
Abstract
Cellulases are among the most important groups of industrial enzymes that are widely consumed in biofuel production, pulp and paper, textile, and detergent industries. The methylotrophic yeast Pichia pastoris was used for heterologous expression of a thermophilic cellulase collection. P. pastoris cells were transformed by the codon-optimized polycistronic EBG construct. This construct included egxA gene (from Ampullaria crossean, with endo- and exoglucanase activities), cglT gene (from Thermoanaerobacter brockii, with β-glucosidase activity), and zsgreen (a fluorescent marker). Gene expression was examined at mRNA level using RT-PCR technique. The results indicated successful transcription of all transgenes. CglT and ZsGreen recombinant proteins were respectively detected by enzymatic assay and fluorescent microscope, while endo- and exoglucanase activities were not determined by enzymatic assays. The highest β-glucosidase activity was measured at 65 ºC and pH 5.5. CglT is a good candidate for completing cellulase collections with low β-glucosidase activity. These cellulase sets could be used in biofuel production because of the high glucose tolerance property of CglT.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Classic cellulase enzyme system consists of three main enzyme groups, exoglucanases or cellobiohydrolases (EC 3.2.1.91), endoglucanases or CMCase (EC 3.2.1.4), and β-glucosidases (EC 3.2.1.21). Synergistic cooperation of these enzymes could decompose cellulose (homopolymer of glucose units) to a hexacarbon sugar, glucose. First, endoglucanase and exoglucanase enzymes act on cellulose, and their activity for β-1,4-glycosidic bonds hydrolyzing leads to production of cellobiose molecules which are then cleaved by β-glucosidases into glucose [1]. Cellulases have a very wide range of applications in different industries (such as food and animal feed, textile, and biofuel industries) individually or in combined groups [2,3,4,5]. They could be applied for industrial and agricultural waste management and environmental conservation [6,7,8].
The thermal stability of cellulase enzymes is an important parameter for many utilizations due to biotechnological processes and enzymes’ storage conditions. It is suggested that the possibility of conducting the processes at higher temperatures would lead to reduced enzyme requirements, increased reaction rate, and a decrease in the risk of contaminations with mesophilic microorganisms. Besides, thermophilic enzymes can be transferred and stocked easier than mesophilic enzymes [9].
Cellulase enzymes could be provided by cellulase-producing microorganisms or recombinant protein technology. We planned to have a complete set of recombinant cellulases; and therefore, a gene cassette was designed including egxA, cglT, and zsgreen genes which encoded endoglucanase and exoglucanase (EGXA protein) [10], β-glucosidase (CglT protein), and a green fluorescent marker (ZsGreen protein), respectively. egxA gene was obtained from Ampullaria crossean snail encoding a 560 amino acid thermophilic enzyme with endoglucanase and exoglucanase activities, while cglT gene was provided from Thermoanerobacter brockii bacterium that encodes a 450 amino acid thermophilic β-glucosidase enzyme [10, 11].
Pichia pastoris (Komagataella phaffii) is a suitable eukaryotic host for secreted recombinant protein production at academic or industrial levels due to obtaining high cell density and secreting low levels of endogenous proteins into culture media [12, 13]. A strong and tightly regulated promoter PAOX1 is widely used for foreign protein production in P. pastoris which could separate the growth phase from the production phase; therefore, biomass density could reach proper levels before initiating the expression of gene of interest [13]. Furthermore, codon optimization was applied for improving cellulase gene expression in this host [14].
There are several strategies for co-expression of multiple genes in eukaryotic cells including introducing different plasmids into the host cell, using a plasmid with multiple promoters, designing proteolytic sites, internal ribosome entry sites, or self-processing 2A sequences between genes. 2A sequences are short peptides that allow co-translation of all open reading frames (ORFs) presented on a polycistronic mRNA by ribosomal skipping. It terminates the translation at the carboxyl terminus of the first 2A peptide and releases the polypeptide chain, while ribosome starts the translation of the second ORF. GDVEXNPGP is the conserved oligopeptide among different 2A sequences, and cleavage occurs before the last proline. 2A peptide is normally added to the C-terminal of the first and P at N-terminal of the next polypeptide chain. The main advantages of this technique are relatively high cleavage efficiency and almost equal expression levels of different ORFs of the construct [15,16,17]. 2A sequences have been used for heterologous protein production in fungi such as Trichoderma reesei [18], Saccharomyces cerevisiae [19, 20], and Pichia pastoris [21]. We used this technique for the co-expression of three genes (two cellulase genes and a fluorescent marker) in this study. We aimed to have a cellulase enzyme set consisting of EGXA and CglT proteins, investigated the expression of these genes in P. pastoris for the first time, and compared the ability of P. pastoris in expression of these genes with other hosts. Although P. pastoris is a suitable host for expression of many heterologous genes, it is necessary to examine its possible efficiency in new cases.
2 Materials and methods
2.1 Microorganism strains and vector
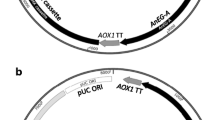
Escherichia coli JM109 was used for pPICZαA vector amplification, and P. pastoris GS115 (Invitrogen) was utilized as the host for foreign protein production. The construct EBG including egxA coding sequence (CDS) (GenBank accession number, FJ183727.1) from Ampullaria crossean encoding endoglucanase and exoglucanase enzymes, cglT CDS from Thermoanaerobacter brockii (GenBank accession number, Z56279.1) encoding β-glucosidase enzyme, and zsgreen CDS (GenBank accession number, AF168422.1) encoding fluorescent marker protein was synthesized by Genray company (China) (Fig. 1). It should be mentioned that these three genes were connected by two 2A sequences (P2A and T2A), and a signal peptide sequence (MF4I) was placed before cglT CDS. A nine-nucleotide segment (GGATCTGGT) was added before 2A sequences to improve their functions [17, 22, 23]. The company exerted the codon optimization (proper to P. pastoris codon usage) and cloned it into the expression vector pPICZαA using EcoRI and NotI restriction enzymes.

Scheme of EBG construct. EBG construct was designed and cloned in pPICZαA vector to produce a recombinant cellulase enzyme set by yeast P. pastoris cells
2.2 Culture media
Escherichia coli (E. coli) JM109 were cultured in lysogeny broth (LB) medium (Sigma-Aldrich). Different media including yeast extract-peptone-dextrose (YPD; 1% yeast extract, 2% peptone, 2% dextrose or glucose) (Sigma-Aldrich), yeast extract-peptone-dextrose-sorbitol (YPDS; 1% yeast extract, 2% peptone, 2% dextrose or glucose, 1 M sorbitol), buffered glycerol complex (BMGY; 1% yeast extract, 2% peptone, 100 mM potassium phosphate, 1.34% yeast nitrogen base w/o amino acids, 4 × 10−5% biotin, 1% glycerol), and buffered methanol complex (BMMY; 1% yeast extract, 2% peptone, 100 mM potassium phosphate, 1.34% yeast nitrogen base w/o amino acids, 4 × 10−5% biotin, 0.5% methanol) were used for various phases of experiments. YPDS, BMGY, and BMMY media were prepared according to EasySelect Pichia Expression Kit Manual. All required chemicals were purchased from Merck company.
