20 min [26]. Novel thermostable xylanase (GenBank NO., MH685571) from camel rumen metagenome retained less than 50% of its maximum activity after incubation at 50 ℃ for 40 min. In comparison to other xylanases, XynDRTY1 exhibited thermostability and wide pH range finds. These indicate that XynDRTY1 has great potential application in various industrial processes.

Effects of temperature and pH on the activity and stability of the recombinant XynDRTY1. A, Temperature effect on the activity of XynDRTY1. B, pH effect on the activity of XynDRTY1. C, The effect of temperature on stability at different temperatures (60 ℃, 65 ℃, and 70 ℃) for 0, 20, 40, 60, 80, 100, and 120. D, The effect of pH on stability. The primary activity was taken as 100%. Each value in the figure represents the mean ± SD (n = 3). 100% = 21.2 ± 3 U/mg
3.4 Effect of metal ions and chemical agents on XynDRTY1
As shown in Supplementary Table S1, XynDRTY1 activity was activated by Fe3+ (115.2 ± 0.5%), slightly deactivated by K+, Mg2+, Fe2+, Ca2+, Ni2+, Ba2+, Pb2+, Zn2+, and Al3+ and strongly inhibited by Co2+ and Ag2+ at 10 mM metal ions. It almost lost all activity at present of 10 mM Cu2+ or Mn2+. Metal ions such as Co2+, Mn2+, and Cu2+ have an inhibitory effect on the activity of the majority of GH10 family xylanases [27]. Tween 20, Tween 80, and PFMS had no influence on xylanase activity at the concentration of 1%, but were slightly inhibited by EDTA and Tween 60. Organic solvents and SDS can inhibit most enzyme activity of xylanases [20]. Like most xylanases, XynDRTY1 was highly suppressed by 1% SDS and 10% methyl alcohol. While it still retained over 60% of the maximal activities in the presence of 10% ionic liquid and isopropyl alcohol, and over 45% of the maximal activities in the presence of 10% ethyl alcohol and 1% DTT. These suggested that Co2+, Ag2+, Cu2+, Mn2+, SDS, and methyl alcohol should be avoided during the application of XynDRTY1.
3.5 Substrate specificity and kinetic analysis of XynDRTY1
Substrate specificity of XynDRTY1 was shown in Table 1. It exhibited activities for beechwood xylan (21.2 ± 3 U/mg), oat xylan (8.2 ± 0.3 U/mg), and avicel (1.2 ± 0.3 U/mg), but no activity for beta-1,3;1,4-glucan, CMC, soluble starch, and pNPX. The Km, Vmax, and Kcat of recombinant XynDRTY1 for beechwood xylan were 15 ± 0.4 mg/ml, 48.8 ± 3 μmol/min/mg, and 38.2 ± 2.3 S−1, respectively. In general, xylanases from the GH10 exhibited not only activities for different source xylan but also a variety of substrates like avicel and CMC [28]. This indicates that XynDRTY1 is a multifunctional enzyme.
3.6 TLC analysis of Beechwood xylan hydrolysis by XynDRTY1
As shown in Fig. 4, hydrolytic products of beechwood xylan with XynDRTY1 were analyzed by TLC. The result showed that the major hydrolytic products by XynDRTY1 were X1, X2, and X4. Many studies have shown that xylooligosaccharides, as an emerging prebiotic, play an important role in promoting the growth of probiotics and balancing the stability of human gut microbiota [29, 30]. This indicates that XynDRTY1 has potential application value in the production of prebiotics.

Thin-layer chromatography (TLC) of hydrolyzation products of xylo-oligosaccharides by XynDRTY1. Lane 1, standards: xylose (X1), xylobiose (X2), xylotriose (X3), and xylotetraose (X4); lane 2, beechwood xylan without enzyme; lane 3, beechwood xylan hydrolysis by purified XynDRTY1
4 Conclusions
In summary, a novel xylanase gene (xynDRTY1) encoding thermostable GH10 xylanase was synthesized and heterologously expressed in E. coli BL21 (DE3). To our knowledge, this is the first report of metagenome-derived xylanase identified from Diretiyan of Tengchong hot spring. A detailed enzymatic characterization of XynDRTY1 was performed. XynDRTY1 was found to be thermophilic and thermostable. Furthermore, XynDRTY1 exhibited the activity of endo-1,4-beta-xylanase. Overall, in this work, XynDRTY1 had different specificities and characteristics, rendering it an ideal candidate for use in the hydrolysis of lignocellulose and the production of prebiotics.
