
Abstract
Elacridar (GF120918) is a highly potent inhibitor of both P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2), the main efflux transporters expressed at the blood-brain barrier (BBB). Elacridar shows very low aqueous solubility, which complicates its formulation for i.v. administration. An intravenous infusion protocol would be preferred to achieve high and controlled plasma concentrations of elacridar in large animals, including nonhuman primates. Formulation of elacridar for i.v. infusion was achieved using a co-solvent strategy, resulting in an aqueous dispersion with a final concentration of 5 g L−1 elacridar with tetrahydrofuran (5% w/v) in aqueous D-glucose solution (2.5%, w/v). Particle size (mean = 2.8 ± 0.9 μm) remained stable for 150 min. The preparation was i.v. administered as a continuous infusion (12 mg kg−1 h−1 for 90 min) to three baboons. Arterial and venous plasma pharmacokinetics (PK) of elacridar were monitored using a newly developed and validated HPLC-UV method. Elacridar concentration increased rapidly to reach a plateau at 9.5 μg mL−1 within 20 min after the start of infusion. Elacridar PK in venous plasma did not differ from arterial plasma facing the BBB, indicating the absence of an arteriovenous concentration gradient. Intravenous infusion of elacridar allows for controlled exposure of the BBB and offers a useful tool to assess the impact of ABCB1/ABCG2 on drug disposition to the brain in nonhuman primates, a relevant animal model for the study of transporter function at the BBB.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The distribution of many drugs from the blood to the brain parenchyma is limited by the activity of ATP-binding cassette (ABC) transporters at the luminal membrane of brain capillary endothelial cells, which form the blood-brain barrier (BBB). These multidrug transporters were shown to limit the BBB permeation and control the brain clearance of many structurally diverse drugs by active and unidirectional efflux transport into the blood [1].
The most abundant ABC transporters at the human BBB are P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) [2, 3]. Studies in transporter knockout mice have shown that Abcb1a and Abcg2 work together in limiting brain distribution of shared (dual) ABCB1/ABCG2 substrates. When only Abcb1a alone or Abcg2 alone is knocked out, the other transporter still significantly restricts its brain access, so that dual substrates can only gain substantial brain access when both Abcb1a and Abcg2 are knocked out. This mutual functional compensation between Abcb1a and Abcg2 at the BBB has been clearly demonstrated in mice for various anticancer drugs that are dual ABCB1/ABCG2 substrates [4].
Most members of the therapeutic class of tyrosine kinase inhibitors (TKIs) are dual ABCB1/ABCG2 substrates, including axitinib, cediranib, dabrafenib, dasatinib, erlotinib, gefitinib, imatinib, pazopanib, regorafenib, rucaparib, sorafenib, sunitinib, tandutinib, trametinib, vandetanib, veliparib, and vemurafenib [5, 6]. Growth of a considerable percentage of brain metastases and primary tumors is dependent on tyrosine kinase activity, but inadequate brain delivery of TKIs severely limits the treatment of central nervous system (CNS) malignancies with TKIs [7]. Pharmacological inhibition is an appealing strategy to investigate the impact of ABCB1/ABCG2-mediated efflux on the brain distribution of their common substrates [5].
Elacridar (N-(4-(2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl)phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide, GF120918) is a potent dual ABCB1/ABCG2 inhibitor which inhibits ABCB1 and ABCG2 with comparable potencies [5]. Elacridar has been shown to improve the brain delivery of a large number of dual ABCB1/ABCG2 substrates, such as TKIs, in mice and rats (for review, see Durmus et al. [5]. Compared to other prototypical dual ABCB1/ABCG2 inhibitors, elacridar benefits from available tolerance and safety data in humans [8]. So far, only oral formulations of elacridar have been tested in humans, which can in all likelihood not be used to inhibit ABCB1/ABCG2 at the BBB due to very low oral bioavailability leading to insufficiently high plasma concentrations [9, 10].