2.3 Plasmid verification, linearization and microorganism transformation
Recombinant plasmid (pPICZαA-EBG) was digested by two restriction enzymes (EcoRI and NotI, NEB) for verification of EBG fragment insertion in pPICZαA plasmid. DNA fragments obtained from enzymatic double digestion were examined on agarose gel (0.7%) by gel electrophoresis [24].
To propagate the original and recombinant plasmids (pPICZαA and pPICZαA-EBG, respectively), competent E. coli JM109 cells were prepared and transformed following Inoue method [25]. Transformed colonies were selected on LB-Agar medium containing 25 μg/ml Zeocin antibiotic. GeneJET Plasmid Miniprep Kit (Thermo Fisher Scientific) was applied for plasmid extraction from E. coli and P. pastoris cells.
Original and recombinant plasmids were linearized by SacI restriction enzyme (Thermo Fisher Scientific) and transferred to P. pastoris GS115 cells by transformation technique [24]. P. pastoris electrocompetent cells were prepared based on EasySelect Pichia Expression Kit Manual and were transformed using manufacturer instructions of the BioRad electroporation system. After transformation, the cells were incubated in YPDS medium for 60 min and then transferred on YPDS plates containing 100 μg/ml Zeocin antibiotic and incubated at 30 ºC for 4–5 days. Appeared colonies were used for protein expression cultures.
2.4 P. pastoris transformants cultivation
To investigate recombinant protein expression, transformed colonies were cultured in BMGY medium (at 30 ºC and 230 rpm) overnight (OD600 = 2–6), then yeast cells were pelleted by centrifugation at 1500 g for 5 min (room temperature). Pellets were resuspended in BMMY medium (10 ml and OD600 = 1), transferred into 100-ml flasks, and incubated at 30 ºC and 230 rpm for 6 days. To induce the AOX1 promoter, methanol (500 μl, 0.5% final concentration) was added to each flask every 24 h.
2.5 Detecting the expression of recombinant genes at mRNA level
Our construct included three genes; two of which (egxA and cglT) encoding enzyme proteins, and zsgreen gene that encodes a green fluorescent protein. Recombinant colonies of P. pastoris were cultured in BMGY and then BMMY media. Cell pellets were obtained by centrifugation (8000 g and 4 ºC for 10 min), rinsed with PBS, and grinned in liquid nitrogen for 5 min. Thermo Fisher Scientific Kits were used for RNA extraction, DNase treatment, and cDNA synthesis. Forward and reverse primers for recombinant genes and act1 gene from P. pastoris (as a control gene) were designed by Allel ID v.6 software (Table 1). mRNA expression of egxA, cglT, zsgreen, and act1 genes (GenBank accession numbers: FJ183727.1, Z56279.1, AF168422.1, and AF216956.1, respectively) was examined using RT-PCR technique.
2.6 Detecting the expression of recombinant genes at protein level
Transgenic P. pastoris colonies were grown in BMGY medium overnight and then cultured in BMMY medium (50 ml containing 0.5% methanol). After 6 days, the cells were separated by centrifugation at 10,000 g and 4 ºC for 10 min. The supernatants were utilized for enzyme assays. Solid ammonium sulfate was dissolved in supernatants (85% saturation) and kept overnight at 4 ºC. Centrifugation at 10,000 g and 4 ºC for 10 min pelleted the proteins which were then dissolved in 1 ml citrate buffer (100 mM) and used for recombinant enzyme activity measurements [26]. Extraction of intracellular proteins of pelleted yeast cells was performed using glass beads and lysis buffer (NaCl, 100 mM., 2-mercaptoethanol, 10 mM., EDTA, 1 mM) [27]. Extracted proteins were used for enzyme assays.
2.6.1 ZsGreen protein, fluorescent marker
P. pastoris samples (containing recombinant vector as main samples and parental vector as control samples) were taken from BMMY media and used for preparing yeast slides. These slides were observed by a fluorescent microscope (Olympus EX51 microscope).
2.6.2 Endo- and exoglucanase assays
Carboxymethyl cellulose (CMC) (2% w/v) (Sigma-Aldrich) in citrate buffer (50 mM) and avicel PH 101 (2% w/v) (Merck) in citrate buffer (100 mM) were used as substrates for endoglucanase and exoglucanase activity detection, respectively. Reaction mixtures including substrate solution and enzyme solution (1:1) were prepared with various pHs (3–9) and incubated in different temperatures (30–60 ºC) for several durations (30 min to overnight). Then, enzyme activities were assayed by dinitrosalicylic acid (DNS) method, and glucose concentrations were determined by measuring the absorbances at 540 nm [28,29,30].
2.6.3 β-glucosidase assay
The β-glucosidase activity assay was performed by 4-nitrophenyl-β-D-glucopyranoside (pNPG) (Sigma-Aldrich) as a substrate of the enzyme. Substrate solutions (in phosphate buffer, 100 mM) and enzyme solutions were mixed (1:9) in 100 μl final volume and incubated at 75 ºC for 30 min. Sodium bicarbonate (1 M, 100 μl, Merck) was utilized for stopping the enzyme reactions. Then, optical densities were read at 410 nm [29, 31].
Different pHs (3.5–8.5) and temperatures (30–75 ºC) were tested to find conditions that showed the highest enzyme activity of the recombinant protein. Enzyme activities were calculated based on the standard curve of p-nitrophenol, and one unit of enzyme activity was defined as the amount of enzyme required to produce 1 μmol p-nitrophenol within 1 min under the reaction conditions.
2.6.4 Optimum methanol concentration for induction
The induction of the BMMY medium could be done by using different concentrations of methanol. Among the recombinant colonies, the GR1 colony, with the highest β-glucosidase activity, was selected for optimizing methanol concentration. Cultures of GR1 colonies in BMMY medium were prepared and then induced daily with 0.5%, 1%, and 2% methanol. The GR1 colony (transgenic P. pastoris colony which had received pPICZαA-EBG vector), GRP (transgenic P. pastoris colony that received parental pPICZαA vector), and GS115 (control colony of P. pastoris) were grown in these media for 6 days. Then, yeast cells were pelleted, and the supernatants were used for β-glucosidase assays.
Each concentration of methanol and each enzyme assay were tested three times.
2.6.5 SDS-PAGE analysis
Samples used for SDS-PAGE analysis included concentrated culture media proteins (provided by solid ammonium sulfate, 85%). Stacking (5%) and running gels (12%) were prepared at pH 6.8 and pH 8.8, respectively, and protein bands were stained with Coomassie Brilliant Blue R-250 [32, 33].
