
Abstract
Autism spectrum disorder (ASD) is a group of the neurodevelopment disorders presenting as an isolated ASD or more complex forms, where a broader clinical phenotype comprised of developmental delay and intellectual disability is present. Both the isolated and complex forms have a significant causal genetic component and submicroscopic genomic copy number variations (CNV) are the most common identifiable genetic factor in these patients. The data on microarray testing in ASD cohorts are still accumulating and novel loci are often identified; therefore, we aimed to evaluate the diagnostic efficacy of the method and the relevance of implementing it into routine genetic testing in ASD patients. A genome-wide CNV analysis using the Agilent microarrays was performed in a group of 150 individuals with an isolated or complex ASD. Altogether, 11 (7.3%) pathogenic CNVs and 15 (10.0%) variants of unknown significance (VOUS) were identified, with the highest proportion of pathogenic CNVs in the subgroup of the complex ASD patients (14.3%). An interesting case of previously unreported partial UPF3B gene deletion was identified among the pathogenic CNVs. Among the CNVs with unknown significance, four VOUS involved genes with possible correlation to ASD, namely genes SNTG2, PARK2, CADPS2 and NLGN4X. The diagnostic efficacy of aCGH in our cohort was comparable with those of the previously reported and identified an important proportion of genetic ASD cases. Despite the continuum of published studies on the CNV testing in ASD cohorts, a considerable number of VOUS CNVs is still being identified, namely 10.0% in our study.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autism spectrum disorders (ASD) are one of the most common neurodevelopmental disorders with a globally estimated prevalence of around 60/10.000 (Elsabbagh et al. 2012). The clinical presentation of ASD is highly heterogeneous, with main characteristics being impairment of communication and social interactions, restricted interests and (stereo)typical behavioural patterns. The first signs may be observed in the early childhood and definite first clinical identification and in the diagnosis established on average between 2 and 4 years of age (Egger et al. 2014). The complex, multifactorial and not fully understood etiological background of ASD likely combines the effects of multiple genes and environmental factors. The current heritability estimates, based on the study of more than 14,000 children with ASD, range up to 50% and the recurrence risk is significantly higher in monozygotic twins, as compared to dizygotic twins. In the latter group, the recurrence risk estimates are comparable to the general sibling’s risk (Sandin et al. 2014). Obtaining the definite genetic cause in the individual with ASD provides a relevant information about the disease, its progress, the most appropriate management interventions and the recurrence risk.
Within the group of individuals, which fulfil the clinical ASD criteria, there are approximately 75% of individuals with isolated ASD (Rosti et al. 2014). The remaining 25% of individuals present with ASD within the complex, syndromic clinical expression/phenotype. Estimated 10% of patients in the latter group present with the specific clinical phenotype which is mostly efficiently recognized, such as fragile X syndrome, tuberous sclerosis complex and Angelmann and Rett syndromes. The majority of remaining complex ASD cases represent a heterogeneous group of numerous clinically diverse presentations, many of which still need to be etiologically explained. With the implementation of the molecular karyotyping (array CGH (aCGH)) and whole-exome sequencing into the genetic diagnostics, the genome copy number variation (CNV) and a single-gene mutations are being recognized as significant causal factors in ASD. The molecular karyotyping is currently accepted as the first tier diagnostic tool for both the isolated and complex forms of ASD (Miller et al. 2010) as it provides significantly higher diagnostic yields (5–10%), as compared to the conventional karyotype analysis and/or fragile X testing, both of which were golden standards before the advent of an aCGH (Roberts et al. 2014; Shen et al. 2010; Sanders et al. 2011). The discovery rate of causative CNVs has been lower in the so-called simplex family studies with a single high-functioning proband with at least one unaffected sibling and the parents were analysed. In accordance with that, the studies on complex cases, which most often include the accompanying features of the developmental delay and intellectual disability, report a higher diagnostic yield, up to a 20% (Battaglia et al. 2013). Despite the numerous studies and databases of genomic variation in healthy individuals and those with simple or complex ASD, there clearly remains an important gap in our capabilities of explaining a genomic CNV in relation to the disease.
We performed an aCGH in 150 individuals with isolated and complex ASD with the aim to evaluate the diagnostic efficacy of aCGH and the relevance of its implementation in a routine genetic testing in ASD patients.
