
Abstract
Background
Breast cancer, a genetically intricate disease with diverse subtypes, exhibits heightened incidence globally. In this study, we aimed to investigate blood-based microRNAs (miRNAs) as potential biomarkers for breast cancer. The primary objectives were to explore the role of miRNAs in cancer-related processes, assess their differential expression between breast cancer patients and healthy individuals, and contribute to a deeper understanding of the molecular underpinnings of breast cancer.
Methods
MiRNA extraction was performed on 40 breast cancer patients and adjacent normal tissues using a commercial RNA isolation kit. Total RNA quantification and quality assessment were conducted with advanced technologies. MiRNA profiling involved reverse transcription, labeling, and hybridization on Agilent human miRNA arrays (V2). Bioinformatics analysis utilized the DIANA system for target gene prediction and the DIANA-mirPath tool for pathway enrichment analysis. Selected miRNAs underwent validation through quantitative real-time PCR.
Results
Principal component analysis revealed overlapping miRNA expression patterns in primary and malignant breast tumors, underscoring the genetic complexity involved. Statistical analysis identified 54 downregulated miRNAs in malignant tumors and 38 in primary tumors compared to controls. Bioinformatics analysis implicated several pathways, including Wnt, TGF-b, ErbB, and MAPK signaling. Validation through qRT-PCR confirmed altered expression of hsa-miR-130a, hsa-miR-21, hsa-miR-223, and hsa-let-7c key miRNAs, highlighting their significance in breast cancer. The results from microarray were further validated by qPCR and the expression of which are downregulated in breast cancer was detected.
Conclusion
This study provides significant insights into distinct miRNA expression patterns in normal and malignant breast tissues. The overlapping miRNA profiles in primary and malignant tumors underscore the complexity of genetic regulation in breast cancer. The identification of deregulated miRNAs and affected pathways contributes to our understanding of breast cancer pathogenesis. The validated miRNAs hold potential as diagnostic and prognostic markers, offering avenues for further clinical exploration in breast cancer research.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer’s complexity is attributed to its heterogeneous genetic alterations, gene expression signatures, and phenotypic differences. Breast cancer has been classified into six primary subtypes, comprising luminal A, luminal B, basal-like, normal-like, claudin-low subtype, and tumors enriched with human epidermal growth factor receptor 2 (Her2) (Sorlie et al. 2001; Perou et al. 2000). This disease also exhibits marked diversity apart from morphological features and clinical outcomes (Al Tamimi et al. 2010; Tarone 2006). There has been an increase of 3.1% breast cancer cases in developing nations (Al-Foheidi et al. 2013; Forouzanfar et al. 2011). Breast cancer is a heterogeneous disease comprising distinct molecular subtypes, including luminal A, luminal B, HER2-enriched, and basal-like (triple-negative) subtypes. Each subtype exhibits unique miRNA expression profiles, reflecting underlying differences in tumor biology and clinical behavior. However, comprehensive characterization of miRNA profiles across breast cancer subtypes is still incomplete (Volinia et al. 2012a, b). Identifying subtype-specific miRNA signatures could enhance our understanding of disease heterogeneity and facilitate the development of personalized therapeutic approaches.
The frequency of breast cancer is low in the Saudi Arabian population in comparison with the Western world, yet it has the highest occurring malignancy among Saudi females with a frequency of 21.1% (Cancer Incidence Report, 2002).
Blood-based microRNAs (miRNAs) have emerged as a promising field of research in the quest for effective biomarkers across various cancer types, including breast cancer. These tiny RNA molecules, approximately 22 nucleotides in length, have garnered attention due to their potential role in cancer-related processes such as invasiveness, development of therapeutic resistance, and adverse clinical outcomes, as highlighted by studies such as those by Iorio et al. in 2008 and van Schooneveld et al. in 2012. One of the key areas of investigation involves exploring the differences in miRNA expression levels between breast cancer patients and healthy individuals. This comparison is integral to the exploration of blood-based miRNAs as potential diagnostic and prognostic biomarkers, as elucidated by Lagendijk et al. 2018.