2.6.6 Possibility of recombinant enzymes digestion by Kex2 protease
The dipeptides Lys-Arg or Arg-Arg (as the recognition sites of Kex2 enzyme) were searched throughout EGXA and CglT proteins. According to [34] study, an octapeptide sequence including these recognition sites was scored for possible digestion of recombinant proteins by Kex2 enzyme.
3 Results
3.1 Plasmid verification and linearization
Electrophoresis of double digestion (by EcoRI and NotI restriction enzymes) of pPICZαA-EBG plasmid showed two DNA fragments related to pPICZαA plasmid (3.6 kb) and EBG construct (4.2 kb). This observation confirmed that EBG construct was correctly inserted in pPICZαA plasmid. Linearization of recombinant plasmid (by SacI restriction enzyme) is also confirmed by agarose gel electrophoresis (Fig. 2).

Enzymatic digestions of recombinant plasmid pPICZαA-EBG. Enzymatic digestion of recombinant plasmid was performed by EcoRI and NotI restriction enzymes. Two DNA fragments (3.6 and 4.2 kb) were observed on 0.7% agarose gel. Digestion of recombinant plasmid with SacI restriction enzyme resulted in linear plasmid. DNA size marker (1 kb) (lane 1), products of double enzymatic digestion (lane 2), circular recombinant vector pPICZαA-EBG (relaxed and supercoiled forms) (lane 3), linear recombinant vector pPICZαA-EBG (lane 4)
3.2 Expression of recombinant genes at mRNA level
We utilized the RT-PCR technique to examine the proper transcription of recombinant genes (egxA, cglT, and zsgreen) in yeast P. pastoris GS115 strain. The RT-PCR was done for recombinant yeast cells that received pPICZαA-EBG vector, and those cells receiving parental vector pPICZαA (as control). The cells were cultured in two different non-induced BMGY medium and induced BMMY medium. Electrophoresis of RT-PCR products showed that recombinant genes were transcribed in recombinant yeast cells that received pPICZαA-EBG vector and cultured in induced medium. Negative controls indicated no transcription of recombinant genes (Fig. 3).

Expression of recombinant cellulase and marker genes in transgenic P. pastoris GS115 cells at mRNA level. DNA fragments with 178, 186, 120, and 135 bp lengths were obtained from RT-PCR which related to recombinant egxA, cglT, and zsgreen genes, as well as host act1 gene (as positive control), respectively. DNA size marker, 100 bp (lanes 1), P. pastoris cells which received pPICZαA-EBG vector and cultured in induced BMMY medium (lanes 2 and 6), P. pastoris cells which received pPICZαA-EBG vector and cultured in non-induced BMGY medium (lanes 3 and 7), P. pastoris cells which received parental pPICZαA vector and cultured in induced BMMY medium (lanes 4 and 8), and negative controls (lanes 5 and 9)
3.3 Expression of recombinant genes at protein level
3.3.1 Fluorescent marker protein
Slides of yeast cells that received recombinant and parental vectors (pPICZαA-EBG and pPICZαA vectors, respectively) were observed by fluorescent microscope. The former cells expressed bright green ZsGreen protein (Fig. 4).

Expression of fluorescent ZsGreen protein in P. pastoris GS115 cells. The yeast cells that had received recombinant pPICZαA-EBG vector expressed the marker protein (ZsGreen) and were seen as bright green dots on the slide (A). The yeast cells that had received parental vector (pPICZαA vector) were used as negative control (B). Scale bar represents 200 μm
3.3.2 Endoglucanase and exoglucanase activities
Endo- and exoglucanase assays using culture media and intracellular extracted proteins of genetically engineered yeast cells (cultures in induced BMMY medium) led to no detection of enzyme activities. Enzyme assays were performed under different reaction conditions (various pHs, temperatures, and reaction times) and repeated for concentrated culture media (prepared by solid ammonium sulfate) as well; absorbances of reaction mixtures were almost 0 at 540 nm. Therefore, enzyme activities were considered 0.
3.3.3 β-Glucosidase activity
Recombinant colonies obtained from yeast transformation with pPICZαA-EBG vector were cultured in induced BMMY medium. After 6 days, culture media were harvested and tested for β-glucosidase activity. Enzyme reactions were performed at 75 ºC and pH 5.5 (optimum temperature and pH for CglT activity from T. brockii bacterium) [11]. The best colony was selected based on p-nitrophenol production during enzyme assays and used for the optimization phase. It was observed that most of the colonies produced p-nitrophenol in the same range and only two colonies (GR6 and GR7) indicated significantly lower levels of p-nitrophenol production during the β-glucosidase reaction. However, the GR1 colony was chosen for the next phases of the experiment because it showed the highest level of p-nitrophenol production.
3.3.4 Optimization of methanol concentration in BMMY medium
Different concentrations of methanol (0.5%, 1%, and 2%) were used for BMMY media preparation. GR1 colonies were cultured in these media for 6 days, and then culture media were used for assessment of β-glucosidase activity. Recombinant CglT activities did not show significant differences between media with 0.5% and 1% methanol, whereas 2% methanol in BMMY medium led to a significant reduction of β-glucosidase activity (Fig. 5). Induced medium (BMMY medium) with 0.5% methanol was selected for the next phases.

Effect of methanol concentrations on recombinant CglT production by yeast P. pastoris. Comparing the relative activities of recombinant β-glucosidase enzymes produced by GR1 yeast colonies in BMMY media with different concentrations of methanol (0.5%, 1%, and 2%). Reaction enzymes were carried out at 75 °C and pH 5.5. GR, recombinant P. pastoris GS115 yeasts received pPICZαA-EBG vector, GRP, recombinant P. pastoris GS115 colonies containing parental pPICZαA vector, GS115, non-recombinant P. pastoris GS115 colonies
3.3.5 Optimum temperature and pH for recombinant CglT enzyme
GR1 colonies were cultured in BMMY medium (with 0.5% methanol) for 6 days. Then, culture media were harvested and used for β-glucosidase assays at different temperatures (30–80 ºC) and various pHs (3.5–8.5). CglT enzyme activities were compared in varied reaction conditions and indicated that 65 ºC temperature and pH 5.5 were the best conditions for this recombinant β-glucosidase activity (Fig. 6A, B).

Optimum temperature and pH for recombinant CglT enzyme activity. The graphs indicate the relative β-glucosidase activities of recombinant CglT enzyme at temperature range, 30–80 ºC (A) and various pHs from 3.5 to 8.5 (B)
3.3.6 SDS-PAGE analysis of recombinant proteins production by P. pastoris
SDS-PAGE analysis was utilized for examination of recombinant protein production by P. pastoris. Samples were prepared from concentrated induced culture media of transformed yeasts which had received pPICZαA-EBG and parental pPICZαA vectors. A comparison of these two samples showed two extra bands for yeast cells containing pPICZαA-EBG. These two bands were possibly related to CglT protein. It seems that the polypeptide chain of recombinant β-glucosidase has been hydrolyzed in P. pastoris cells or culture media because we expected a 52-kD protein band for this enzyme, instead of the two observed protein bands (about 39 and 37 kD) by SDS-PAGE (Fig. 7). These protein bands are in agreement with recognition of two potential cleavage sites (PGKRTEMG and LLKRLDRE) on CglT polypeptide chain by Kex2 enzyme (Table 2). It seems that these truncated proteins keep their enzyme activities, because we detected β-glucosidase activities in recombinant yeast culture media.