Methods
Study subjects
In the period between January 2012 and June 2016, a cohort of 150 individuals with isolated or complex ASD were tested for the genome-wide copy number variation at the Laboratory for Molecular Cytogenetics, Clinical Institute of Medical Genetics, UMC Ljubljana. Patients were referred from the Clinical Institute of Medical Genetics and the Department of Child, Adolescent and Developmental Neurology, University Children’s Hospital, UMC Ljubljana. Diagnosis of ASD was established using the Autism Diagnostic Observation Schedule (ADOS). Developmental delay and intellectual disability were evaluated using Bayley Scales of Infant and Toddler Development and the Wechsler Intelligence Scale for Children (WISC), depending on the patient’s age.
The patients were designated to one of the three groups, according to the complexity of their clinical phenotype. The first group consisted of the patients diagnosed with ASD with or without the additional behavioural characteristics (group ASD), the second group included patients with ASD and a developmental delay (group ASD + DD) and the third group included the children with ASD and other complex health problems (intellectual disability, epilepsy and congenital anomalies; group ASD + other). Congenital anomalies and epilepsy were diagnosed and monitored within complex clinical evaluation using other diagnostic methods, depending on the type of anomaly, including ultrasound, MRI, EEG, hearing loss evaluation and ophthalmological evaluation.
Genetic testing
The DNA was isolated from peripheral blood samples, according to the manufacturer’s protocol using the Qiagen Mini kit (Qiagen, Valencia, CA). The quality and concentration parameters of the DNA were measured on a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific Inc.) and a Qubit 2.0 fluorometer (Life Technologies Inc.).
Following the sample extraction, the DNA was processed according to the Agilent protocol (Version 7.3 March 2014) using commercially available male and female genomic DNA (Agilent Technologies, Human Reference DNA, Male and Female) or in-house DNA reference mix as a reference DNA. The Agilent SurePrint G3 Unrestricted CGH ISCA v2 8x60K microarrays were used which provide a practical average resolution of 100 kb. The array images were acquired using the Agilent laser scanner G2565CA. The image files were quantified using the Agilent Feature extraction software for Cytogenomics 3.0 and analysed with the Agilent Cytogenomics 3.0 software (Agilent Technologies).
Classification of results
The Slovene genomic population database (our in-house database) and the Database of Genomic Variants (DGV; http://cdgv.tcag.ca/dgv/app/home) were used as a reference set for a normal copy number variation.
The discovered CNVs were compared with known aberrations in the different available databases ClinGen (http://dbsearch.clinicalgenome.org/search/), Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl (DECIPHER; https://decipher.sanger.ac.uk/) and ClinVar (National Center for Biotechnology Information, U.S. National Library of Medicine; http://www.ncbi.nlm.nih.gov/clinvar/). The CNVs were classified into the three groups, pathogenic, a variant of unknown significance (VOUS) and benign, according to the ACMG standards and guidelines (Kearney et al. 2011). Benign CNVs were those reported in the abovementioned databases as benign or present in our in-house database in more than 1% of cases. The class of pathogenic CNVs included either the already known microdeletion/microduplication syndromes or the larger losses/gains of the DNA, which have already been described as pathogenic in the literature (PubMed) and/or the abovementioned databases. In addition, we compared our results with a SFARI gene autism database, which holds 2222 different CNV regions and 752 genes, associated with ASD in the different types of studies (collected until February 2018; Simons Foundation Autism Research Initiative; https://gene.sfari.org/autdb/CNVHome.do).
Results
In the group of 150 patients, there were 117 boys and 33 girls, with male-to-female ratio of 3.7:1. Altogether, there were 61 diagnosed with ASD with or without additional behavioural characteristics, 61 diagnosed with ASD and developmental delay and 28 patients with ASD and the other complex diagnoses.
Details about age at genetic testing, standard deviation and the distribution of diagnostic subgroups according to gender are presented in Table 1. Some of the patients were already in their adulthood and came back to clinical geneticists after many years, being referred from general practitioners or neurologists based on new genetic diagnostic workflow for autism.