Recent quantitative proteomics work has shown that ABCG2 is relatively more important at the human than at the rodent BBB with ABCG2/ABCB1 expression ratios of approximately 1.3 in humans and of 0.3 in mice [2, 3]. On the other hand, ABC transporter expression profiles in human brain microvessels have been shown to closely resemble those of nonhuman primates [11]. This observation justifies the use of nonhuman primates to test the influence of ABCB1/ABCG2-mediated efflux on the brain distribution of dual ABCB1/ABCG2 substrates in the perspective of clinical translation. To that end, an intravenous (i.v.) formulation of elacridar would be preferred in order to set up an optimal administration protocol, define the target plasma concentrations for adequate ABCB1/ABCG2 inhibition at the nonhuman primate BBB and control the plasma concentration of elacridar and its subsequent exposure to the BBB. Unfortunately, such an i.v. formulation is not available because elacridar shows unfavorable physicochemical properties such as poor water solubility (12.3 × 10−5 mg mL−1) [12] and high lipophilicity (log P = 5.67) [13] which complicate its formulation in aqueous solvent for i.v. administration.
In this work, we report an original protocol to prepare an i.v. formulation of elacridar which was administered to nonhuman primates. Corresponding pharmacokinetics (PK) of elacridar in venous and arterial plasma was assessed by a newly developed HPLC-UV method.
Methods
Solvents and chemicals
Elacridar hydrochloride (C34H33N3O5.HCl; molecular weight 600.1 g mol−1) was purchased from Syncom (The Netherlands). Tariquidar dimesylate (C38H38N4O6.C2H8O6S2; molecular weight 838.94 g mol−1), used as an internal standard, was purchased from Eras Labo (France). Ultrapure water was obtained using an ultraviolet purification system (Purelab, France). Tetrahydrofuran (THF) stabilized with 2,6-di-tert-butyl-4-methylphenol was purchased from Carlo Erba Reagents (France). Hydrochloric acid (37%, v/v), ammonium hydroxide solution (28%, v/v), as well as HPLC-grade solvents were purchased from Sigma-Aldrich (France).
Injectable formulation
To elacridar hydrochloride (504 ± 20 mg) THF (50 g) was added and the mixture sonicated for 5 min to allow for solubilization. Ultrapure water (50 mL) was then added and the mixture was again sonicated for 5 min. Once elacridar was solubilized, THF was evaporated under Rotavap vacuum (35 min, 34 °C, 180 mbar). This stock preparation could be stored at room temperature and no precipitate could be observed after 3 h storage. Immediately before infusion, the stock preparation (48 ± 5 mL) was mixed with aqueous D-glucose solution (5%, w/v, 50 mL) to obtain a ready-to-inject dispersion of elacridar in aqueous D-glucose (2.5%, w/v, 100 mL) with a final elacridar concentration of 5 g L−1.
Determination of residual THF in the preparation
Analysis of residual organic solvent (i.e. THF) in the i.v. formulation of elacridar was performed on a gas chromatograph (GC) Clarus 580 (Perkin Elmer, France) equipped with a headspace sampler TurboMatrix 40 (T°C needle: 120 °C; T°C transfer: 180 °C; T°C oven: 80 °C, Perkin Elmer) and a flame ionization detector (FID) (250 °C, air flow rate: 450 mL min−1; hydrogen flow rate 45 mL min−1). The initial temperature of the capillary column CP-select 624 (30 m × 0.53 mm, 3 μm, Varian, France) was 40 °C for 5 min and was then increased to 140 °C at 10 °C min−1, where it was maintained for 5 min. The carrier gas was helium with a pressure of 5 psi, delivered at a flow rate of 7.33 mL min−1. The preparation (10 μL) was diluted with water (9.99 mL) and analyzed by GC-FID. The assay was sensitive and linear over a THF concentration range of 0.5–6.0 g mL−1 corresponding to 0.5 to 6% (w/v) THF in the formulation of elacridar for i.v. injection.