SDS-PAGE analysis of recombinant proteins produced by P. pastoris GS115. Total protein presented in culture media of recombinant yeast P. pastoris cells containing pPICZαA-EBG vector (lane 1) and parental pPICZαA vector (lane 2). Two protein bands related to recombinant proteins are shown with arrows. Molecular weight marker (lane 3)
3.3.7 Possibility of recombinant enzymes digestion by Kex2 protease
Scoring EGXA and CglT proteins for likely digestion by Kex2 is summarized in Table 2. Five and six potential cleavage sites were observed for CglT and EGXA polypeptide chains, respectively. Two cleavage sequences of each protein obtained a high score. It seems that the Kex2 function is the strong potential cause of these two proteins digestion. It appears that this possible digestion did not influence on enzyme activity of some CglT copies, while EGXA proteins lost their activity as a result of polypeptide chain cleavage.
There is a possibility for secretory recombinant proteins to be hydrolyzed by internal or external proteases of the host cells, partially or completely. It seems that partial hydrolysis may have occurred on the recombinant CglT protein in our study. To further clarify, a β-glucosidase enzyme was found in Thermoanaerobacter sp. (GenBank accession number: HBW59855.1, 165 amino acids), and a protein blast of Thermoanaerobacter brokii CglT (β-glucosidase, 450 amino acids, GenBank accession number: CAA91220.1) with this protein showed 99% identity between amino acids 4–168 of CglT with amino acids 1–165 of this enzyme (Fig. 8). Therefore, it seems that the existence of N terminal fragment of CglT is enough for its β-glucosidase activity. Thus although some copies of recombinant CglT are hydrolyzed partially, they could keep their catalytic activity.

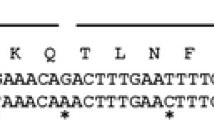
Protein blast of CglT protein and a β-glucosidase from Thermoanaerobacter sp. Protein blast of Thermoanaerobacter brokii CglT (β-glucosidase, 450 amino acids, GenBank accession number: CAA91220.1) and a β-glucosidase from Thermoanaerobacter sp. (GenBank accession number: HBW59855.1, 165 amino acids) showed a 99% identity between amino acids 4–168 of CglT and amino acids 1–165 of this enzyme
4 Discussion
Cellulase enzymes play roles as critical components of various industries such as paper, detergents, food, and feed industries [35]. Besides, these enzymes can degrade biomass and could be utilized for renewable biofuel production [36]. Therefore, they have the potential to be converted to the largest group of industrial enzymes [37].
Researchers follow two pathways to produce cost-effective cellulase enzymes. (1) They design investigations to increase fungal cellulase production levels in natural hosts and (2) conduct experiences intending to optimize the recombinant expression systems of microorganisms. Heterologus hosts permit the development of microbial strains that are capable of producing a set of synergistic cellulase enzymes. These enzyme sets could be produced by a single cell [38] or by different strain combination [39].
4.1 Recombinant β-glucosidase enzyme activity
We designed the EBG construct that included egxA and cglT cellulase genes and a fluorescent marker gene (zsgreen) and examined the expression of this cellulase set in P. pastoris. These three genes were connected by two 2A sequences (P2A and T2A). Therefore, we expected the whole construct to be transcribed as a polycistronic mRNA and two cellulase genes to be translated equally. We anticipated that the two signal peptides (MF-α and MF4I) would conduct the two cellulase polypeptides towards the endoplasmic reticulum (ER) and Golgi apparatus and thus the final recombinant proteins could be secreted to the culture medium.
We detected recombinant β-glucosidase (CglT) activity in the culture medium of P. pastoris and performed optimization for its better production and activity. The highest CglT activity (821 mU/ml) was obtained in induced culture medium containing 0.5% methanol while the reaction enzyme was performed at 65 ºC and pH 5.5. Primary studies on this enzyme were done with the intracellular recombinant expression of cglT gene in bacterial hosts (E. coli and Bacillus subtilis). The recombinant enzyme activity was measured as 873 mU/ml (mU activity per ml of cell extract). This β-glucosidase enzyme is classified in the GH1 family of glycoside hydrolase enzymes. Two main advantages of this enzyme are high tolerance against glucose inhibition (ki = 200 mM), and its activity at high temperatures [11]. Therefore, CglT, as a good candidate, could be applied to compensate for deficiency of β-glucosidase activity in cellulase sets during biomass decomposition. This enzyme was successfully applied for improving cellulase activities of Clostridium thermocellum S14 cellulosomes. When the bacterial cellulosomes were used for lignocellulosic material decomposition, cellobiose aggregation occurred and led to endoglucanase and exoglucanase inhibition. Adding recombinant CglT to these cellulase sets (cellulosomes) resulted in cellobiose degradation and removing the inhibition of endoglucanase and exoglucanase enzymes. On the other hand, CglT showed high tolerance against glucose and remained active at high glucose concentrations. It was indicated that improved C. thermocellum S14 cellulosomes functioned more efficiently (10 times) than commercial cellulase collection (Celluclast 1.5 L and Novozyme-188) in biomass material degradation [40]. Another research entered CglT into the cellulase enzyme sets of C. thermocellum (CelD, CBHA, CBH48Y) and was succeeded to find a cellulase combination with optimum activity at 60 ºC which degraded 70% of corn stover to glucose after fermentation for 24 h [41]. Such cellulase sets could be used in enzymatic digestion of biomass for biofuel production. Besides, there are various applications for β-glucosidase enzymes in the food industry. These enzymes hydrolyze bitter compounds of fruit juices especially citrus juices [42, 43], release flavors from glycosylated materials presented in plant tissues such as tea [44], and function in extraction pathways of medical compounds (for example isoflavones and genistein) from plant materials [45, 46]. β-Glucosidase enzymes are also applied in the feed industry and help better digestion of cellulosic nutrition by animals and poultry [47, 48].