References
Ghadikolaei KK, Sangachini ED, Vahdatirad V, Noghabi KA, Zahiri HS (2019) An extreme halophilic xylanase from camel rumen metagenome with elevated catalytic activity in high salt concentrations. AMB Express 9(1):86. https://doi.org/10.1186/s13568-019-0809-2
Takkellapati S, Li T, Gonzalez MA (2018) An overview of biorefinery-derived platform chemicals from a cellulose and hemicellulose biorefinery. Clean Technol Environ Policy 20(7):1615–1630. https://doi.org/10.1007/s10098-018-1568-5
Samanta AK, Jayapal N, Kolte AP, Senani S, Sridhar M, Dhali A, Suresh KP, Jayaram C, Prasad CS (2014) Process for enzymatic production of xylooligosaccharides from the xylan of corn cobs. J Food Process Preserv 39(6):729–736. https://doi.org/10.1111/jfpp.12282
Tenkanen M, Puls J, Poutanen K (1992) Two major xylanases of Trichoderma reesei. Enzyme Microb Technol 14(7):566–574. https://doi.org/10.1016/0141-0229(92)90128-b
López-López O, Cerdán M, González-Siso M (2013) Hot spring metagenomics. Life 3(2):308–320. https://doi.org/10.3390/life3020308
Knapik K, Becerra M, González-Siso M-I (2019) Microbial diversity analysis and screening for novel xylanase enzymes from the sediment of the lobios hot spring in Spain. Sci Rep 9(1):11195. https://doi.org/10.1038/s41598-019-47637-z
Kurazono H, Pal A, Bag PK, Balakrish Nair G, Karasawa T, Mihara T, Takeda Y (1995) Distribution of genes encoding cholera toxin, zonula occludens toxin, accessory cholera toxin, and El Tor hemolysin Vibrio cholerae of diverse origins. Microb Pathog 18(3):231–235. https://doi.org/10.1016/s0882-4010(95)90076-4
Uchiyama T, Miyazaki K (2009) Functional metagenomics for enzyme discovery: challenges to efficient screening. Curr Opin Biotechnol 20(6):616–622. https://doi.org/10.1016/j.copbio.2009.09.010
Tiwari R, Nain L, Labrou NE, Shukla P (2017) Bioprospecting of functional cellulases from metagenome for second generation biofuel production: a review. Crit Rev Microbiol 44(2):244–257. https://doi.org/10.1080/1040841x.2017.1337713
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18(5):821–829. https://doi.org/10.1101/gr.074492.107
Tatusov RL (2001) The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 29(1):22–28. https://doi.org/10.1093/nar/29.1.22
Nakaya A, Katayama T, Itoh M, Hiranuka K, Kawashima S, Moriya Y, Goto S (2012) KEGG OC: a large-scale automatic construction of taxonomy-based ortholog clusters. Nucleic Acids Res 41(D1):D353–D357. https://doi.org/10.1093/nar/gks1239
Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Bateman A (2007) The Pfam protein families database. Nucleic Acids Res 36(Database), D281–D288. https://doi.org/10.1093/nar/gkm960
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882. https://doi.org/10.1093/nar/25.24.4876
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874. https://doi.org/10.1093/molbev/msw054
Šali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234(3):779–815. https://doi.org/10.1006/jmbi.1993.1626
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23(21):2947–2948. https://doi.org/10.1093/bioinformatics/btm404
Gouet P, Robert X, Courcelle E (2003) ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res 31(13):3320–3323. https://doi.org/10.1007/s10404-008-0309-1
Yin YR, Meng ZH, Hu QW, Jiang Z, Xian WD, Li LH, Li WJ (2017) The Hybrid Strategy of Thermoactinospora rubra YIM 77501T for utilizing cellulose as a carbon source at different temperatures. Front Microbiol 8:942. https://doi.org/10.3389/fmicb.2017.00942
Yin YR, Hu QW, Xian WD, Feng Z, Zhou EM, Hong M, Min X, Zhi XY, Li WJ (2016) Characterization of a neutral recombinant xylanase from Thermoactinospora rubra YIM 77501T. Antonie Van Leeuwenhoek 110:429–436. https://doi.org/10.1007/s10482-016-0798-y
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31(3):426–428. https://doi.org/10.1021/ac60147a030