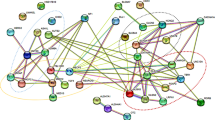
We discovered 11 (7.3%) clinically relevant CNVs and 15 (10.0%) VOUS CNVs in 150 patients (Table 1). The highest proportion of pathogenic CNVs was identified in the group of patients, categorized as ASD + other (4/28; 14.3%), but these numbers are too low to be statistically significant. Further details are presented in Fig. 1 and Table 2.

The distribution of CNVs among all patient groups
Patients were divided into three groups. Group 1 represents patients diagnosed with ASD with or without additional behavioural characteristics (group ASD), group 2 represents patients with ASD and developmental delay (group ASD + DD) and group 3 represents patients with ASD and other complex health problems (intellectual disability, epilepsy and congenital anomalies; group ASD + other). Different colours present different categories of discovered CNVs.
Among the pathogenic CNVs that were discovered in our patients, the ones that most commonly appeared in the literature were loss at 16p11.2 (68 studies according to SFARI database), followed by the loss at 22q11.21 (58 studies), gain at 7q11.23 (42 studies), loss at Xq28 (36 studies), gain at 6q22.31 (20 studies) and loss at Xq24 (10 studies). In our cohort of ASD patients, deletion in the region 22q11.21 occurred twice; all other pathogenic CNVs were only found once. A partial UPF3B gene (OMIM*300298) deletion was identified in one patient. No similar deletions have been reported before.
Among less common pathogenic CNVs, we identified a 10p15.3 deletion in a girl with ASD, mild dysmorphic features and mild global developmental delay, with a more pronounced delay in speech and language development and gross motor skills. The proband has had growth parameters in the normal range and no significant congenital anomaly (normal head MRI, normal heart and abdominal ultrasound). Similar cases have been described under a 10p15.3 microdeletion syndrome and ZMYND11 gene (OMIM*608668) has been shown to be the cause for major clinical characteristics in this syndromic intellectual disability (Cobben et al. 2014; Tumiene et al. 2017). Another rare pathogenic CNV was a 16q23.2q23.3 deletion in a girl with ASD, ADHD and mild global developmental delay, with a more significant delay in speech and language development. The haploinsufficiency of the CMIP gene (OMIM*610112) in the deleted region has been reported in four cases with similar phenotype (Luo et al. 2017).
Similarly, telomeric 20p13 deletions have been rarely described. Herein, we identified a pathogenic terminal 20p13 deletion in a boy with ASD, developmental delay, microcephaly and stereotypic behaviour. At the reevaluation, astigmatism and dental caries were noted. No major anomalies were identified. This deletion includes the smallest region of overlap, reported previously in a group of patients with 20p13 deletion (An et al. 2013).
The group of VOUS was further scrutinized by detailed analysis of all affected genes. Four out of 15 VOUS contained gene that was previously reported as significantly correlated with ASD, syntrophin-γ2 (SNTG2, OMIM*608715), located in 2p25.3, RING domain-containing E3 ubiquitin ligase parkin (PARK2, OMIM*602544) located in 6q26, calcium-dependent activator protein for secretion (CADPS2, OMIM*609978) located in 7q31.32 and neuroligin 4 (NLGN4X, OMIM*300427) located in Xp22.32p22.31.
Discussion
The presented results of molecular karyotyping in the group of individuals diagnosed with isolated or complex ASD indicate an important diagnostic efficacy of aCGH (7.3%). All of the reported clinically relevant CNVs would not be detected using classical chromosome analysis, which was the first tier test in the past. In addition, 10% of individuals were identified as carriers of the potentially relevant CNVs, classified currently as a VOUS.
The current guidelines for the diagnostic genetic testing in individuals with ASD with or without additional clinical features, such as developmental delay, seizures or other congenital anomalies, recommend the aCGH as a first tier test. The reported diagnostic yield using molecular karyotyping is between 5 and 20%, depending on the inclusion criteria in different published reports (Roberts et al. 2014; Shen et al. 2010; Sanders et al. 2011; Battaglia et al. 2013). The higher numbers are reported in the studies where individuals with a more complex, severe phenotype are included. Our cohort included both the isolated and complex ASD cases and we identified 7.3% pathogenic CNVs and additional 10.0% of VOUS which is in accordance with the previous data. When the patients were stratified according to the severity of their clinical presentation, the diagnostic efficacy was significantly higher in the group of patients with complex phenotype, being 11.1% for pathogenic CNVs and additional 13.3% for VOUS.