4.2 Recombinant EGXA protein
We placed egxA gene in EBG construct to obtain endoglucanase and exoglucanase activities and complete cellulase activities of this construct. RT-PCR analysis showed that this gene was transcribed in yeast host cells successfully, whereas we could not detect expected enzyme activities related to EGXA protein in yeast cells and BMMY culture medium. This might be caused by the following reasons:
(1) MF-α signal peptide sequence positioned before egxA gene (as a part of pPICZαA vector) was expected to conduct EGXA protein towards ER to be finally secreted from host cells. MF-α pre-pro signal peptide sequence is the most extensively used secretion signal for recombinant expression of proteins in P. pastoris [49], including fungal proteins [50,51,52], green fluorescent proteins [53], human insulin [54], or human C-reactive protein [55]. The MF-α signal peptide consists of a pre-region (19 amino acids), a pro region (64 amino acids), and a spacer (6 amino acids). The pre region guides the polypeptide chain towards the ER and is deleted by signal peptidase [56,57,58]. Pro region and spacer are required for the final processing of protein in Golgi apparatus and its secretion to the culture medium [56, 59]. Kex2 protease cleaves the pro region after the Lys84-Arg85 sequence [60], four extra amino acids remain at the N-terminal of protein which are deleted by STE13 protease, and mature protein can be secreted to the extracellular space [61]. This signal peptide acts with a post-translational translocation mechanism [62, 63]. Hsp40/Hsp70 chaperons are very important in this mechanism and maintain complete polypeptide chains at a loosely folded state which is a translocation state of polypeptide chains [64]. The expression of recombinant proteins could load additional stress on the translocation pathway of ER and reduce its efficiency. Misfolded recombinant proteins in the cytosol would be sent to the proteasome for degradation.
Because MF-α signal peptide directs post-translational translocation of the related protein into the ER, it might not be suitable for recombinant proteins with intracellular native position, because these proteins are prone to fold in cytosol completely, and their translocation into the ER and subsequently secretion may be performed inefficiently. However, this possibility is weak about EGXA recombinant protein, because it natively is a secretory protein and its folding is done in the lumen of ER. MF-α factor might result in aggregation of EGXA protein at a high-level in the ER lumen. However, an investigation was performed for recombinant expression of E2-Crimson by MF-α factor and a mutated MF-α signal peptide of pPICZα vector (replacement of Leu42 with Ser42) in P. pastoris. It showed that although MF-α factor caused E2-Crimson aggregation in the ER lumen, mutated MF-α factor removed this problem, and no aggregation was observed [65]. We utilized pPICZαA vector, and this mutated signal peptide for EGXA expression, given that E2-Crimson is a dimeric protein and more complex than EGXA protein, we expected that recombinant EGXA could be translocated to the Golgi apparatus efficiently, and it seems that the probability of EGXA aggregation in the ER lumen is weak. However, replacement of the MF-α sequence with another signal peptide such as Ost-1 might improve egxA gene expression in P. pastoris. We will explain that protein digestion by the Golgi proteases is a stronger possibility than wrong signal peptide for EGXA protein.
(2) The secretion of some recombinant proteins could be affected by improper folding in yeast ER [66, 67]. EGXA polypeptide might have been translocated to the ER properly, but misfolding in the ER lumen led to its degradation by endoplasmic reticulum–associated degradation (ERAD) mechanism [68, 69]. In some cases, misfolded proteins were transferred to the yeast Golgi apparatus and then to the vacuole and were degraded by the vacuolar protease complex [70, 71]. Yeast cells possess a quality control system that detects the misfolding proteins by vacuolar protein sorting receptors (VPSRs) at the Golgi apparatus and targets them to the vacuole [72]. It was indicated that these VPSRs could distinguish recombinant proteins with suitable folding and direct them towards vacuole [53, 73]. This proposed that these receptors extended their ligand range to proteins which their conformation was different from normal protein conformation of the yeast secretory pathway [53]. Yeast cells without VPSRs secreted foreign proteins more efficiently [53, 74, 75]. It must be mentioned that VPSR deletion led to vacuolar hydrolases to be presented in culture medium of yeast cells [76]. These hydrolases might contribute to secreted recombinant protein degradation in culture medium.
(3) We attempted to investigate the likely digestion of EGXA by P. pastoris proteases. Most likely candidates which could influence recombinant EGXA are the Golgi apparatus proteases (including Kex2 and Yapsin 1). Kex2 is responsible for cleavage of the pro part of α-factor signal peptide and produces a mature secretory protein in the trans-Golgi saccules. In the absence of the Kex2 enzyme, Yapsin 1 can carry out this function. These enzymes identify Lys-Arg or Arg-Arg residues and hydrolyze the peptide bond after Arg. Therefore, a mature secretory protein could be targeted out of the cell [77,78,79]. These enzymes can potentially identify the Lys-Arg or Arg-Arg residues that exist throughout polypeptide chains. Of course, other residues surrounding the dipeptide (two residues before and three residues after the Lys-Arg) are important for recognition. Bader et al. [34] scored four positions of X-X-Lys-Arg or X-X-Arg-Arg (P4, P3, P2, and P1, respectively) according to Kex2 substrates. They tried to predict the potential substrates of this enzyme. In addition to this scoring, they stated that the presence of small or acidic amino acids (Ala, Val, Ile, Asp or Glu) at positions P1′, P2′, and P4′ (first, second, and fourth residues after Lys-Arg or Arg-Arg dipeptide) increased the chance of dipeptide recognition by Kex2 enzyme and presence of basic amino acids (Arg, Lys, or His) at these positions decreased the possibility of digestion site recognition by the enzyme. It seems that proline presence at P1′ position inhibits the enzyme activity [80]. We utilized this information to predict the possible cleavage of our recombinant proteins which were expressed in P. pastoris. Six potential cleavage sites for EGXA polypeptide chain were detected from which two cleavage sequences obtained a high score (Table 2). It seems that the Kex2 function is the strongest potential cause of EGXA protein digestion. It appears that EGXA proteins lost their activities as a result of polypeptide chain cleavage.
Although enzyme activities of EGXA were not detected in culture media and intracellular-extracted proteins of recombinant P. pastoris cells that had received pPICZαA-EBG vector, insect Tn5 cells [81] and mammalian cells including cell lines PK15, HEK293A, 3T3, and CHO 1–15 [82] were suitable hosts for the heterologous expression of this protein.
5 Conclusion
The expression of polycistronic EBG construct (including cellulase activities combination) partly succeeded in yeast P. pastoris cells and led to secretion of recombinant β-glucosidase (CglT) enzyme (from T. brockii) to the culture medium. It seems that P. pastoris is not a suitable host cell for recombinant expression of egxA gene from Ampularia crossean, and its expression is forced with some post-translational problems and degradation in P. pastoris cells. Recombinant CglT is a good candidate for enforcing thermophilic cellulase enzyme sets which could not function properly because of inefficient β-glucosidase activity.
