
Abstract
Carrot (Daucus carota L.) is acknowledged as a highly valuable vegetable crop. Despite having high demand, limited breeding efforts have been made to develop the varieties and hybrids suitable to wider climatic conditions due to improper characterization of the available germplasm. An accession panel (AP) consisting of 144 accessions of five different root colors representing Asiatic and Western gene pools collected from different parts of India was utilized in the present study. This diverse AP was used to assess the population structure and genetic diversity from 80 polymorphic DNA markers distributed throughout the genome. Population structure, neighbor-joining (NJ) tree, and principal coordinate analysis (PCoA)-based diversity assessment divided the AP into three subpopulations/clusters. Greater than ninety percent polymorphism and the higher average polymorphic information content (͂> 0.50) coupled with higher gene diversity (He) indicating the broad genetic base of the population. Moderate to high Fst and gene flow (Nm) between the subpopulations revealed a moderate genetic differentiation among Indian carrot accessions owing to the highly outcrossing nature of carrot. Analysis of molecular variance (AMOVA) exhibited higher variation among individuals within the subpopulations (69.00%) or total populations (19.00%) than among the subpopulations (13%) as expected in the single Daucus species used here. The information obtained in the study would benefit the carrot breeders to explore the genetic diversity of the Indian carrots in the carrot breeding program for widening the genetic base and multi-color target trait improvement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Carrot (Daucus carota L.) is one of the highest valued vegetable crops of the Apiaceae family. It is consumed in various ways by almost all the age groups across the globe for its nutritionally rich carotenoids, vitamins, minerals, proteins, and phenolic compounds with zero cholesterol and zero fat (Maksylewicz and Baranski 2013). Carrots have incredible antioxidants, detoxifying properties, anti-aging, anti-cancerous properties, good for the heart, and dental health, and also act as a natural liver cleanser (Alasalvar et al. 2001; Simon et al. 2008; Maksylewicz and Baranski 2013). Globally, China is the leading producer of carrot, and in India, it is growing in an area of 86 thousand hectares with the production of 13.79 lakh metric tons (http://nhb.gov.in/statistics). Carrot origin dates back to the tenth century in Persia and Asia Minor, which was then of a thin-rooted purple or white color (Simon et al. 2008; Iorizzo et al. 2013). The center of diversity has reported being Central Asia and Afghanistan region, where it was cultivated approximately 1100 years ago (Mackevic 1929). The domestication of carrot has first started in Central Asia and Asia Minor with purple- and yellow-colored roots followed by its distribution to Eastern Europe and England during the eleventh to fifteenth century (Banga 1957), and today, these wild carrots have been widely distributed in different regions of the world. Based on the origin and color, carrots were classified as Eastern/Asiatic carrots and Western/carotene types. The Asiatic carrot is either of black purple/anthocyanin type or red-colored root mainly grown in Egypt and Asian countries, including India, whereas the Western carrot is of orange-, yellow-, or white-colored tap root probably derived from the Asiatic types either by mutation or by selection in the yellow type hybrid progenies (Maksylewicz and Baranski 2013; Bradeen and Simon 2007; Rong et al. 2010). The present-day attractive, β carotene-rich dark orange-colored, improved flavored, smooth-textured carrot is the result of secondary domestication event (Iorizzo et al. 2013). Majority of the breeding efforts for attractive dark orange carrots (western types) concentrated in colder climates due to its long day requirement and flowering ability in the temperate region. Despite having greater genetic diversity for geographical adaptation, or root colors in the Indian carrot gene pool, to date, very few attempts have been made in the proper characterization of the germplasm lines for the tropical suitability (Asiatic types) compared with the temperate carrot improvement. Moreover, because of the popularity and consumer preference of domesticated dark orange carrot, other colored carrots have almost become obsolete despite having rich pigments like anthocyanin in purple/black, lycopene in red, and lutein in yellow.