At a molecular level, miRNAs are intriguing non-coding RNA molecules that play a pivotal role in gene regulation. They can influence gene expression by either cleaving the messenger RNA (mRNA) molecule or inhibiting its translation, as described by Bartel in 2004. This dual capacity for gene regulation suggests that miRNAs have the potential to be both oncogenic and tumor suppressors, depending on the specific target genes they influence. For instance, miR-10b has been identified as overexpressed in breast cancer patients experiencing regional and distant relapse, as well as local recurrence, as demonstrated by Ma et al. in 2007. A notable example is miR-21, an “oncomir” in breast cancer, which acts on genes like BCL2, PTEN, and tropomyosin 1, all of which are involved in the intricate processes of cell cycle progression and the regulation of apoptosis. This dual role of miRNAs, as oncogenic drivers or tumor suppressors, further underscores their significance in breast cancer pathogenesis, as observed in studies by Frankel et al. in 2007 and Si et al. in 2007.
MicroMRNAs (miRNAs) are small, endogenous RNA molecules that play critical roles in post-transcriptional gene regulation. They are involved in diverse cellular processes, including proliferation, differentiation, and apoptosis. Dysregulation of miRNAs has been implicated in various diseases, particularly cancer, where they can act as oncogenes or tumor suppressors.
Extensive research has demonstrated the significance of miRNAs in cancer pathogenesis. They are involved in regulating key signaling pathways implicated in tumorigenesis and metastasis. For instance, miR-21 is commonly upregulated in many cancers, promoting cell proliferation and invasion (Fang et al. 2020). Conversely, miR-34a functions as a tumor suppressor by targeting genes involved in cell cycle progression and apoptosis (Hermeking 2012).
While the precise mechanisms and relationships between deregulated miRNAs and cancer remain subjects of ongoing investigation, there is growing evidence that a significant portion of human miRNAs reside within genomic regions associated with cancer. These regions include those implicated in loss-of-function events, gain-of-function events, and genomic rearrangements, as elucidated by Calin et al. in 2004. This insight underscores the complex interplay between miRNAs and cancer-related genomic alterations and highlights the potential of miRNAs as crucial players in the molecular landscape of breast cancer.
Current diagnostic and prognostic strategies for breast cancer rely primarily on clinicopathological features, such as tumor size, lymph node status, and hormone receptor expression. However, these parameters have limitations in accurately predicting disease outcome and treatment response. Moreover, conventional biomarkers, such as HER2 status, may not fully capture the complexity of breast cancer biology. Integrating miRNA expression profiles into existing diagnostic and prognostic models holds promise for improving risk stratification and treatment decision-making (Yan et al. 2021).
The primary objective of this research was to investigate the complexities of miRNA expression patterns within the context of breast cancer. To accomplish this, a comprehensive investigation was conducted, encompassing the analysis of miRNA profiles in a cohort of 24 breast tumors. These tumor samples were methodically compared with 16 corresponding adjacent normal tissues, forming the basis for a comparative exploration of miRNA expression in the context of malignancy. To support the reliability and robustness of the findings, the differential expression of miRNAs was rigorously examined and validated. This validation process involved an independent dataset, comprising a distinct set of normal and tumor tissue samples. This methodological approach was undertaken to ensure that the observed differences in miRNA expression were not coincidental or influenced by potential confounding factors, but rather reflective of genuine variations associated with the presence of breast cancer. In breast cancer, the role of miRNAs can be a multifaceted area of investigation by which molecular mechanisms underlying tumorigenic, progression of disease, and therapeutic resistance can be evaluated (liang et al. 2020). In conclusion, this comprehensive study sought to shed light on the distinctive miRNA expression patterns inherent to breast cancer, thereby contributing to a deeper understanding of the molecular underpinnings of this complex disease.