References
Béguin P, Aubert JP (1994) The biological degradation of cellulose. FEMS Microbiol Rev 13(1):25–58. https://doi.org/10.1111/j.1574-6976.1994.tb00033.x
Ando S, Ishida H, Kosugi Y, Ishikawa K (2002) Hyperthermostable endoglucanase from Pyrococcus horikoshii. Appl Environ Microbiol 68(1):430–433. https://doi.org/10.1128/AEM.68.1.430-433.2002
Dhiman T, Zaman M, Gimenez R, Walters J, Treacher R (2002) Performance of dairy cows fed forage treated with fibrolytic enzymes prior to feeding. Anim Feed Sci Technol 101(1–4):115–125. https://doi.org/10.1016/S0377-8401(02)00177-3
Olofsson K, Bertilsson M, Lidén G (2008) A short review on SSF–an interesting process option for ethanol production from lignocellulosic feedstocks. Biotechnol Biofuels 1(1):7. https://doi.org/10.1186/1754-6834-1-7
Baffi MA, Tobal T, Lago JHG, Boscolo M, Gomes E, Da-Silva R (2013) Wine aroma improvement using a β-glucosidase preparation from Aureobasidium pullulans. Appl Biochem Biotechnol 169(2):493–501. https://doi.org/10.1007/s12010-012-9991-2
Bayer EA, Lamed R, Himmel ME (2007) The potential of cellulases and cellulosomes for cellulosic waste management. Curr Opin Biotechnol 18(3):237–245. https://doi.org/10.1016/j.copbio.2007.04.004
Menetrez MY (2012) An overview of algae biofuel production and potential environmental impact. Environ Sci Technol 46(13):7073–7085. https://doi.org/10.1021/es300917r
Pandey K, Singh B, Pandey AK, Badruddin IJ, Pandey S, Mishra VK et al (2017) Application of microbial enzymes in industrial waste water treatment. Int J Curr Microbiol App Sci 6(8):1243–1254. https://doi.org/10.20546/ijcmas.2017.608.151
Kumar S, Nussinov R (2001) How do thermophilic proteins deal with heat? Cell Mol Life Sci 58(9):1216–1233. https://doi.org/10.1007/PL00000935
Ding M, Teng Y, Yin Q, Zhao J, Zhao F (2008) The N‐terminal cellulose‐binding domain of EGXA increases thermal stability of xylanase and changes its specific activities on different substrates (2008). Acta Biochim Biophys Sin 40(11):949–954. https://doi.org/10.1111/j.1745-7270.2008.00481.x
Breves R, Bronnenmeier K, Wild N, Lottspeich F, Staudenbauer WL, Hofemeister J (1997) Genes encoding two different β-glucosidases of Thermoanaerobacter brockii are clustered in a common operon. Appl Environ Microbiol 63(10):3902–3910. https://doi.org/10.1128/AEM.63.10.3902-3910.1997
Cregg JM, Vedvick TS, Raschke WC (1993) Recent advances in the expression of foreign genes in Pichia pastoris. Nat Biotechnol 11(8):905. https://doi.org/10.1038/nbt0893-905
Ahmad M, Hirz M, Pichler H, Schwab H (2014) Protein expression in Pichia pastoris: recent achievements and perspectives for heterologous protein production. Appl Microbiol Biotechnol 98(12):5301–5317
Mellitzer A, Weis R, Glieder A, Flicker K (2012) Expression of lignocellulolytic enzymes in Pichia pastoris. Microb Cell Factories 11(1):61. https://doi.org/10.1007/s00253-014-5732-5
Ryan MD, King AM, Thomas GP (1991) Cleavage of foot-and-mouth disease virus polyprotein is mediated by residues located within a 19 amino acid sequence. J Gen Virol 72(11):2727–2732. https://doi.org/10.1099/0022-1317-72-11-2727
Donnelly ML, Hughes LE, Luke G, Mendoza H, Ten Dam E, Gani D, Ryan MD (2001) The ‘cleavage’activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring ‘2A-like’sequences. J Gen Virol 82(5):1027–1041. https://doi.org/10.1099/0022-1317-82-5-1027
de Felipe P, Luke GA, Hughes LE, Gani D, Halpin C, Ryan MD (2006) E unum pluribus: multiple proteins from a self-processing polyprotein. Trends Biotechnol 24(2):68–75. https://doi.org/10.1016/j.tibtech.2005.12.006
Subramanian V, Schuster LA, Moore KT, Taylor LE, Baker JO, Vander Wall TA, Linger JG, Himmel ME, Decker SR (2017) A versatile 2A peptide-based bicistronic protein expressing platform for the industrial cellulase producing fungus Trichoderma reesei. Biotechnol Biofuels 10(1):1–15. https://doi.org/10.1186/s13068-017-0710-7
Liu Z, Chen O, Wall JBJ, Zheng M, Zhou Y, Wang L, Vaseghi HR, Qian L, Liu J (2017) Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci Rep 7(1):1–9. https://doi.org/10.1038/s41598-017-02460-2
Jiao X, Sun W, Zhang Y, Liu X, Zhang Q, Wang Q, Zhang S, Zhao ZK (2018) Exchanging the order of carotenogenic genes linked by porcine teschovirus-1 2A peptide enable to optimize carotenoid metabolic pathway in Saccharomyces cerevisiae. RSC Adv 8(61):34967–34972. https://doi.org/10.1039/c8ra06510a
Geier M, Fauland P, Vogl T, Glieder A (2015) Compact multi-enzyme pathways in P. pastoris. Chem Comm 51(9):1643–1646. https://doi.org/10.1039/c4cc08502g
Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF et al (2004) Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide–based retroviral vector. Nat Biotechnol 22(5):589–594. https://doi.org/10.1038/nbt957
Xiong AS, Yao QH, Peng RH, Han PL, Cheng ZM, Li Y (2005) High level expression of a recombinant acid phytase gene in Pichia pastoris. J Appl Microbiol 98(2):418–428. https://doi.org/10.1111/j.1365-2672.2004.02476.x
Green M, Sambrook J (2012) Molecular cloning: a laboratory manual, 4th edn. Cold Spring Harbor Laboratory Press, New York
Inoue H, Nojima H, Okayama H (1990) High efficiency transformation of Escherichia coli with plasmids. Gene 96(1):23–28. https://doi.org/10.1016/0378-1119(90)90336-p
Burgess RR (2009) Protein precipitation techniques. Methods Enzymol 463:331–342. https://doi.org/10.1016/S0076-6879(09)63020-2
Çağlayan M, Wilson SH (2014) Enzymatic activity assays in yeast cell extracts. Bio-protocol 4(23):e1312. https://doi.org/10.21769/BioProtoc.1312
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31(3):426–428. https://doi.org/10.1021/ac60147a030
Zhang YP, Hong J, Ye X (2009) Cellulase assays. Methods Mol Biol 581:213–231. https://doi.org/10.1007/978-1-60761-214-8_14
Kim YK, Lee SC, Cho YY, Oh HJ, Ko YH (2012) Isolation of cellulolytic Bacillus subtilis strains from agricultural environments. ISRN Microbiol 2012. https://doi.org/10.5402/2012/650563