The availability of genetically diverse germplasm of carrot suitable to geographically diverse climate necessitates exploration, collection, evaluation, and proper characterization of the accessions to utilize in crop breeding. India is a country wherein the climate has been broadly grouped into tropical wet (humid), tropical dry, humid subtropical climate, and mountain climate (temperate) according to the Koeppen system of climate classification (Geiger 1954, 1961). Characterization of the gene pool is possible by traditional morphological descriptors and evaluation of quantitative traits by subjecting them to multi-season and multi-location field experimentation. It is easier and economical to characterize the germplasm at the species level based on morphological markers; however, identification of genotypes within a species based on morphology alone is rather difficult. Furthermore, the reliability of phenotypic data could be questionable for the traits that are highly influenced by the environment; besides, an inadequate number of variable morphological traits limit the understanding of genetic diversity. With the availability of high-quality genome sequence of carrot (Iorizzo et al. 2016), characterization of the germplasm using DNA markers would be more rewarding as they are environmentally neutral and abundant covering whole genome (Maksylewicz and Baranski 2013; Simon et al. 2008; and Cavagnaro et al. 2011). A colossal number of DNA markers in carrot are developed and utilized for genetic mapping, fine mapping, and genetic diversity analysis in carrot (Cavagnaro et al. 2011; Ellison et al. 2017)
Among the various kinds of DNA markers, microsatellites or simple sequence repeats (SSR) being robust, locus-specific, and multi-allelic nature are proved to be valuable in assessing the genetic diversity of crop species to understand the genetic relationships between crop plants and their wild relatives (Varshney et al. 2005; Kalia et al. 2011). The genetic composition or the gene flow within the population is being elucidated by population structure by most of the genetic studies. Assessment of structure of the gene pool based on genetic data offers insights into the whole population and the role of evolutionary forces such as mutation, migration, selection, random drift, and geographical barriers that creates diverse clusters often termed as subpopulations (Greenbaum et al. 2016). The number of subpopulations from a given population is based on two different approaches, viz., model-based method, and based on the genetic distance matrices obtained from the genetic data (Pritchard et al. 2000). The former method relies on the likelihood of the genetic data randomly chosen from the predefined population models by assuming that the K-subpopulation each is in Hardy-Weinberg equilibrium. Whereas the genetic distance matrix-based grouping defines the pair-wise genetic distances among the individuals of the population visualized from multidimensional scaling (Greenbaum et al. 2016). In Daucus, assessment of genetic diversity and population structure to date could not uncover a clear population structure owing to its highly cross-pollinating nature (Maksylewicz and Baranski 2013). However, the demarcation between wild and cultivated gene pools revealed in an investigation involving a few carrot accessions using AFLP markers (Shim and Jorgensen 2000) and SNP markers (Iorizzo et al. 2013). There is a moderate genetic differentiation between the cultivated carrot gene pool 138 using SSR markers as observed in previous study (Baranski 139 et al. 2012). Collection and proper characterization of the germplasm lines is a crucial step in any crop improvement programs. Carrot being the cool season crop, collection of accessions from diverse geographical regions and their individual description would offer a greater scope in understanding the genetic base of the population and their suitability to wider climatic conditions, which in turn provide a gateway to design the breeding strategies. A concerted effort was made to collect a large number of diverse colored carrot accessions from different parts of India for their detailed characterization by exploring the publicly available DNA markers of carrot. The objective of the present study was to understand the composition and structure of the entire population of carrot accessions consisting of five diverse root colors (dark orange, light orange, yellow, red, black/purple) collected from different parts of India, a country with greater geographical diversity. The investigation of the genetic diversity of these accessions would help the carrot breeders to utilize the diverse germplasm lines in the future carrot breeding program under the circumstances of nutritional security and climate change challenges.
Material and methods
Plant material
A set of carrot germplasm consisting of 144 accessions hereafter mentioned as accession panel (AP) is used in the study. These accessions were collected from different parts of India representing temperate region or Western types and tropical regions or Asiatic types which were belonging to five different root colors, viz., light orange, dark orange, yellow, red, and black/purple anthocyanin types (Supplementary file 1). AP comprised of local cultivars, open-pollinated varieties (OPV), hybrids, varieties, and unexplored germplasm accessions is collected from local farmers, public, private organizations, and gene banks, viz., National Bureau of Plant Genetic Research, New Delhi and Indian Institute of Vegetable Research, Varanasi, India.
DNA isolation and quantification
Total genomic DNA was isolated from an individual plant selected from each accession using a standard CTAB method (Briard et al. 2000). The DNA pellet was dissolved using 1X TE (Tris-EDTA) buffer. The DNA purity and quality were ensured in 1.0% agarose gel. Quantification with a nano-spectrophotometer (Nano Drop 2000, Thermo Scientific) was followed by its dilution to working concentration of 50 ng/μl using TE buffer and stored at − 20 °C deep freezer until its further use for polymerase chain reaction (PCR).
Genotyping
Initially, publicly available 140 carrot DNA markers were used for genotyping of AP. The markers consisted of genomic SSR (Cavagnaro et al. 2011; Rong et al. 2010 and Niemann 2001), genic microsatellites (Iorizzo et al. 2011), sequence characterized amplified region (SCAR) (Cavagnaro et al. 2009), EST-based gene-specific markers (Cavagnaro et al. 2009; Kawahara et al. 1992), and indels (Iorizzo et al. 2016). For statistical analysis, only the markers showing > 80% amplification were selected. The DNA markers that recorded > 20% null alleles or no amplification and dominant type (presence/absence polymorphism) were dropped from the statistical analysis. With these criteria, genotypic data of 80 DNA markers out of 140 DNA markers across AP were analyzed. The details of 80 polymorphic markers with their melting temperate (Tm), repeat type, the expected allele size, and the linkage group (previously mapped by different authors) are provided in supplementary file 2.