Materials and methods
MicroRNA extraction
Extraction of total RNA from 40 breast cancer patients and their adjacent normal part was done using commercial RNA isolation kit (Qiagen, Germany). Quantification of the extracted mRNA was conducted with the NanoDrop ND-1000 Spectrophotometer from Thermo Fisher Scientific, USA, while the assessment of RNA sample quality was performed using the Agilent 2100 Bioanalyzer, a product of Agilent Technologies, also based in the USA.
MicroRNA arrays
A total of 2 μg of each sample, derived from both breast cancer and normal tissue, underwent reverse transcription to produce cDNA. Subsequently, this cDNA was labeled with a miRNA labeling reagent and subjected to hybridization on the Agilent human miRNA arrays (V2), a cutting-edge technology developed by Agilent Technologies in the USA. The miRNA array utilized in our study encompassed a comprehensive array of features drawn from the Sanger database v.10.1, spanning 923 human miRNAs and 76 viral miRNAs. Following the hybridization process, the microarray chips were subjected to a thorough washing procedure using the Wash Buffer Kit, ensuring the removal of non-specific binding and enhancing the specificity of the results. Finally, the processed microarray chips were scanned, employing advanced technology from Agilent Technologies in the USA, to extract valuable data from the hybridized samples.
Bioinformatics analysis
The microarray data analysis generated list of enriched miRNAs was further studied to identify potential human miRNA target genes by the DNA Intelligent Analysis (DIANA) system viz. DIANA-microT-CDS server v5.0. Target genes prediction through this system involves databases like TargetScan, miRanda, RNAhybrid, and PicTar 4-way. It performs functional analysis and provides full outcome of each target (Paraskevopoulou et al. 2013). Genes predicted under the relevant category by two or more tools were included under the potential candidate category. Additionally, we compiled a catalog of potential target genes based on the existing understanding of miRNAs’ roles in breast cancer. This comprehensive list of putative targets has been conveniently summarized in supplementary Table S1 for reference. To further explore the impact of miRNAs on cellular processes, we connected the power of the DIANA-mirPath web-based computational tool, as described by Papadopoulos and colleagues in 2009. This tool facilitated the identification of molecular pathways that might undergo alterations in response to changes in the expression levels of multiple miRNAs, providing deeper insights into the regulatory mechanisms at play in breast cancer.
cDNA synthesis and quantitative real-time polymerase chain reaction (qRT-PCR)
To validate the findings from the microarray and bioinformatics analyses, a subset of deregulated miRNAs exhibiting significant up and downregulation were randomly selected. These chosen miRNAs underwent further validation through a SYBR-based qRT-PCR assay, conducted using the Qiagen kit (Germany). The reverse transcription process to generate complementary DNA (cDNA) was carried out utilizing the miScript Reverse Transcription (RT) kit, also from Qiagen in Germany. In this procedure, 1 μg of total RNA was combined with 1 μL of miScript Reverse Transcriptase Mix and 4 μL of miScript RT buffer. The mixture was incubated at 37 °C for 60 min, followed by a denaturation step at 95 °C for 5 min.
Quantitative RT-PCR was performed using the Applied Biosystems 7500 Fast Sequence Detection System, an advanced instrument from Applied Biosystems, USA. The miScript SYBR Green PCR Kit from Qiagen USA was employed for this purpose. The amplification protocol included an initial denaturation step at 95 °C for 10 min, followed by 40 amplification cycles, consisting of 94 °C for 15 s, 55 °C for 30 s, and 70 °C for 30 s. It is important to note that all real-time PCR reactions were performed in triplicates to ensure precision, and the resultant products were subjected to a melting curve analysis.