Lo Leggio L, Kalogiannis S, Bhat MK, Pickersgill RW (1999) High resolution structure and sequence of T. aurantiacus xylanase I: implications for the evolution of thermostability in family 10 xylanases and enzymes with (beta)alpha-barrel architecture. Proteins 36:295–306. https://doi.org/10.1002/(sici)1097-0134(19990815)36:3%3c295::aid-prot4%3e3.0.co;2-6
Niderhaus C, Garrido M, Insani M, Campos E, Wirth S (2018) Heterologous production and characterization of a thermostable GH10 family endo-xylanase from Pycnoporus sanguineus bafc 2126. Process Biochem 67(APR.):92–98. https://doi.org/10.1016/j.procbio.2018.01.017
Bhalla A, Bansal N, Kumar S, Bischoff KM, Sani RK (2013) Improved lignocellulose conversion to biofuels with thermophilic bacteria and thermostable enzymes. Bioresource Technol 128(Complete):751–759. https://doi.org/10.1016/j.biortech.2012.10.145
He J, Tang F, Chen D, Yu B, Luo Y, Zheng P, Mao X, Yu J, Yu F (2019) Design, expression and functional characterization of a thermostable xylanase from Trichoderma reesei. PLoS One 14(1):e0210548. https://doi.org/10.1371/journal.pone.0210548
Bai W, Xue Y, Zhou C, Ma Y (2012) Cloning, expression and characterization of a novel salt-tolerant xylanase from Bacillus sp. SN5. Biotechnol Lett 34(11):2093–2099. https://doi.org/10.1007/s10529-012-1011-7
Jacomini D, Bussler L, Corrêa JM, Kadowaki MK, Simo R (2020) Cloning, expression and characterization of C. crescentus xyna2 gene and application of xylanase II in the deconstruction of plant biomass. Mol Biol Rep 47(6):4427–4438. https://doi.org/10.1007/s11033-020-05507-2
Loaces I, Bottini G, Moyna G, Fabiano E, Martínez A, Noya F (2016) Endog: a novel multifunctional halotolerant glucanase and xylanase isolated from cow rumen. J Mol Catal B Enzym S1381117716300042.https://doi.org/10.1016/j.molcatb.2016.01.004
Chapla D, Pandit P, Shah A (2012) Production of xylooligosaccharides from corncob xylan by fungal xylanase and their utilization by probiotics. Bioresource Technol 115(none):215–221. https://doi.org/10.1016/j.biortech.2011.10.083
Finegold SM, Li Z, Summanen PH, Downes J, Thames G, Corbett K et al (2014) Xylooligosaccharide increases bifidobacteria but not lactobacilli in human gut microbiota. Food Funct 5(3):436. https://doi.org/10.1039/c3fo60348b
Chen M, Liu S, Imam K, Sun L, Wang Y, Gu T et al (2020) The effect of xylooligosaccharide, xylan, and whole wheat bran on the human gut bacteria. Front Microbiol 11:2936–2947. https://doi.org/10.3389/fmicb.2020.568457
Acknowledgements
We gratefully acknowledge Prof. Wen-Jun Li and his team from Sun Yat-sen University for their guidance on our metagenomic data compilation and analysis.
Funding
This research was supported by the National Natural Sciences Foundation of China Regional Program (Grant Nos. 31660015 and 31860243) and Yunnan Applied Basic Research Projects (Grant Nos. 202101AU070138, 2017FB024, and 2017FH001-032).
Author information
Authors and Affiliations
Contributions
YRY, PS, and LQY conceived the study. YRY and WH cloned the gene and cultured strains. RFY and HYL purified the recombinant protein. LL measured enzymatic activity. XWL and ZLL performed the data analysis and mapping. YRY, WH, PS, and LQY wrote the manuscript. All authors discussed the results and commented on the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
This article does not contain any studies related to human participants or animals.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yin, YR., Li, L., Yang, RF. et al. Characterization of a metagenome-derived thermostable xylanase from Tengchong hot spring. Biomass Conv. Bioref. 14, 10027–10034 (2024). https://doi.org/10.1007/s13399-022-03296-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13399-022-03296-1