PCR amplification and allele sizing
PCR components consisted of 50 ng/μl DNA template of each individual, 5 pmol each of forward and reverse primer mix, 2X PCR master mix (AMPLIQUIN) consisted of dNTPs, Taq polymerase enzyme, Taq buffer, and MgCl2 were used for amplification. PCR protocol and annealing temperature (Tm) were followed, as mentioned in Supplementary file 2. PCR product was run in 4% high-quality agarose by following the procedure of Cavagnaro et al. (2011). Allele size recorded by using the GeneTools analysis software version 4.03.05.0 with the help of a 100-bp ladder (Takara). The following statistical analyses were performed using the diploid data set.
Statistical analysis
Marker-wise genetic diversity parameters
Genetic diversity parameters, viz., number of alleles (Na), number of effective alleles (Ne), gene diversity (He), observed heterozygosity (Ho), major allele frequency (MAF), polymorphic information content (PIC), Wright’s inbreeding coefficient (F), and Shannon’s information index (I), were estimated for each of the 80 polymorphic markers across the AP. Wherein, “Ho” is the actual amount of heterozygosity measured at a particular locus or population, and “He” is the proportion of heterozygosity expected for a particular locus or population undergoing random mating and acting within Hardy-Weinberg equilibrium. The decline in heterozygosity due to subdivision within a population was quantified using an index known as Wright’s “F”-statistics (F) (Wright 1965). “PIC” and “I” are the marker informativeness and discriminating power of the markers based on the allelic frequency of each locus. Three subpopulations obtained in the NJ tree from AP were compared for the above parameters using a mean ± standard error (SE) values, considering the respective number of accessions in each subpopulations. The private alleles specific to each subpopulation was estimated to know the genetic diversity of the subpopulations. Statistical analysis was performed using the software packages GenAlex 6.5 (Peakall and Smouse 2012) and PowerMarker 3.25 (Liu and Muse 2005).
Among the subpopulations of NJ tree, the pair-wise fixation index (Fst), as a measure of genetic differentiation, was estimated. It explains the genetic variance of the population structure based on Wright’s F-statistics (Wright 1965) and Nei’s pair-wise genetic distance (D) as a measure of genetic diversity between the subpopulations was also estimated using the GenAlex 6.5. Locus wise F-statistics (Fst, Fis, and Fit) and level of gene flow (Nm = (1/Fst)−1)/4) were estimated for the subpopulations to find out the efficiency of 80 DNA markers in genetic discrimination of the subpopulations using the software package Popgene (Yeh et al. 1999).
Population structure assessment
To understand the composition/structure and gene flow within the AP across 80 polymorphic DNA markers, population structure analysis was carried out by using the STRUCTURE v 2.3.4 software package (Pritchard et al. 2000) with the use of Bayesian model of clustering. Parameters were set to consider the admixture model with correlated allelic frequencies among populations. The number of subpopulations (K value) was set from 2 to 10 with 100,000 replicates for burn-in-periods and 100,000 number of Markov chain Monte Carlo (MCMC) repetitions after burn-in with ten independent runs for each K. The most likely value of K was determined based on the ad hoc statistic ΔK value estimated according to Evanno’s procedure (Evanno et al. 2005) by using Structure Harvester tool (Earl and von Holdt 2012). Each accession was assigned with the posterior membership coefficients (Q) in the structure-partitioned subpopulations with K = 3. Accessions are assigned to the respective subpopulations based on Q membership value >0 .50. The subpopulations obtained by structure analysis were named as S1, S2, and S3.
Cluster analysis
Two methods of cluster analyses were performed; the first method was based on genetic distance matrix estimated by the software GenAlex v6.5 (Peakall and Smouse 2012), for AP followed by the construction of unweighted neighbor-joining (NJ) tree using the software DARWIN version 6.0 (Perrier and Jacquemoud-Collet 2006) with 10,000 bootstrap values. The subpopulations obtained in the NJ tree were named C1, C2, and C3. In the second method, principal coordinate analysis (PCoA) was conducted using the GenAlex v6.5 from the set of 80 polymorphic marker data based on the pair-wise genetic distance matrix between the accessions.