Statistical analysis
Microarray images were automatically processed using Agilent Feature Extraction Software v.9.5.3 from Agilent Technologies, USA. The resulting raw expression data was then imported into the R package via the Bioconductor’s LIMMA package. Differential miRNA expression was analyzed using the empirical Bayes method, and preprocessing was performed to normalize intensity distribution within and between arrays using the quantile-scaling algorithm in LIMMA. Data filtering based on flags was applied to identify differentially expressed features among study groups, reducing the number of uninformative features. The Benjamini–Hochberg method controlled false discovery rates (FDR) by adjusting p-values. To account for potential variations in miRNA quantities, qRT-PCR data was utilized to understand relative miRNA expression levels through the 2-ΔΔCt method.
Results
MicroRNA expression pattern
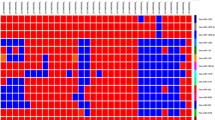
The PCA of all miRNAs from primary and malignant breast cancer was done to study pattern in regulation and the entire dataset was mapped on a three-axis graph as principal components, i.e., PC1, PC2, and PC3. The analysis detected both primary and malignant tumors to exhibit similar pattern of miRNA expression in comparison to the control group and thus indicates the complexity of genetic regulation. PCA of microarray data detected overlapping of miRNA expression pattern in both primary and malignant tumor in comparison to controls (Fig. 1a). The heat map of miRNA expression differentiated into normal and overlapped primary and malignant breast tumors by hierarchical clustering analysis (Fig. 1b).

Unsupervised clustering of the microRNA (miRNA) microarray data. A PCA of the miRNA expression patterns in primary tumor, malignant tumor, and healthy controls. The PCA (in red, healthy controls; in blue primary tumors and in green malignant tumors) show a distinct directionality in different groups based on similarities and differences. The axes correspond to principal component 1 (PC1, x-axis), PC2 (y-axis), and PC3 (z-axis). B Unsupervised hierarchical clustering and heatmap representation of miRNA expression pattern in primary tumor, malignant tumor and healthy control. The color-coded scale (green = downregulation and red = upregulation) for the normalized expression value is indicated at the top of the figure
Analysis of deregulated miRNAs
The restrictive statistical analysis and fold change for comparing the combination of the primary, malignant, and control group identified key miRNAs with altered expression pattern. In the malignant tumor, two miRNAs were upregulated and 54 miRNAs were downregulated in comparison to controls. In primary tumor group, a total of two miRNAs were upregulated, and 38 miRNAs were downregulated in comparison to the control group (Table 1). The miRNAs exhibiting similar expression pattern in primary and malignant tumor were further analyzed. The miRNome expression pattern between primary and malignant tumors was insignificant (Fig.S1) (Fig. 2).

The pathway histogram of the altered miRNAs in breast cancer that were incorporated using DIANA-mirPath. Statistically significant biological pathways potentially affected by the differentially expressed microRNAs in breast cancer
Bioinformatic analysis to identify putative miRNA targets and pathways
To unveil the role of miRNA-mediated pathways in breast cancer, we employed an enriched set of deregulated miRNAs from malignant cases and compared them with those from healthy controls. This comparative analysis allowed us to identify and incorporate potentially affected pathways using the DIANA-mirPath tool. The examination revealed that several pathways were prominently impacted, including the Wnt signaling pathway, transforming growth factor-beta (TGF-b) signaling pathway, ErbB signaling pathway, MAPK signaling pathway, mammalian target of rapamycin (mTOR) signaling pathway, insulin signaling pathway, Jak-STAT signaling pathway, p53 pathway, calcium signaling pathway, and the cell cycle pathway (for a detailed summary of these findings, please refer to Table S1).
Validation of deregulated miRNAs
Microarray results on miRNAs expression and overexpression of miR-149 on both the primary and malignant tumor was validated by SYBR-based qRT-PCR. The upregulated miR-4298 and miR-923 were discarded because of lack of reference towards their pathological role in breast cancer. Only functionally relevant miRNAs were analyzed in our study. The results from microarray were further validated by qPCR and the expression of hsa-miR-130a, hsa-miR-21, hsa-miR-223, and hsa-let-7c which are downregulated in breast cancer was detected (Fig. 3). The validity of our microarray results was further tested by qRT-PCR in primary and malignant tumors in comparison to controls. The major finding of the study states that the expression pattern of miRNAs is similar in primary and malignant tumors.