Characterization of the preparation
Mean particle size (Z-average) and polydispersity index (PDI) of elacridar particles were determined by dynamic light scattering (DLS, weighted by intensity, 173° scattering angle), using a Malvern Zetasizer Nano ZSs (Malvern Instruments Ltd., Malvern, UK). This apparatus allows for size measurement of particles in the range between 0.3 nm and 10 μm. Before measurement, appropriate dilution (1/100) of the preparation was performed in a THF/water mixture (5/95). All measurements were carried out at 25 °C in triplicate. The dispersion stability has been evaluated during its storage at room temperature by regular size measurements at 5, 60, 110, 120, 130, 140, 150, 160, 170, and 180 min. At each time-point, visual inspection of the preparation was performed to detect any precipitate.
Animal use
All animal use procedures were in accordance with the recommendations of the European Community (86/809/CEE) and the French National Committees (law 87/848) for the care and use of laboratory animals. The experimental protocol was approved by a local ethics committee for animal use (CETA/APAFIS#892).
Three adult male Papio Anubis baboons (24–27 kg) were used for this study. During elacridar infusion, animals underwent a concomitant positron emission tomography (PET) imaging scan, which results have already been reported [14]. This PET protocol aimed at investigating the ABCB1/ABCG2 potency of the preparation. The present article focuses on the on the pharmaceutical aspects of the project (formulation, analytics, PK) that have not yet been reported.
PK study in nonhuman primates
The animals first received ketamine (10 mg kg−1, i.m.) to induce anesthesia. After being intubated, a catheter was inserted into a sural vein for the elacridar infusion. Another sural vein catheter was dedicated to propofol infusion. A catheter was inserted in the right femoral artery and the right femoral vein for blood sampling. Anesthesia was maintained using an i.v. bolus of propofol (2 mL) followed by a 16 to 22 mL h−1 i.v. infusion under oxygen ventilation. The tidal volume was adjusted to achieve stable end-tidal carbon dioxide tension between 38 and 42 mmHg. Heart rate and rectal temperature were monitored throughout the experiment.
Freshly prepared elacridar for i.v. infusion (12 mg.kg−1 h−1) was infused over 90 min. At selected time points after start of elacridar infusion (0, 2, 5, 15, 20, 25, 30, 35, 40, 45, 60, and 90 min), arterial and venous blood samples (3 mL) were simultaneously collected for determination of elacridar concentration.
Quantification of elacridar in baboon plasma
HPLC method
HPLC-UV method was developed for the determination of elacridar in plasma. HPLC was performed on an Alliance® 2996 system, equipped with an autosampler, a binary pump (Waters, France) and a photodiode array detector (PDA, 2996, Waters). Separation was achieved using an AtlantisT3® C18 column (4.6 mm × 150 mm, 5 μm, Waters). The mobile phase was composed of water containing 0.1% (v/v) trifluoroacetic acid (solvent A) and acetonitrile containing 0.1% (v/v) trifluoroacetic acid (solvent B) delivered in a gradient elution mode at a flow rate of 1.2 mL min−1: solvent B increased linearly from 20 to 65% from 0 to 15 min. Elacridar and IS were detected at a UV wavelength of 250 nm. Stock solutions of elacridar and IS were prepared in water/acetonitrile (27/73 and 80/20, v/v, respectively) at concentrations of 1 mg mL−1. The stock solutions were stored at − 20 °C.