Analysis of molecular variance and genetic diversity indices
Genetic differentiation of the subpopulations can be studied by various parameters, among which, analysis of molecular variance (AMOVA) estimates the extent of variance among and within subpopulations and total populations. In order to estimate the variance among and within the total and subpopulations of the Indian carrot accessions, AMOVA was carried out by considering the respective number of accessions belonging to each of the subpopulations obtained in cluster analysis using the software package GenAlex 6.5.
Other genetic diversity parameters included were pair-wise Fst values among the subpopulations, which explain the amount of genetic differentiation among the subpopulations and Nm, an indirect measure that explains the gene flow or the number of migrants successfully entering populations per generation (Whitlock and McCauley 1999).
Results
Genetic diversity parameters
The AP consisting of 144 accessions across 80 polymorphic DNA markers, a total of 305 alleles, could be extracted, ranging from 2.0 to 9.0 in each marker (Table 1 and Supplementary file 3) with an average of 3.81 ± 0.20 alleles per locus, whereas the average “Ne” was 2.17 ± 0.10. The average PIC value was 0.51 ± 0.04, with a range of 0.08 (yY-W Indel) to 0.78 (ESSR058). The marker-wise gene diversity (He) value ranged from 0.01 to 0.79 with an average of 0.46 ± 0.02, MAF 0.584 ± 0.043, and the average “I” was 0.84 ± 0.04 with a maximum of 1.68 and a minimum of 0.03. The average “F” value was 0.72 ± 0.03, with a range of − 0.04 to as high as 1.00 (Supplementary file 3). When the subpopulations were compared for number of private alleles (Np), a total of 64 private alleles was recorded among the three subpopulations of which the highest number of private alleles was recorded by subpopulation I (28), followed by subpopulation II (19), and subpopulation III (17) (Supplementary file 4). The highest mean genetic diversity parameters were observed in subpopulation I as shown by the total number of alleles (254), Na (3.18 ± 0.16), Ne (1.98 ± 0.09), He (0.41 ± 0.03), and “I” (0.72 ± 0.04) than other two subpopulations (Table 1).
Population structure and cluster analysis
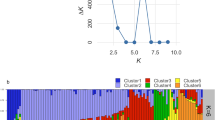
Population structure analysis was carried out to study the genetic relationships among the 144 Indian carrot accessions belonging to five different root colors of carrots. The AP showed optimum K value of “three” based on peak ΔK (Fig. 1a), suggesting three main subpopulations, viz., S1, S2, and S3, consisted of 40, 48, and 46 accessions, respectively (Fig. 1b, supplementary file 5). Ten admixtures not assigned to any of the subpopulations were observed, as shown by low q membership value (< 0.80). When the subpopulations of the population structure were scanned for accessions belonging to different root colors, the majority of the dark orange types were grouping in S1, whereas light orange were in S2 and S3 comprised of black/purple and red colors. Yellow carrots were distributed randomly in all three clusters. Many of the temperate accessions were grouped within S1, and the majority of the tropical types distributed in S2 and S3 with few exceptions. The grouping pattern of the population structure analysis was almost in congruence with the botanical classification as Western (orange/yellow colors) or Eastern (black/purple/red colors) from 80 marker loci. However, the accessions were not structured based on either the origin or external root color criteria as expected in the highly cross-pollinating character of carrot.

Optimum K value shown by ΔK value graph and corresponding population structure depicting a number of subpopulations in Indian carrot accessions panel (AP) consisting of 144 accessions. a Figure depicting ΔK value obtained by Evanno’s procedure extracted from a software tool Structure Harvester. b Three subpopulations obained in Population Structure Analysis Values in the vertical line of population structure indicate the membership coefficient Q (0.00 to 1.00) for each of the accessions along with the length of color codes; black vertical line separates subpopulations
Unweighted NJ tree separated the population clearly into three clusters (C1, C2, and C3), which are almost similar to the composition of subpopulations of population structure (Fig. 2). The 1st cluster consisted of 50 accessions, out of which 40 were similar to the S1 in a population structure, and the remaining four were belonging to cluster 2 (UHSBC-01, UHSBC-02, UHSBC-03, and UHSBC-116). The number of accessions in cluster 2 and 3 was 46 and 48, respectively, and was in correspondence with population structure except few (Supplementary file 5). When the accessions belonging to different root color were observed in three clusters, many of the light orange, yellow, red, and black/purple accessions were distributed in clusters II and III; however, most of the dark orange (> 80%) were in the cluster I.