Real-time polymerase chain reaction validation of miRNA microarray data
Discussion
Breast cancer is a heterogeneous disease and several studies have identified abnormally expressed miRNAs in breast cancer tissue compared to healthy breast tissue (Iorio et al. 2005; Farazi et al. 2011; Sempere et al. 2007; Volinia et al. 2012). Carcinogenesis though is primarily attributed to aberrations in oncogenes and the tumor suppressors, its role of miRNA in cancer onset and progression is also being increasingly recognized. MicroRNAs linked to tumorigenesis are termed oncomirs and are so termed because of their proximity to chromosome breakpoints (Calin et al. 2004). The first group of oncomirs to be identified was the let-7 family that regulates the expression of the RAS oncogene and thus indirectly alters cell proliferation through the downstream MAP signaling pathway (Johnson et al. 2005). MicroRNA expression profiling in different cancer types has revealed extensive deregulation of miRNA thus increases the understanding related to cancer biology and aiding in the development of new diagnostic and therapeutic tools (Di Leva and Croce 2013; Huh et al. 2013; Guo et al. 2013).
Our current study was focused on documenting the expression profile of mRNAs from primary and metastatic breast cancer in comparison to controls and to identify the deregulated miRNA patterns. The PCA analysis detected both primary and malignant breast cancer to exhibit similar pattern of miRNA expression in comparison to control group. This overlap of miRNA expression pattern between the primary and malignant tumor group indicates the complexity of genetic regulation involved in cancer. The interesting highlight of our study was that we detected a total of 54 miRNAs to be downregulated in the malignant tumor in contrast to 38 miRNAs in the primary tumor when compared with the control group under the same selection criteria. Previous studies on miRNA expression profiling of breast cancer have documented miR-10b, miR-125, miR-145, miR-21, and miR-155 to be highly deregulated. The upregulation of miR-21 and miR-155 and downregulation of miR-10b, miR-125, and miR-145 indicate the role of miRNAs as oncogenes (Blenkiron and Miska 2007).
Development and progression of cancer has been detected to be an outcome of altered cell signaling and a broad range of pathways related to proliferation, differentiation, and apoptosis which can be targeted by co-expressed miRNAs predicted through pathway enrichment analysis (Hanahan and Weinberg 2000). From the set of enriched deregulated miRNAs detected through microarray in our study, the number of genes associated with each of the pathway include 91 genes from the Wnt signaling pathway, 57 genes from the TGF-b pathway, 56 genes from the ErbB pathway, 126 genes from the MAPK signaling pathway, 29 genes within the mTOR pathway, 66 genes from the insulin signaling pathway, 70 genes from the Jak-STAT pathway, 34 genes within the p53 pathway, 71 genes from the calcium signaling pathway, 49 genes within the cell cycle pathway, 33 genes in the VEGF pathway, and 30 genes within the apoptosis pathway indicating that the inhibition of these miRNAs would result in concomitant activation of the corresponding key pathways. The activation of key pathways then drives the entire cascade in breast cancer including affecting cell growth, differentiation, proliferation, and transformation.
The observed miRNA expression patterns in primary and malignant breast tumors were consistently downregulated across studies, it reinforces their potential significance in breast cancer. Recent study by Li et al. (2020) demonstrated that downregulation of miR-21, miR-200 family, and miR-205 in breast cancer tissues correlates with aggressive tumor behavior and poor prognosis.
In the current study, identification of miRNAs and implication in breast cancer highlight the complexity of miRNA regulation in this disease. For instance, recent findings by Wu et al. (2021) suggest that miR-34a-5p is significantly downregulated in breast cancer and acts as a tumor suppressor by targeting oncogenic pathways.