Within the identified pathogenic CNVs, we discovered a previously not reported partial UPF3B (UPF3, yeast homolog of B, OMIM*300298) gene deletion as clinically relevant in the patient. The UPF3B gene is associated with an X-linked syndromic mental retardation, type14 (MRXS14, OMIM#300676). The clinical features of MRSX14 are macrocephaly with dysmorphic facial features, high stature, pectus deformities and scoliosis or kyphosis, long upper limbs and fingers, and big feet. The muscular hypotonia, mild to moderate developmental delay and ASD or ADHD have been described as a part of the clinical spectrum, as well (Xu et al. 2013; Laumonnier et al. 2010). Recently, some cases with developmental delay only have also been reported (Jolly et al. 2013). Truncating point mutations and 1–4-bp deletions have been found in patients; so far, there are no reports on partial gene deletions. The proposed mechanism of action was a loss of function due to the frameshift and nonsense mutations. We discovered a partial gene deletion, encompassing intron 1 and exon 2, in the UPF3B gene in an 8-year-old boy with the diagnosis of autism and developmental delay.
We also report on three cases with rare pathogenic CNV (10p15.3 deletion, 16q23.2q23.3 deletion and 20p13 deletion). Each one of these was only reported in a few cases in the literature; therefore, herein reported cases give further insight into the clinical spectrum of microdeletion syndromes.
Four of the identified VOUS contain the genes previously associated with ASD. The syntrophin-γ2 (SNTG2, OMIM*608715) interacts with neuroligins, as a crucial binding factor at the inhibitory synapses (Yamakawa et al. 2007). Mutations in both binding neural cell adhesion molecules, neuroligin 3 (NLGN3, OMIM*300336) and neuroligin 4 (NLGN4, OMIM*300427) have been observed in the autism and mental retardation (Jamain et al., 2003). Our patient had a partial duplication of the gene and only ASD in his clinical phenotype. The parents of this proband were unavailable for genetic testing, which would provide some additional information about the discovered CNV. The second VOUS was partial PARK2 (OMIM*602544) deletion. The parkin, a RING domain-containing E3 ubiquitin ligase, is a cytoskeleton-associated protein, shown to be expressed in the central nervous system (Yoshii et al. 2011). Partial heterozygous PARK2 microdeletions and microduplications have been reported in the patients with ASD (Scheuerle and Wilson 2011) and functional studies showed decreased protein expression in carriers of CNVs in the PARK2 gene (Yin et al. 2016). Some authors suggest that this might function as a second-hit model, where individuals carry both partial PARK2 deletion and some yet unidentified single point mutation, not detectable by aCGH analysis. True enough, our patient with ASD and mild developmental delay inherited the deletion from his healthy mother; therefore, second-hit model might be a plausible explanation for this VOUS.
A partial duplication of CADSP2 (OMIM*609978, the third VOUS) was also not tested for the parental origin in our patient. The gene deletions have been shown to induce autistic-like phenotype in mouse model and both CNV and point mutations were identified in individuals with ASD and intellectual disability (Sadakata et al. 2013; Bonora et al. 2014). Even more, some latest results suggest imprinting effect, as the gene is only expressed from maternal allele in specific tissues.
The fourth VOUS of a partial NLGN4X gene duplication was identified in a boy with ASD and mild developmental delay. NLGN4X belongs to a family of neuronal cell surface proteins, splice site-specific ligands for beta-neurexins, suggested to be involved in the synapse formation and remodelling. Point mutations have already been associated with the autism, Asperger syndrome and mental retardation (Jamain et al. 2003) and no partial duplications have been reported so far. Interestingly, there is a NLGN4Y gene and females express NLGN4X from one copy of the X chromosome, while males express simultaneously both NLGN4X and NLGN4Y (Ross et al. 2015). A study on the group of 47 XYY males speculated that higher incidence of behavioural problems and learning and language disabilities might be partially due to the increased copy number of neuroligins.