Cluster diagram showing 3 subpopulations (C1, C2, and C3) among 144 Indian carrot accessions by unweighted neighbor-joining tree (NJ) drawn in Darwin 6.0 based on 80 polymorphic markers. The numbers in blue color within the tree branches are the bootstrap values (> 50), and the numbers at the edge of each line are the serial numbers of accessions as mentioned in supplementary file5
PCoA was also in ordinance with the population structure and NJ tree with 3 clusters (C1, C2, and C3). The cumulative variance explained by two principal coordinates accounted for 16.17% with the maximum variance of 7.29% by the first principal coordinate, followed by 5.40% by the second principal coordinate (Fig. 3). When the accessions of different colors were observed in PCoA distribution, the greater part of C1 was dark orange (21). The cluster C2 was dominant with light orange and yellow, whereas the cluster C3 consisted of > 50% of the red and black purple accessions. The details of the composition of the clusters with the respective accessions obtained in STRUCTURE, NJ tree, and PCOA are presented in supplementary file 5.

Two dimensional principal coordinate analysis (PCoA) plot generated by the GenAlex 6.5 showing the composition of three clusters and distribution of five root colors and two adaptable subpopulations in these three main clusters. X and Y value indicates the principal coordinates with the variance explained in the bracket
Genetic differentiation
The estimates of F-statistics, gene flow index (Nm), and AMOVA were used as measures of genetic differentiation of the subpopulations obtained from the NJ tree.
AMOVA revealed significant variance among the subpopulations with 13.00% estimated variance (p > 0.001), whereas among the individuals within the subpopulations (69.0%) and among the individuals within the total population (19.0%) explained higher variance (Table 2). The Fit value of 0.814 was higher than Fis (0.787) or Fst (0.126).
Pair-wise Fst values and Nei’s genetic distance (D) across the subpopulations were assessed in order to know the level of genetic differentiation or diversity between the subpopulations of AP (Table 3). The highest Fst value was observed between subpopulation I and III (0.148) followed by subpopulation II and III (0.121). Whereas higher genetic distance was observed between subpopulation II and III (0.216) followed by I and III (0.165) (Table 3).
Locus wise F-statistics (Fis, Fit, Fst) and Nm have estimated across the subpopulations that showed an average Fst of 0.138, indicating the moderate to high genetic differentiation of the subpopulations, which is supported by average gene flow (Nm = 1.569) (Supplementary File 6). The highest Fst (0.722) coupled with the lowest Nm value (0.096) was recorded for a locus GSSR63, followed by GSSR14 (Fst = 0.588 and Nm = 0.175) (Supplementary file 6). In contrast, the highest free gene flow (Nm) coupled with the lowest Fst was observed by GSSR124 marker loci (Nm = 585.033 and Fst = 0.000).
Discussion
In recent years, the most proficient way of understanding the genetic diversity and the population structure of plant genetic resources is to characterize them with reliable DNA markers. Insights into the whole given population with its derived subpopulations help to learn about the genetic differentiation between the subpopulations, their uniqueness, and genetic diversity. This knowledge would be useful to design the breeding strategies to incorporate the desirable variations into a cultivar. In this study, we attempted to assess genetic diversity and population genetic structure of diverse Indian carrot accessions using genome-wide DNA markers. Microsatellites were found to be the most useful markers due to their high resolving power in analyzing the population structure than SNP (Chen et al. 2007; DeFaveri et al. 2013) as these markers are co-dominant and multi-allelic natured and also can be easily transferred across the species or genus (Cholin et al. 2019). Hence, the majority of the markers selected in the present study are microsatellites representing genic and genomic regions.
Assessment of genetic diversity by various diversity indices, viz., “Na,” “Ne,” “Ho,” “He,” “I,” “PIC,” and “F,” helped us to understand the nature and extent of variability among and within subpopulations. Estimation of PIC value is a simple way to understand the discriminating power of markers across the germplasm accessions (Maksylewicz and Baranski 2013). According to Botstein et al. (1980), the “PIC” value of > 0.50 indicates high polymorphism, whereas the PIC value between 0.25 and 0.50 is moderate, and < 0.25 indicates low polymorphism. Higher average PIC value (> 0.50) across the DNA markers studied in AP indicated their effectiveness in genetic diversity assessment of the carrot accessions. Total number alleles for AP or the subpopulations and the average values of “Na,” “Ne,” “MAF,” “F,” and “I” indicated that the DNA markers used in the study are informative to characterize the diverse AP (Supplementary file 3 and Table 1).
In carrot, two major gene pools, viz., Western (orange/yellow) and Asiatic types (red/purple/black), have been suggested based on molecular evidence of population structure using microsatellites (Clotault et al. 2010; Baranski et al. 2012). Despite this taxonomic differentiation, there was no clear geographical isolation or population structure in carrot germplasm as revealed by non-structured genetic diversity using dominant markers (Bradeen et al. 2002; Grzebelus et al. 2002; Nakajima et al. 1998) or moderate separation between the Asian- and Western-cultivated carrot using co-dominant SSR markers (Baranski et al. 2012). Other reports on wild and domesticated accessions of carrots also suggested an insignificant reduction in genetic variability among these subpopulations based on isozyme markers (St. Pierre and Bayer 1991; St. Pierre et al. 1990).