Downregulated miRNAs identified in the study may serve as potential biomarkers for breast cancer diagnosis, prognosis, and therapeutic stratification. For instance, miR-155 has been implicated in breast cancer progression and resistance to chemotherapy (Gao et al. 2019). For example, miR-155 is downregulated in malignant tumors in the current study suggests its potential as a predictive biomarker for treatment response.
Furthermore, understanding the regulatory roles of dysregulated miRNAs can inform the development of targeted therapies. Recent advances in miRNA-based therapeutics, such as miRNA mimics or inhibitors, offer promising avenues for personalized treatment strategies in breast cancer (Khan et al. 2021).
Deregulated miRNAs result in complex modulation of multiple targets involved in signaling pathways including those for tumorigenesis. Several studies have identified key signaling pathways as a target of multiple downregulated miRNAs that impact proliferation of the cancer cell as well as tumor invasiveness (Chen et al. 2009; Johnson et al. 2005; Yu et al. 2010). The commonly deregulated miRNAs in primary and malignant breast cancer indicate that these common pathways could be important in tumor progression. Thus, coordinated downregulated miRNAs found in breast tumors could be impacting not only the oncogenes, but also other key genes involved in signaling pathways to activate tumorigenic downstream signals.
Conclusion
Our study demonstrates significant differences in miRNA expression patterns between normal and malignant breast tissue, with evidence of generalizability to unseen samples in cross-validation tests. Interestingly, our findings indicate that primary and malignant tumor subgroups exhibit overlapping miRNA expression patterns. Nevertheless, for comprehensive validation, further evaluation in a larger patient cohort is needed. In summary, we have identified four miRNAs with distinct expression patterns in normal and breast cancer tissue, offering valuable insights into breast cancer pathophysiology. These results underscore the potential clinical significance of these miRNAs for diagnosing and understanding the pathogenesis of breast cancer.
Data availability
Data is available with authors on request with justification.
Change history
30 April 2024
A Correction to this paper has been published: https://doi.org/10.1007/s13353-024-00866-z
References
Al Tamimi DM, Shawarby MA, Ahmed A, Hassan AK, Alodaini AA (2010) Protein expression profile and prevalence pattern of the molecular classes of breast cancer - a Saudi population based study. BMC Cancer 10:223. https://doi.org/10.1186/1471-2407-10-223
Al-Foheidi M, Al-Mansour MM, Ibrahim EM (2013) Breast cancer screening: review of benefits and harms, and recommendations for developing and low-income countries. Med Oncol 30(2):471. https://doi.org/10.1007/s12032-013-0471-5
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297. https://doi.org/10.1016/S0092-8674(04)00045-5
Blenkiron C and Miska EA. miRNAs in cancer: approaches, aetiology, diagnostics and therapy. Hum Mol Genet 2007; 16 Spec No 1: R106 - R113. https://doi.org/10.1093/hmg/ddm056
Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM (2004) Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA 101(9):2999–3004. https://doi.org/10.1073/pnas.0307323101