Sample preparation
Arterial and venous blood samples obtained from the animal experiments were collected in tubes containing lithium iodoacetate as an anticoagulant (BD Vacutainer, France) and were immediately centrifuged for 10 min (2054 g, 4 °C). The plasma was stored at − 80 °C until analysis (< 1 month). For analysis, samples were thawed at room temperature. To the plasma sample (600 μL), IS solution diluted with water to a concentration of 60 μg.mL−1 (100 μL) and water containing 4% hydrochloric acid (v/v, 500 μL) were added. After vortexing, 1 mL of this mixture was deposited on a cation exchange solid-phase extraction cartridge (MCX, 30 mg, Oasis®, Waters, France) which had been pre-conditioned with 1 mL of methanol and 1 mL of water. The cartridge was washed with water containing 0.4% (v/v) hydrochloric acid (1 mL) followed by acetonitrile containing 0.4% (v/v) hydrochloric acid (1 mL). Elacridar and IS were finally eluted with acetonitrile containing 4% (v/v) ammonia (3 × 1 mL). The combined alkaline acetonitrile fractions were evaporated during 120 min (SPD1010, SpeedVac® system, Thermo Scientific, France). The residue was dissolved in water/acetonitrile/trifluoroacetic acid (80/20/0.5, v/v/v, 100 μL), vortexed for 30 s and sonicated for 5 min. This solution (20 μL) was then injected into the HPLC system.
Results
Preparation of elacridar for i.v. infusion
In preliminary experiments, different solvents were tested to solubilize elacridar and showed the low solubility of this compound in most clinically approved solvent (Table 1). However, the solubility of elacridar in THF at room temperature prompted us to develop of a co-solvent strategy for the formulation of elacridar for i.v. injection.
Ready-to-inject formulation of elacridar consisted in a yellow dispersion with no apparent precipitate (Fig. 1). In the final preparation, residual THF concentration was 4.3 ± 0.8 g per 100 mL.

Ready-to-inject dispersion (mean particle size of 2.8 ± 0.7 μm) of elacridar for i.v. infusion in nonhuman primates. The preparation was achieved using a co-solvent strategy, resulting in a final concentration of 5 g L−1 elacridar with tetrahydrofuran (5% w/v) in aqueous D-glucose solution (2.5%, w/v)
The preparation was characterized using dynamic light scattering. The results showed the presence of particles with a mean size of 2.8 ± 0.9 μm with a PDI of 0.5 ± 0.2. The formulation could be qualified as a dispersion. The stability of the dispersion at room temperature was evaluated by following the size of particles as a function of time using the same technique. The dispersion exhibited no evolution of size and PDI during the first 150 min (mean size of 2.8 ± 0.7 μm with a PDI of 0.5 ± 0.1 at 150 min). Between 150 and 180 min, a precipitation occurred relative to the increase in the size and PDI of particles (mean size of 4.7 ± 2.2 μm with a PDI of 0.7 ± 0.5 at 160 min; > 7 μm at 170 min). After 180 min, the mean size exceeded the limit of the apparatus (10 μm), thus showing the destabilization of the dispersion. These data showed that this formulation was suitable for perfusion in animals during 90 min.
Elacridar PK in baboons
Elacridar administration to baboons proved to be well tolerated and did not influence physiologic parameters (heart rate and rectal temperature) as well as the time to wake up after anesthesia.
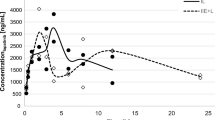
After the start of the i.v. infusion, elacridar concentration in venous plasma rapidly reached a plateau at a value of 9.5 ± 0.6 μg mL−1 within 20 min (Fig. 2). There was a significant correlation between the venous and the arterial plasma concentrations of elacridar measured in all animals (P < 0.001) indicating the absence of an arteriovenous concentration gradient for this compound (Fig. 2).