The genotypic matrix in the present study divided the total population into three subpopulations with a few admixtures based on the Bayesian method of population structure analysis. The result was in congruence with the number and composition of the clusters observed in the NJ tree and PCoA, with few exceptions, indicating a high amount of genetic diversity in the Indian carrot gene pool, most of which are unexplored in the breeding program. There was no clear demarcation of clusters based on root colors as the accessions belonging to five root colors were distributed in all the three subpopulations/clusters, which is expected as carrot is highly outcrossing nature of the crop. Nevertheless, the majority of the western carrots (dark orange) were distributed in C1/S1, most of the light orange types were in C2/S2, and C3/S3 consisted of mainly eastern types (red and black/purple) with few exceptions, and yellow types distributed randomly across all 3 clusters. These findings are in support of the previous hypothesis of carrot domestication that spontaneous hybridization among wild and earlier yellow-colored carrots resulted in the origin of present-day orange carrot (Heywood 1983; Banga 1957; Maksylewicz and Baranski 2013; Rong et al. 2010). After the selection and domestication of attractive dark orange types, mere importance was given to other nutritionally rich-colored carrots such as black, purple, yellow, and red for many decades or even centuries. In recent years, due to the increased awareness about the health benefits of these multi-colored carrots, efforts are also being undertaken to develop varieties or hybrids rich in these pigments both for raw consumption and industrial use (Arscott and Tanumihardjo 2010; Bradeen and Simon 2007; Simon et al. 2008; Kammerer et al. 2003). In this regard, the accessions used in the present study would be the better source for targeted multi-color carrot improvement.
Estimated variance obtained in AMOVA showed higher genetic variance among the individuals within subpopulations than between the subpopulations or the individuals among the total population. The reason could be attributed to higher genetic diversity within subpopulations than between subpopulations as expected from the highly cross-pollinated single-cultivated species, viz., Daucus carota, used in the present study. Fis and Fit values obtained here are much higher compared with the previous studies of intra-population genetic diversity of carrot by Maksylewicz and Baranski (2013).
If the Fst value ranges from 0.15 to 0.25, the subpopulations are assumed to show highly significant genetic differentiation (Balloux and Lugon-Moulin 2002). Moderate genetic differentiation was observed among the subpopulations, as demonstrated by pair-wise Fst values among the subpopulations (0.109 to 0.148) obtained in the present study. Moderate genetic differentiation among the subpopulations can be attributed to the mating system and probable bidirectional gene flow in carrot. The results of gene flow between wild and domesticated types were confirmed in the previous reports using SNP markers (Simon 2000; Rong et al. 2010; Iorizzo et al. 2013, Umehara et al. 2005).
Conclusion
The present study highlights the characterization of Indian germplasm accessions with genome-wide markers to assess the population structure, and genetic diversity existed in the AP consisted of diverse root colors. The higher genetic diversity and a structured population with moderate genetic differentiation among the three subpopulations indicate the broad genetic base of the Indian carrot accessions. Markers used in the study proved informative for characterization and diversity assessment. The germplasm accessions used in the present study collected from broad geographical regions of India would also be beneficial for the development of climate-resilient varieties/hybrids of carrot and widen the carrot gene pool.