Chen X, Guo X, Zhang H, Xiang Y, Chen J, Yin Y, Cai X, Wang K, Wang G, Ba Y, Zhu L, Wang J, Yang R, Zhang Y, Ren Z, Zen K, Zhang J, Zhang C-Y (2009) Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene 28(10):1385–1392. https://doi.org/10.1038/onc.2008.474
Di Leva G, Croce CM (2013) miRNA profiling of cancer. Curr Opin Genet Dev 23(1):3–11. https://doi.org/10.1016/j.gde.2013.01.004
Fang Y, Sun B, Li Z, Chen Z (2020) Recent advances in nanomedicine for breast cancer therapy. Cancer Treat Rev 86:102016
Farazi TA, Horlings HM, Ten Hoeve JJ, Mihailovic A, Halfwerk H, Morozov P, Brown M, Hafner M, Reyal F, van Kouwenhove M, Kreike B, Sie D, Hovestadt V, Wessels LF, van de Vijver MJ, Tuschi T (2011) MicroRNA sequence and expression analysis in breast tumors by deep sequencing. Cancer Res 71(13):4443–4453. https://doi.org/10.1158/0008-5472.CAN-11-0608
Forouzanfar MH, Foreman KJ, Delossantos AM, Lozano R, Lopez AD, Murray CJ, Naghavi M (2011) Breast and cervical cancer in 187 countries between 1980 and 2010: a systematic analysis. Lancet 378(9801):1461–1484. https://doi.org/10.1016/S0140-6736(11)61351-2
Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH (2007) Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem 283:1026–1033. https://doi.org/10.1074/jbc.M707224200
Gao YC, Wu J, MicroRNA X (2019) MicroRNA-155 regulates the proliferation, cell cycle, apoptosis, and migration of colon cancer cells and targets CBL. Exp Ther Med 18(2):1225–1232
Guo L, Zhao Y, Yang S, Cai M, Wu Q, Chen F (2013) Genome-wide screen for aberrantly expressed miRNAs reveals miRNA profile signature in breast cancer. Mol Biol Rep 40(3):2175–2186. https://doi.org/10.1007/s11033-012-2277-5
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Hermeking H (2012) MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer 12(9):613–626
Huh JH, Kim TH, Kim K, Song J-A, Jung YJ, Jeong J-Y, Lee MJ, Kim YK, Lee DH, An HJ (2013) Dysregulation of miR-106a and miR-591 confers paclitaxel resistance to ovarian cancer. Br J Cancer 109(2):452–461. https://doi.org/10.1038/bjc.2013.305
Iorio MV, Ferracin M, Liu C-G, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Menard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM (2005) MicroRNA gene expression deregulation in human breast cancer. Cancer Res 65(16):7065–7070. https://doi.org/10.1158/0008-5472.CAN-05-1783
Iorio MV, Casalini P, Tagliabue E, Menard S, Croce CM (2008) MicroRNA profiling as a tool to understand prognosis, therapy response and resistance in breast cancer. Eur J Cancer 44(18):2753–2759. https://doi.org/10.1016/j.ejca.2008.09.037
Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ (2005) RAS in regulated by the let-7 microRNA family. Cell 120(5):635–647. https://doi.org/10.1016/j.cell.2005.01.014
Khan AQ, Ahmed EI, Elareer NR, Junejo K, Steinhoff M, Uddin S (2021) Role of miRNA-regulated cancer stem cells in the pathogenesis of human malignancies. Cells 10(7):1632
Lagendijk M, Sadaatmand S, Koppert LB, Tilanus-Linthorst MMA, de Weerd V, Ramirez-Moreno R, Smid M, Sieuwerts AM, Martens JWM (2018) MicroRNA expression in pre-treatment plasma of patients with benign breast diseases and breast cancer. Oncotarget 9(36):24335–24346. https://doi.org/10.18632/oncotarget.25262
Li J, Wang Y, Song Y, Fu Z, Yu W, Zhou M (2020) miR-21, miR-200 family, and miR-205 are promising diagnostic and prognostic miRNAs for breast cancer. BMC Cancer 20(1):1–12
Liang Y, Zhang H, Song X. and Yang, Q. Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets. In Seminars in cancer biology . 2020 60: 14–27). Academic Press.