Pharmacokinetics of elacridar in arterial and venous plasma measured during intravenous constant infusion (12 mg kg−1 h−1 over 90 min) in baboons. Elacridar concentrations are presented as mean ± SD of three different animals
Discussion
In the present study, we developed an original formulation allowing for the continuous i.v. infusion of the potent ABCB1/ABCG2 inhibitor elacridar in large animals. The formulation of elacridar was achieved using a co-solvent strategy based on the solubility of elacridar in THF. Not many i.v. formulations of elacridar have so far been reported in the literature. This is probably due to the fact that elacridar is practically insoluble in aqueous solvents [12] and highly lipophilic (log P = 5.67) [13]. So far reported i.v. formulations required either the injection of organic solvent (dimethyl sulfoxide or propylene glycol) in large quantities [13] or an acidification of the preparation for injection to a pH of 3.75 [15], which may result in poor tolerance at the site of injection as well as in acidosis when the injected volume is too large. We additionally tested several other injectable organic solvents but failed to dissolve elacridar, leading to the choice of THF for the preparation of the i.v. injection. Starting from 50 g THF, it was decided to evaporate THF in order to reduce the content of this solvent in the preparation. No precipitate could be visually observed in this stock preparation. One stock preparation was used for stability testing and a precipitate could be observed after 5 h storage, only. The stock preparation could be further diluted in injectable D-glucose solution (5%, w/v), thus reducing the proportion of THF in the preparation. Residual THF content in the preparation was 4300 ± 0.8 mg 100 mL−1, corresponding to a total injected dose in baboons of approximately 200 mg kg−1. For comparison, the acute toxicity of THF was defined at a median lethal dose (LD50) of 2900 mg kg−1 in rats [17].
Only very few data on the PK of elacridar are available from the literature. In humans, oral doses of 1000 mg elacridar have been administered to patients treated with paclitaxel. The maximum plasma concentration (Cmax) of elacridar was 0.434 μg mL−1 which was reached at 7.7 h after intake [8]. In another study in which a new amorphous solid dispersion tablet of elacridar was tested in healthy volunteers, a Cmax of 0.326 μg mL−1 was reached at 9 h after intake [9]. In our experiments, a steady-state concentration of elacridar of 9.5 μg mL−1 was reached within only 20 min after the start of the infusion. This rapid equilibrium of the plasma concentrations justifies the use of a continuous infusion instead of bolus injection to maintain a suitable and controlled exposure of the BBB to elacridar.
The capillaries forming the BBB, where ABCB1 and ABCG2 are expressed, carry arterial blood [18]. Therefore, the exposure of the BBB to elacridar depends on the concentration of elacridar in the arterial plasma which may differ from venous plasma concentrations. Indeed, differences between arterial and venous plasma concentrations have been reported for many drugs, especially in the distribution phase [19]. Comparison of the venous and arterial plasma kinetics of elacridar indicated that venous blood sampling, which is preferentially performed during PK studies, accurately predicted the exposure of the luminal side of the BBB to elacridar. Moreover, maintained plateau concentrations, reached within only 20 min of infusion, allow for a relatively stable and controlled exposure of the BBB to elacridar.
Our continuous infusion protocol of elacridar was tested in nonhuman primates. Oral administration of elacridar has been reported in monkeys at the dose of 20 mg/kg as a well triturated suspension in 0.5% aqueous methylcellulose [20]. However, elacridar bioavailability was probably too low to achieve sufficient exposure to the BBB and suitable ABCB1/ABCG2 inhibition [9].
The ABCB1 and ABCG2 inhibition protocol proposed in this work, based on the i.v. infusion of elacridar, has already been evaluated in vivo [14]. PET imaging with [11C]erlotinib, a dual ABCB1/ABCG2 substrate, showed that the selected elacridar dose (12 mg kg−1 h−1) and administration route were efficient at inhibiting ABCB1/ABCG2-mediated efflux of erlotinib at the BBB in baboons. This work provided evidence of the possibility to achieve sufficient ABCB1/ABCG2 inhibition using elacridar at the nonhuman primate BBB [14]. The present protocol of elacridar infusion, allowing for constant and controlled arterial plasma concentrations, will be useful to define the target and IC50 elacridar plasma concentrations for suitable inhibition of ABCB1/ABCG2 at the BBB.