Finally, the male predominance in ASD is well known and a significantly higher proportion of males (3.7:1 ratio) was referred to genetic testing in the cohort reported here, as well. An intriguing explanation for male predominance was suggested by Sanders et al. (2011), where authors investigated the correlation of gender versus size and gene content of CNV in ASD cohort. Based on their study, they suggested that the females are more resistant to the genetic perturbation as a higher impact on IQ per gene was shown in males. Bearing this in mind, it might be that females with CNV from the CNV risk factor group are just above the clinical/diagnostic threshold for the ASD or mild intellectual disability and are therefore not being evaluated and tested. This may explain some of the discovered VOUS CNVs in boys with ASD, inherited from apparently healthy mothers. In the series of patients reported here, six VOUS variants were inherited, five of them being present in boys. Our group of patients is too small to make further conclusions and additional studies are needed on big cohorts to clarify suggested interpretation.
In conclusion, the genetic variation has been acknowledged to have a relevant impact in ASD. Using the molecular karyotyping, the causative CNVs were identified in 7.3% of individuals with isolated or complex ASD. In addition, 10.0% of individuals harbour a VOUS, which contributes to the current knowledge and suggests a potential risk factors for ASD. Their true clinical relevance is expected to be clarified in the future with additional cohort reports and functional studies.
Abbreviations
- aCGH:
-
array comparative genomic hybridization
- ASD:
-
autism spectrum disorder(s)
- CNV:
-
copy number variant
- VOUS:
-
variant of unknown significance
References
An Y, Amr SS, Torres A, Weissman L, Raffalli P, Cox G, Sheng X, Lip V, Bi W, Patel A, Stankiewicz P, Wu BL, Shen Y (2013) SOX12 and NRSN2 are candidate genes for 20p13 subtelomeric deletions associated with developmental delay. Am J Med Genet B Neuropsychiatr Genet 162B(8):832–840
Battaglia A, Doccini V, Bernardini L, Novelli A, Loddo S, Capalbo A, Filippi T, Carey JC (2013) Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur J Paediatr Neurol 17(6):589–599
Bonora E, Graziano C, Minopoli F, Bacchelli E, Magini P, Diquigiovanni C, Lomartire S, Bianco F, Vargiolu M, Parchi P, Marasco E, Mantovani V, Rampoldi L, Trudu M, Parmeggiani A, Battaglia A, Mazzone L, Tortora G, IMGSAC ME, Seri M, Romeo G (2014) Maternally inherited genetic variants of CADPS2 are present in autism spectrum disorders and intellectual disability patients. EMBO Mol Med 6(6):795–809
Cobben JM, Weiss MM, van Dijk FS, De Reuver R, de Kruiff C, Pondaag W, Hennekam RC, Yntema HG (2014) A de novo mutation in ZMYND11, a candidate gene for 10p15.3 deletion syndrome, is associated with syndromic intellectual disability. Eur J Med Genet 57:636–638
Egger G, Roetzer KM, Noor A, Lionel AC, Mahmood H, Schwarzbraun T, Boright O, Mikhailov A, Marshall CR, Windpassinger C, Petek E, Scherer SW, Kaschnitz W, Vincent JB (2014) Identification of risk genes for autism spectrum disorder trough copy number variations analysis in Austrian families. Neurogenetics 15(2):117–127
Elsabbagh M, Divan G, Koh YJ, Kim YS, Kauchali S, Marcín C, Montiel-Nava C, Patel V, Paula CS, Wang C, Yasamy MT, Fombonne E (2012) Global prevalence of autism and other pervasive developmental disorders. Autism Res 5(3):160–179
Jamain S, Quach H, Betancur C, Råstam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T (2003) Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet 34(1):27–29
Jolly LA, Homan CC, Jacob R, Barry S, Gecz J (2013) The UPF3B gene, implicated in intellectual disability, autism, ADHD and childhood onset schizophrenia regulates neural progenitor cell behaviour and neuronal outgrowth. Hum Mol Genet 22(23):4673–4687
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST, A working group of the American college of medical genetics ACMG laboratory quality assurance committee (2011) American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 13(7):680–685