References
Alasalvar C, Grigor JM, Zhang D, Quantick PC, Shahidi F (2001) Comparison of volatiles, phenolics, sugars, antioxidant vitamins, and sensory quality of different colored carrot varieties. J Agric Food Chem 49(3):1410–1416
Arscott SA, Tanumihardjo SA (2010) Carrots of many colors provide basic nutrition and bioavailable phytochemicals acting as a functional food. Compr Rev Food Sci Food Saf 9(2):223–239
Balloux F, Lugon-Moulin N (2002) The estimation of population differentiation with microsatellite markers. Mol Ecol 11(2):155–165
Banga O (1957) Origin of the European cultivated carrot. Euphytica 6(1):54–63
Baranski R, Maksylewicz-Kaul A, Nothnagel T, Cavagnaro PF, Simon PW, Grzebelus D (2012) Genetic diversity of carrot (Daucus carota L.) cultivars revealed by analysis of SSR loci. Genet Resour Crop Ev 59:163–170
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331
Bradeen JM, Simon PW (2007) Carrot. In: Genome mapping and molecular breeding in plants. Springer Berlin Heidelberg, In, pp 161–184
Bradeen JM, Bach IC, Briard M, Le Clerc V, Grzebelus D, Senalik DA, Simon PW (2002) Molecular diversity analysis of cultivated carrot (Daucus carota L.) and wild Daucus population reveals a genetically non- structured composition. J American Soc Hort Sci 127:383–391
Briard M, Le Clerc V, Grzebelus D, Senalik DA, Simon PW (2000) Modified protocols for rapid carrot genomic DNA extraction and AFLP™ analysis using silver stain or radioisotopes. Plant Mol Biol Rep 18:235–241
Cavagnaro PF, Chung S, Szklarczyk M, Grzebelus D, Senalik D, Atkins AE (2009) Characterization of a deep-coverage carrot (Daucus carota L.) BAC library and initial analysis of BAC-end sequences. Mol Gen Genomics 281(3):273–288
Cavagnaro PF, Chung SM, Manin S, Yildiz M, Ali A, Alessandro MS, Iorizzo M, Senalik DA Simon PW (2011) Microsatellite isolation and marker development in carrot genomic distribution, linkage mapping, genetic diversity analysis and marker transferability across Apiaceae. BMC Genomics 12:386
Chen C, Durand E, Forbes F, François O (2007) Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol EcolNotes 7(5):747–756
Cholin SS, Poleshi CA, Manikanta DS, Christopher C (2019) Exploring the genomic resources of carrot for cross-genera transferability and phylogenetic assessment among orphan spices and vegetables of Apiaceae family. Hortic Environ Biote 60(1):81–93
Clotault J, Geoffriau E, Linneton E, Briard M, Peltier D (2010) Carotenoid biosynthesis genes provide evidence of geographical subdivision and extensive linkage disequilibrium in the carrot. Theor Appl Genet 121:659–672
DeFaveri J, Viitaniemi H, Leder E, Merilä J (2013) Characterizing genic and nongenic molecular markers: comparison of microsatellites and SNPs. Mol Ecol Resour 13(3):377–392
Earl DA, von Holdt BM (2012) Structure harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Ellison S, Senalik D, Bostan H, Iorizzo M, Simon P (2017) Fine-mapping, transcriptome analysis, and marker development for Y2, the gene that conditions β-carotene accumulation in carrot (Daucus carota L.). G3-Genes Genom Genet 7(8):2665–2675
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Geiger R (1954) Klassifikation der Klimate nach W. Köppen. Landolt-Börnstein − Zahlenwerte und Funktionen aus Physik, Chemie, Astronomie, Geophysik und Technik, alte Serie, vol 3. Springer, Berlin, pp 603–607
Geiger R, (1961) Berarbeitete Neuausgabe von Geiger R, Koppen-Geiger/Klima der Erde. Wandkarte (wall map). 1: 16
Greenbaum G, Templeton AR, Bar-David S (2016) Inference and analysis of population structure using genetic data and network theory. Genetics 202(4):1299–1312
Grzebelus D, Baranski R, Kotlinska T, Michalik B (2002) Assessment of genetic diversity in a carrot (Daucus carota L.) germplasm collection. Plant Genet Resour Newsl:51–53
Heywood VH (1983) Relationships and evolution in the Daucus carota complex. Isr J Plant sci 32(2):51–65
Iorizzo M, Senalik DA, Grzebelus D, Bowman M, Cavagnaro PF, Matvienko M, Ashrafi H, Van Deynze A, Simon PW (2011) De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers, and genetic diversity. BMC Genomics 12(1):389
Iorizzo M, Senalik D, Ellison S, Grzebelus D, Cavagnaro PF, Allender C, Brunet J, Spooner D, Deynze A, Simon PW (2013) Genetic structure and domestication of carrot (Daucus carota subspp sativus) (Apiaceae). Am J Bot 100:930–938
Iorizzo M, Shelby E, Douglas S, Zeng P, Pimchanok S, Jiaying H, Bowman M, Marina I, Walter S, Cavagnaro P, Yildiz A, Moranska E, Grzebelus E, Grzebelus D, Ashrafi H, Zhijun Z, Shifeng C, Spooner D, Allen Van D, Philipp S (2016) A high-quality carrot genome assembly provides new insights into carotenoid accumulation and asteroid genome evolution. Nat Genet. https://doi.org/10.1038/ng.3565