The final preparation consisted in a dispersion with a particle size of ~ 3 μm, suitable for i.v. administration. Particle size of the dispersion was shown stable for 150 min, thus allowing for a 90-min infusion. However, the stability of the stock preparation (~ 3 h) and the final dispersion (~ 150 min) were limited in time, which limit the commercial perspectives of the formulation. Moreover, the proportion of THF in the preparation was over the limit of acceptance for clinical use. Indeed, THF is a class 2 solvent, for which the exposure in humans is limited to 7.2 mg/day [16]. Therefore, the preparation is not likely to be accepted for clinical use in patients. It nonetheless provides a simple and convenient method, allowing for constant infusion of elacridar in animals, including large animal species. Pharmacological inhibition of ABCB1 and ABCG2 using elacridar infusion may thus find application as a probe to study the impact of efflux transporters at the BBB on drug distribution to the brain. In particular, this approach is relevant in nonhuman primates who have ABC transporter expression profiles at the BBB similar to those in humans [11].
Conclusion
A novel formulation for continuous i.v. infusion of the dual ABCB1/ABCG2 inhibitor elacridar was developed and successfully used in nonhuman primates to achieve controlled exposure of the BBB to elacridar. In drug development, this infusion protocol may be useful to address the impact of ABCB1/ABCG2 transport function on drug delivery to the brain in this relevant animal model of the human BBB.
Abbreviations
- ABC:
-
ATP-binding cassette
- BBB:
-
Blood-brain barrier
- CSs:
-
Calibration standards
- CNS:
-
Central nervous system
- IS:
-
Internal standard
- i.v.:
-
Intravenous
- C max :
-
Maximum plasma concentration
- PK:
-
Pharmacokinetics
- PDI:
-
Polydispersity index
- QCs:
-
Quality controls
- THF:
-
Tetrahydrofuran
- TKIs:
-
Tyrosine kinase inhibitors
References
Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37(1):13–25. https://doi.org/10.1016/j.nbd.2009.07.030.
Shawahna R, Uchida Y, Declèves X, Ohtsuki S, Yousif S, Dauchy S, et al. Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol Pharm. 2011;8(4):1332–41. https://doi.org/10.1021/mp200129p.
Uchida Y, Wakayama K, Ohtsuki S, Chiba M, Ohe T, Ishii Y, et al. Blood-brain barrier pharmacoproteomics-based reconstruction of the in vivo brain distribution of P-glycoprotein substrates in cynomolgus monkeys. J Pharmacol Exp Ther. 2014;350(3):578–88. https://doi.org/10.1124/jpet.114.214536.
Kathawala RJ, Gupta P, Ashby CR, Chen Z-S. The modulation of ABC transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother 2015;18:1–17.
Durmus S, Hendrikx JJMA, Schinkel AH, Apical ABC. Transporters and cancer chemotherapeutic drug disposition. Adv Cancer Res. 2015;125:1–41. https://doi.org/10.1016/bs.acr.2014.10.001.
Kort A, Durmus S, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Brain and testis accumulation of regorafenib is restricted by breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1). Pharm Res. 2015;32(7):2205–16. https://doi.org/10.1007/s11095-014-1609-7.
Agarwal S, Sane R, Oberoi R, Ohlfest JR, Elmquist WF. Delivery of molecularly targeted therapy to malignant glioma, a disease of the whole brain. Expert Rev Mol Med. 2011;13:e17. https://doi.org/10.1017/S1462399411001888.
Malingré MM, Beijnen JH, Rosing H, Koopman FJ, Jewell RC, Paul EM, et al. Co-administration of GF120918 significantly increases the systemic exposure to oral paclitaxel in cancer patients. Br J Cancer. 2001;84(1):42–7. https://doi.org/10.1054/bjoc.2000.1543.
Sawicki E, Verheijen RB, Huitema ADR, van Tellingen O, Schellens JHM, Nuijen B, et al. Clinical pharmacokinetics of an amorphous solid dispersion tablet of elacridar. Drug Deliv. Transl. Res. 2017;7;125–131.
van Tellingen O, Yetkin-Arik B, de Gooijer MC, Wesseling P, Wurdinger T, de Vries HE. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat. 2015;19:1–12. https://doi.org/10.1016/j.drup.2015.02.002.