Deshpande MV, Eriksson KE, Pettersson LG (1984) An assay for selective determination of exo-1, 4,-β-glucanases in a mixture of cellulolytic enzymes. Anal Biochem 138(2):481–487. https://doi.org/10.1016/0003-2697(84)90843-1
Simpson RJ (2006) SDS-PAGE of proteins. Cold Spring Harb Protoc. https://doi.org/10.1101/pdb.prot4313
Brunelle JL, Green R (2014) Coomassie blue staining. Methods Enzymol 541:161–167. https://doi.org/10.1016/B978-0-12-420119-4.00013-6
Bader O, Krauke Y, Hube B (2008) Processing of predicted substrates of fungal Kex2 proteinases from Candida albicans, C. glabrata, Saccharomyces cerevisiae and Pichia pastoris. BMC Microbiol 8(1):1–16. https://doi.org/10.1186/1471-2180-8-116
Bhat M (2000) Cellulases and related enzymes in biotechnology. Biotechnol Adv 18(5):355–383. https://doi.org/10.1016/s0734-9750(00)00041-0
Phitsuwan P, Laohakunjit N, Kerdchoechuen O, Kyu KL, Ratanakhanokchai K (2013) Present and potential applications of cellulases in agriculture, biotechnology, and bioenergy. Folia Microbiol 58(2):163–176. https://doi.org/10.1007/s12223-012-0184-8
Wilson DB (2009) Cellulases and biofuels. Curr Opin Biotechnol 20(3):295–299. https://doi.org/10.1016/j.copbio.2009.05.007
Mazzoli R, Lamberti C, Pessione E (2012) Engineering new metabolic capabilities in bacteria: lessons from recombinant cellulolytic strategies. Trends Biotechnol 30(2):111–119. https://doi.org/10.1016/j.tibtech.2011.08.003
Tsai SL, Oh J, Singh S, Chen R, Chen W (2009) Functional assembly of minicellulosomes on the Saccharomyces cerevisiae cell surface for cellulose hydrolysis and ethanol production. Appl Environ Microbiol 75(19):6087–6093. https://doi.org/10.1128/AEM.01538-09
Waeonukul R, Kosugi A, Tachaapaikoon C, Pason P, Ratanakhanokchai K, Prawitwong P et al (2012) Efficient saccharification of ammonia soaked rice straw by combination of Clostridium thermocellum cellulosome and Thermoanaerobacter brockii β-glucosidase. Bioresour Technol 107:352–357. https://doi.org/10.1016/j.biortech.2011.12.126
Geng A, Wu J, Xie R, Li X, Chang F, Sun J (2015) Construction of a bacterial cellulase cocktail for saccharification of regenerated cellulose and pretreated corn stover. BioResources 10(4):7681–92. https://doi.org/10.15376/biores.10.4.7681-7692
Gueguen Y, Chemardin P, Janbon G, Arnaud A, Galzy P (1998) Investigation of the β-glucosidases potentialities of yeast strains and application to bound aromatic terpenols liberation. Stud Org Chem 53:149–157. https://doi.org/10.1016/S0165-3253(98)80018-7
Gupta A, Kumar V, Dubey A, Verma A (2014) Kinetic characterization and effect of immobilized thermostable β-glucosidase in alginate gel beads on sugarcane juice. ISRN Biochem 2014.https://doi.org/10.1155/2014/178498
Su E, Xia T, Gao L, Dai Q, Zhang Z (2010) Immobilization of β-glucosidase and its aroma-increasing effect on tea beverage. Food Bioprod Process 88(2):83–89. https://doi.org/10.1016/j.fbp.2009.04.001
Hu SC, Hong K, Song YC, Liu JY, Tan RX (2009) Biotransformation of soybean isoflavones by a marine Streptomyces sp. 060524 and cytotoxicity of the products. World J Microbiol Biotechnol 25(1):115. https://doi.org/10.1007/s11274-008-9872-6
Pandjaitan N, Hettiarachchy N, Ju Z (2000) Enrichment of genistein in soy protein concentrate with β-glucosidase. J Food Sci 65(3):403–407. https://doi.org/10.1111/j.1365-2621.2000.tb16055.x
Coenen T, Schoenmakers A, Verhagen H (1995) Safety evaluation of β-glucanase derived from Trichoderma reesei: summary of toxicological data. Food Chem Toxicol 33(10):859–866. https://doi.org/10.1016/0278-6915(95)00052-4
Zhang Z, Marquardt RR, Wang G, Guenter W, Crow GH, Han Z et al (1996) A simple model for predicting the response of chicks to dietary enzyme supplementation. J Anim Sci 74(2):394–402. https://doi.org/10.2527/1996.742394x
Aggarwal S, Mishra S (2020) Differential role of segments of α-mating factor secretion signal in Pichia pastoris towards granulocyte colony-stimulating factor emerging from a wild type or codon optimized copy of the gene. Microb Cell Fact 19(1):1–16. https://doi.org/10.1186/s12934-020-01460-8
Rotticci-Mulder JC, Gustavsson M, Holmquist M, Hult K, Martinelle M (2001) Expression in Pichia pastoris of Candida antarctica lipase B and lipase B fused to a cellulose-binding domain. Protein Expr Purif 21(3):386–392. https://doi.org/10.1006/prep.2000.1387
Camarero S, Pardo I, Cañas AI, Molina P, Record E, Martínez A, Martínez MJ, Alcalde M (2012) Engineering platforms for directed evolution of laccase from Pycnoporus cinnabarinus. Appl Environ Microbiol 78(5):1370–1384. https://doi.org/10.1128/AEM.07530-11
Ardila-Leal LD, Alvarado-Ramírez MF, Gutiérrez-Rojas IS, Poutou-Pinales RA, Quevedo-Hidalgo B, Pérez-Flórez A, Pedroza-Rodríguez AM (2020) Low-cost media statistical design for laccase rPOXA 1B production in P. pastoris. Heliyon 6(4):e03852. https://doi.org/10.1016/j.heliyon.2020.e03852
Fitzgerald I, Glick BS (2014) Secretion of a foreign protein from budding yeasts is enhanced by cotranslational translocation and by suppression of vacuolar targeting. Microb Cell Fact 13(1):125. https://doi.org/10.1186/s12934-014-0125-0
Kjeldsen T, Ludvigsen S, Diers I, Balschmidt P, Sørensen AR, Kaarsholm NC (2002) Engineering-enhanced protein secretory expression in yeast with application to insulin. J Biol Chem 277(21):18245–18248. https://doi.org/10.1074/jbc.C200137200
Li J, Sun C, Chen L, Sun L, Duan L, Zheng Q, Hu X (2017) Optimization of the secretory expression of recombinant human C-reactive protein in Pichia pastoris. 3 Biotech 7(5):1–8. https://doi.org/10.1007/s13205-017-0917-0
Fuller RS, Sterne RE, Thorner J (1988) Enzymes required for yeast prohormone processing. Annu Rev Physiol 50(1):345–362. https://doi.org/10.1146/annurev.ph.50.030188.002021
Waters MG, Evans EA, Blobel G (1988) Prepro-alpha-factor has a cleavable signal sequence. J Biol Chem 263(13):6209–6214. https://doi.org/10.1016/S0021-9258(18)68773-3
Paetzel M, Karla A, Strynadka NC, Dalbey RE (2002) Signal peptidases. Chem Rev 102(12):4549–4580. https://doi.org/10.1021/cr010166y