Kalia RK, Rai MK, Kalia S, Singh R, Dhawan AK (2011) Microsatellite markers: an overview of the recent progress in plants. Euphytica 177:309–334
Kammerer D, Carle R, Schieber A (2003) Detection of peonidin and pelargonidin glycosides in black carrots (Daucus carota ssp. sativus var. atrorubensAlef.) by high-performance liquid chromatography/electrospray ionization mass spectrometry. Rapid Commun Mass Spectrom 17:2407–2412
Kawahara R, Sunabori S, Fukuda H, Komamine A (1992) A gene expressed preferentially in the globular stage of somatic embryogenesis encodes elongation-factor 1α in carrot. Eur J Biochem 209(1):157–162
Liu K, Muse SV (2005) Power marker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21(9):2128–2129
Mackevic VI (1929) The carrot of Afghanistan. Bull Appl Bot Genet Plant Breed 20:517–562
Maksylewicz A, Baranski R (2013) Intra-population genetic diversity of cultivated carrot (Daucus carota L.) assessed by analysis of microsatellite markers. Acta Biochim Pol 60:753–760
Nakajima Y, Oeda K, Yamamoto T (1998) Characterization of genetic diversity of nuclear and mitochondrial genomes in Daucus varieties by RAPD and AFLP. Plant Cell Rep 17:848–853
Niemann M (2001) Entwicklung von Mikrosatelliten – Marker bei der Möhre (Daucus carota L.) und die Markierung eines Alternaria-Resistenzgens. Berichte aus der Agrarwissens chaft. Shaker Verlag, Aachen
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in excel. Population genetic software for teaching and research – an update. Bioinformatics 28:2537–2539
Perrier X, Jacquemoud-Collet JP (2006) DARwin software http://darwin.cirad.fr/. Access 08/20/2019
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multi-locus genotype data. Genetics 155:945–959
Rong J, Janson S, Umehara M, Ono M, Vrieling K (2010) Historical and contemporary gene dispersal in wild carrot (Daucus carota ssp. carota) population. Ann Bot 106:285–296
Scientific T (2009) NanoDrop 2000/2000c Spectrophotometer User Manual. Thermo Fisher Scientifc Inc, Delaware, USA
Shim SI, Jorgensen RB (2000) Genetic structure in cultivated and wild carrot (Daucus carota L.) revealed by AFLP analysis. Theor Appl Genet 101:227–233
Simon PW (2000) Domestication, historical development, and modern breeding of carrot. Plant Breed Rev 19:157–190
Simon PW, Freeman RE, Vieira JV, Boiteux LS, Briard M, Nothnagel T, Michalik M, Kwon YS (2008) Carrot. In: Prohens J, Nuez F (eds) Vegetables II: Fabaceae, Liliaceae, Solanaceae, and Umbelliferae. Handbook of plant breeding, vol 2. Springer, pp 327–357
St. Pierre MD, Bayer RJ (1991) The impact of domestication on the genetic variability in the orange carrot, cultivated Daucus carota ssp. sativus and the genetic homogeneity of various cultivars. Theor Appl Genet 82:249–253
St. Pierre MD, Bayer RJ, Weis IM (1990) An isozyme-based assessment of the genetic variability within the Daucus carota complex (Apiaceae: Caucalideae). Canadian J Bot 68:2449–2457
Umehara M, Eguchi I, Kaneko D, Ono M, Kamada H (2005) Evaluation of gene flow and its environmental effects in the field. Plant Biotech 22:497–504
Varshney RK, Graner A, Sorrells ME (2005) Genic microsatellite markers in plants: features and applications. Trends Biotechnol 23(1):48–55
Whitlock M, McCauley D (1999) Indirect measures of gene flow and migration: FST≠1/(4Nm+1). Heredity 82:117–125
Wright S (1965) The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 19(3):395–420
Yeh FC, Yang RC, Boyle T (1999) POPGENE software package version 1.31 for population genetic analysis. University of Alberta, Edmonton
Acknowledgments
CS greatly acknowledges the Indian Institute of Vegetable Research, Varanasi, and National Bureau of Plant Genetic Research, New Delhi, for providing the germplasm accessions and to Dr. Sunil Shebannavar for expert guidance. The authors are greatly thankful to Dr. MVC Gowda for providing valuable suggestions during the manuscript preparations.
Funding
This work was supported by funding from the Department of Biotechnology (DBT), Government of India under DBT BIO-CARe: File No: 102/IFD/SAN/3308/2014-15. DSM is grateful for the financial support to work under the project as a Junior Research Fellow.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by: Izabela Pawłowicz
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(XLSX 53 kb)
Rights and permissions
About this article

Cite this article
Chaitra, K.C., Sarvamangala, C., Manikanta, D.S. et al. Insights into genetic diversity and population structure of Indian carrot (Daucus carota L.) accessions. J Appl Genetics 61, 303–312 (2020). https://doi.org/10.1007/s13353-020-00556-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-020-00556-6