Laumonnier F, Shoubridge C, Antar C, Nguyen LS, Van Esch H, Kleefstra T, Briault S, Fryns JP, Hamel B, Chelly J, Ropers HH, Ronce N, Blesson S, Moraine C, Gecz J, Raynaud M (2010) Mutations of the UPF3B gene, which encodes a protein widely expressed in neurons, are associated with nonspecific mental retardation with or without autism. Mol Psychiatry 15(7):767–776
Luo M, Fan J, Wenger TL, Harr MH, Racobaldo M, Mulchandani S, Dubbs H, Zackai EH, Spinner NB, Conlin LK (2017) CMIP haploinsufficiency in two patients with autism spectrum disorder and co-occurring gastrointestinal issues. Am J Med Genet A 173(8):2101–2107
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP et al (2010) Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 86(5):749–764
Roberts JL, Hovanes K, Dasouki M, Manzardo AM, Butler MG (2014) Chromosomal microarray analysis of consecutive individuals with autism Spectrum disorders or learning disability presenting for genetic services. Gene 535(1):70–78
Ross JL, Tartaglia N, Merry DE, Dalva M, Zinn AR (2015) Behavioral phenotypes in males with XYY and possible role of increased NLGN4Y expression in autism features. Genes Brain Behav 14(2):137–144
Rosti RO, Sadek AA, Vaux KK, Gleeson JG (2014) The genetic landscape of autism spectrum disorders. Dev Med Child Neurol 56(1):12–18
Sadakata T, Shinoda Y, Oka M, Sekine Y, Furuichi T (2013) Autistic-like behavioral phenotypes in a mouse model with copy number variation of the CAPS2/CADPS2 gene. FEBS Lett 587(1):54–59
Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D et al (2011) Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70(5):863–885
Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A (2014) The familial risk of autism. JAMA 311(17):1770–1777
Scheuerle A, Wilson K (2011) PARK2 copy number aberrations in two children presenting with autism spectrum disorder: further support of an association and possible evidence for a new microdeletion/microduplication syndrome. Am J Med Genet B Neuropsychiatr Genet 156B(4):413–420
Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB et al (2010) Clinical genetic testing for patients with autism spectrum disorders. Pediatrics 125(4):e727–e735
Tumiene B, Čiuladaitė Ž, Preikšaitienė E, Mameniškienė R, Utkus A, Kučinskas V (2017) Phenotype comparison confirms ZMYND11 as a critical gene for 10p15.3 microdeletion syndrome. J Appl Genet 58(4):467–474
Xu X, Zhang L, Tong P, Xun G, Su W, Xiong Z, Zhu T, Zheng Y, Luo S, Pan Y, Xia K, Hu Z (2013) Exome sequencing identifies UPF3B as the causative gene for a Chinese non-syndrome mental retardation pedigree. Clin Genet 83(6):560–564
Yamakawa H, Oyama S, Mitsuhashi H, Sasagawa N, Uchino S, Kohsaka S, Ishiura S (2007) Neuroligins 3 and 4X interact with syntrophin-gamma2, and the interactions are affected by autism-related mutations. Biochem Biophy Res Commun 355(1):41–46
Yin CL, Chen HI, Li LH, Chien YL, Liao HM, Chou MC, Chou WJ, Tsai WC, Chiu YN, Wu YY, Lo CZ, Wu JY, Chen YT, Gau SS (2016) Genome-wide analysis of copy number variations identifies PARK2 as a candidate gene for autism spectrum disorder. Mol Autism 7:23
Yoshii SR, Kishi C, Ishihara N, Mizushima N (2011) Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem 286(22):19630–19640
Author information
Authors and Affiliations
Contributions
LL, MV, SB, BGS, DO and MJV carried out the clinical evaluation and provided the clinical genetic consultations to patients. LL and PR analysed and interpreted the data. LL, PR, MV and BP drafted the manuscript. SB, BGS, MJV and DO critically revised the final manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
The study was approved by the National Ethical Review Board in Ljubljana, Slovenia (0120-288/2016-2). All participants (their legal representatives) signed a written, informed consent.
Additional information
Communicated by: Communicated by: Michal Witt
Rights and permissions
About this article

Cite this article
Lovrečić, L., Rajar, P., Volk, M. et al. Diagnostic efficacy and new variants in isolated and complex autism spectrum disorder using molecular karyotyping. J Appl Genetics 59, 179–185 (2018). https://doi.org/10.1007/s13353-018-0440-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-018-0440-y