Singh A, Lugovoy JM, Kohr WJ, Perry LJ (1984) Synthesis, secretion and processing of α-factor-interferon fusion proteins Id yeast. Nucleic Acids Res 12(23):8927–8938. https://doi.org/10.1093/nar/12.23.8927
Germain D, Dumas F, Vernet T, Bourbonnais Y, Thomas DY, Boileau G (1992) The pro-region of the Kex2 endoprotease of Saccharomyces cerevisiae is removed by self-processing. FEBS Lett 299(3):283–286. https://doi.org/10.1016/0014-5793(92)80132-z
Julius D, Blair L, Brake A, Sprague G, Thorner J (1983) Yeast α factor is processed from a larger precursor polypeptide: the essential role of a membrane-bound dipeptidyl aminopeptidase. Cell 32(3):839–852. https://doi.org/10.1016/0092-8674(83)90070-3
Ng DT, Brown JD, Walter P (1996) Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J Cell Biol 134(2):269–278. https://doi.org/10.1083/jcb.134.2.269
Plath K, Mothes W, Wilkinson BM, Stirling CJ, Rapoport TA (1998) Signal sequence recognition in posttranslational protein transport across the yeast ER membrane. Cell 94(6):795–807. https://doi.org/10.1016/s0092-8674(00)81738-9
Ngosuwan J, Wang NM, Fung KL, Chirico WJ (2003) Roles of cytosolic Hsp70 and Hsp40 molecular chaperones in post-translational translocation of presecretory proteins into the endoplasmic reticulum. J Biol Chem 278(9):7034–7042. https://doi.org/10.1074/jbc.M210544200
Barrero JJ, Casler JC, Valero F, Ferrer P, Glick BS (2018) An improved secretion signal enhances the secretion of model proteins from Pichia pastoris. Microb Cell Fact 17(1):1–13. https://doi.org/10.1186/s12934-018-1009-5
Finger A, Knop M, Wolf DH (1993) Analysis of two mutated vacuolar proteins reveals a degradation pathway in the endoplasmic reticulum or a related compartment of yeast. FEBS J 218(2):565–574. https://doi.org/10.1111/j.1432-1033.1993.tb18410.x
Kang HA, Lee KN, Yu MH (1997) Folding and stability of the Z and Siiyama genetic variants of human α1-antitrypsin. J Biol Chem 272(1):510–516. https://doi.org/10.1074/jbc.272.1.510
Hiller MM, Finger A, Schweiger M, Wolf DH (1996) ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science 273(5282):1725. https://doi.org/10.1126/science.273.5282.1725
Werner ED, Brodsky JL, McCracken AA (1996) Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc Natl Acad Sci 93(24):13797–13801. https://doi.org/10.1073/pnas.93.24.13797
Hong E, Davidson AR, Kaiser CA (1996) A pathway for targeting soluble misfolded proteins to the yeast vacuole. J Cell Biol 135(3):623–633. https://doi.org/10.1083/jcb.135.3.623
Holkeri H, Makarow M (1998) Different degradation pathways for heterologous glycoproteins in yeast. FEBS Lett 429(2):162–166. https://doi.org/10.1016/s0014-5793(98)00586-9
Jørgensen MU, Emr SD, Winther JR (1999) Ligand recognition and domain structure of Vps10p, a vacuolar protein sorting receptor in Saccharomyces cerevisiae. FEBS J 260(2):461–469. https://doi.org/10.1046/j.1432-1327.1999.00176.x
Zhang BY, Chang A, Kjeldsen TB, Arvan P (2001) Intracellular retention of newly synthesized insulin in yeast is caused by endoproteolytic processing in the Golgi complex. J Cell Biol 153(6):1187–1198. https://doi.org/10.1083/jcb.153.6.1187
Rakestraw JA, Sazinsky SL, Piatesi A, Antipov E, Wittrup KD (2009) Directed evolution of a secretory leader for the improved expression of heterologous proteins and full-length antibodies in Saccharomyces cerevisiae. Biotechnol Bioeng 103(6):1192–1201. https://doi.org/10.1002/bit.22338
Wang TY, Huang CJ, Chen HL, Ho PC, Ke HM, Cho HY et al (2013) Systematic screening of glycosylation-and trafficking-associated gene knockouts in Saccharomyces cerevisiae identifies mutants with improved heterologous exocellulase activity and host secretion. BMC Biotechnol 13(1):71. https://doi.org/10.1186/1472-6750-13-71
Marcusson EG, Horazdovsky BF, Cereghino JL, Gharakhanian E, Emr SD (1994) The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by the VPS10 gene. Cell 77(4):579–586. https://doi.org/10.1016/0092-8674(94)90219-4
Brake AJ (1990) α-Factor leader-directed secretion of heterologous proteins from yeast. Methods Enzymol 185:408–421. https://doi.org/10.1016/0076-6879(90)85036-n
Brenner C, Fuller RS (1992) Structural and enzymatic characterization of a purified prohormone-processing enzyme: secreted, soluble Kex2 protease. Proc Natl Acad Sci USA 89(3):922–926. https://doi.org/10.1073/pnas.89.3.922
Cawley NX, Olsen V, Zhang CF, Chen HC, Tan M, Loh YP (1998) Activation and processing of non-anchored yapsin 1 (Yap3p). J Biol Chem 273(1):584–591. https://doi.org/10.1074/jbc.273.1.584
Xie YF, Chen H, Huang BR (2007) Expression, purification and characterization of human IFN-λ1 in Pichia pastoris. J Biotechnol 129(3):472–480. https://doi.org/10.1016/j.jbiotec.2007.01.018
Ding M, Teng Y, Yin Q, Zhao J, Zhao F (2008) The N-terminal cellulose-binding domain of EGXA increases thermal stability of xylanase and changes its specific activities on different substrates. Acta Biochim Biophys Sin 40(11):949–954. https://doi.org/10.1111/j.1745-7270.2008.00481.x
Liu Z, Sun Y, Feng T, Ji Q, Cong P, Chen Y, He Z (2014) Mammalian expression levels of cellulase and xylanase genes optimised by human codon usage are not necessarily higher than those optimised by the extremely biased approach. Biotechnol Lett 36(11):2169–2176. https://doi.org/10.1007/s10529-014-1592-4
Acknowledgements
We would like to appreciate Dr. Moein Farshchian for his assistance in gene construct design.
Funding
This work was supported by the Biotechnology Development Council (grant number, 100485) and Ferdowsi University of Mashhad (grant number, 29524).
Author information
Authors and Affiliations
Contributions
Conceptualization: Bahrami, Matin, Javanmard
Methodology: Javanmard
Data analysis: Javanmard
Writing—original draft preparation: Javanmard
Writing—review, and editing: Bahrami, Matin
Supervision: Bahrami, Matin
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Javanmard, A.S., Matin, M. & Bahrami, A.R. Polycistronic cellulase gene expression in Pichia pastoris. Biomass Conv. Bioref. 13, 7151–7163 (2023). https://doi.org/10.1007/s13399-021-01765-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13399-021-01765-7