Ma L, Teruya-Feldstein J, Weinberg RA (2007) Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 449(7163):682–688. https://doi.org/10.1038/nature06174
Papadopoulos GL, Alexiou P, Maragkakis M, Reczko M, Hatzigeorgiou AG (2009) DIANA-mirPath: integrating human and mouse microRNAs in pathways. Bioinformatics 25(15):1991–1993. https://doi.org/10.1093/bioinformatics/btp299
Paraskevopoulou MD, Georgakilas G, Kostoulas N, Vlachos IS, Vergoulis T, Reczko M, Filippidis C, Dalamagas T, Hatzigeorgiou AG (2013) DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res. 41(W1):W169–W173. https://doi.org/10.1093/nar/gkt393
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffery SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D (2000) Molecular portraits of human breast tumors. Nature 406(6797):747–752. https://doi.org/10.1038/35021093
Sempere LF, Christensen M, Silahtaroglu A, Bak M, Heath CV, Schwartz G, Wells W, Kauppinen S, Cole CN (2007) Altered MicroRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res 67(24):11612–11620. https://doi.org/10.1158/0008-5472.CAN-07-5019
Si M-L, Zhu S, Wu H, Lu Z, Wu F, Mo Y-Y (2007) miR-21-mediated tumor growth. Oncogene 26:2799–2803. https://doi.org/10.1038/sj.onc.1210083
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffery SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lonning PE, Borresen-Dale AL (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98(19):10869–10874. https://doi.org/10.1073/pnas.191367098
Tarone RE (2006) Breast cancer trends among young women in the United States. Epidemiology 17(5):588–590. https://doi.org/10.1097/01.ede.0000229195.98786.ee
van Schooneveld E, Wouters MC, van der Auwera I, Peeters DJ, Wildiers H, Van Dam PA, Vergote I, Vermeulen PB, Dirix LY, Van Laere SJ (2012) Expression profiling of cancerous and normal breast tissues identifies microRNAs that are differentially expressed in serum from patients with (metastatic) breast cancer and healthy volunteers. Breast Cancer Res 14(1):R34. https://doi.org/10.1186/bcr3127
Volinia S, Galasso M, Sana ME, Wise TF, Palatini J, Huebner K, Croce CM (2012a) Breast cancer signatures for invasiveness and prognosis defined by deep sequencing of microRNA. Proc Natl Acad Sci USA 109(8):3024–3029. https://doi.org/10.1073/pnas.1200010109
Volinia S, Croce CM, Prognostic S (2012b) Prognostic microRNA/mRNA signature from the integrated analysis of patients with invasive breast cancer. Proc Natl Acad Sci 109(8):2824–2829
Wu D, Zhang J, Fan Z (2021) Targeting miRNAs in breast cancer: Recent advances and future challenges. Mol Cancer 20(1):1–17
Yan L, Li X, X L, & Pan Z (2021) MicroRNA expression patterns in the development, progression, diagnosis, and prognosis of breast cancer. Medicine 100(35):e27151
Yu S, Lu Z, Liu C, Meng Y, Ma Y, Zhao W, Liu J, Yu J, Chen J (2010) miRNA-96 suppresses KRAS and functions as a tumor suppressor gene in pancreatic cancer. Cancer Res 70(14):6015–6025. https://doi.org/10.1158/0008-5472.CAN-09-4531
Acknowledgements
The authors extend their appreciation to the Deputyship for Research and Innovation, “Ministry of Education” in Saudi Arabia for funding this research (IFKSUOR3-601-2).
Funding
This research was funded by Deputyship for Research and Innovation, “Ministry of Education” in Saudi Arabia for funding this research (IFKSUOR3-601-2).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by: Ewa Ziętkiewicz
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: The original article contains an error. The funding statement should be changed from: “This research was funded by the Researchers Supporting Project number (RSPD2024R1005), King Saud University, Riyadh, Saudi Arabia” to: “This research was funded by Deputyship for Research and Innovation, “Ministry of Education” in Saudi Arabia for funding this research (IFKSUOR3-601-2).
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shahid, M., Syed, R., Ansari, M.A. et al. Blood-based microRNA profiling unveils complex molecular dynamics in breast cancer. J Appl Genetics 65, 549–557 (2024). https://doi.org/10.1007/s13353-024-00852-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-024-00852-5