Hoshi Y, Uchida Y, Tachikawa M, Inoue T, Ohtsuki S, Terasaki T. Quantitative atlas of blood-brain barrier transporters, receptors, and tight junction proteins in rats and common marmoset. J Pharm Sci. 2013;102(9):3343–55. https://doi.org/10.1002/jps.23575.
Sane R, Mittapalli RK, Elmquist WF. Development and evaluation of a novel microemulsion formulation of elacridar to improve its bioavailability. J Pharm Sci. 2013;102(4):1343–54. https://doi.org/10.1002/jps.23450.
Sane R, Agarwal S, Elmquist WF. Brain distribution and bioavailability of elacridar after different routes of administration in the mouse. Drug Metab Dispos. 2012;40(8):1612–9. https://doi.org/10.1124/dmd.112.045930.
Tournier N, Goutal S, Auvity S, Traxl A, Mairinger S, Wanek T, et al. Strategies to inhibit ABCB1- and ABCG2-mediated efflux transport of erlotinib at the blood-brain barrier: a PET study in non-human primates. J Nucl Med. 2017;58(1):117–22. https://doi.org/10.2967/jnumed.116.178665.
Tong W-Q, Lee Wells M. Parenteral pharmaceutical compositions containing gf120918a. WO 1996011007 A1. http://www.google.com/patents/WO1996011007A1?cl=en. 1996 avr;
EMA. ICH guideline Q3C (R5) on impurities: guideline for residual solvents. 2015;
Katahira T, Teramoto K, Horiguchi S. [Experimental studies on the acute toxicity of tetrahydrofuran in animals]. Sangyō Igaku Jpn. J. Ind. Health. 1982;24:373–378.
Saubaméa B, Cochois-Guégan V, Cisternino S, Scherrmann J-M. Heterogeneity in the rat brain vasculature revealed by quantitative confocal analysis of endothelial barrier antigen and P-glycoprotein expression. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab 2012;32:81–92, 1, DOI: https://doi.org/10.1038/jcbfm.2011.109.
Goutal S, Auvity S, Legrand T, Hauquier F, Cisternino S, Chapy H, et al. Validation of a simple HPLC-UV method for rifampicin determination in plasma: application to the study of rifampicin arteriovenous concentration gradient. J Pharm Biomed Anal. 2016;123:173–8. https://doi.org/10.1016/j.jpba.2016.02.013.
Karibe T, Hagihara-Nakagomi R, Abe K, Imaoka T, Mikkaichi T, Yasuda S, et al. Evaluation of the usefulness of breast cancer resistance protein (BCRP) knockout mice and BCRP inhibitor-treated monkeys to estimate the clinical impact of BCRP modulation on the pharmacokinetics of BCRP substrates. Pharm Res. 2015;32(5):1634–47. https://doi.org/10.1007/s11095-014-1563-4.
Acknowledgements
We thank Jérôme Cayla for helpful technical assistance during nonhuman primate experiments. Animal experiments were performed on a platform of France Life Imaging network partly funded by the grant “ANR-11-INBS-0006”. The Unité de Technologies Chimiques et Biologiques pour la Santé (UTCBS, CNRS UMR 8258, Inserm U1022. CNRS UMR 8258, Inserm U1022) is acknowledged for kind help regarding the characterization of the preparation.
Author information
Authors and Affiliations
Contributions
Characterization of the preparation was supervised by Karine Andrieux. She participated in the preparation, revision, and approval of the final manuscript. All authors have approved the final manuscript.
Corresponding author
Ethics declarations
All institutional and national guidelines for the care and use of laboratory animals were followed.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Goutal, S., Langer, O., Auvity, S. et al. Intravenous infusion for the controlled exposure to the dual ABCB1 and ABCG2 inhibitor elacridar in nonhuman primates. Drug Deliv. and Transl. Res. 8, 536–542 (2018). https://doi.org/10.1007/s13346-017-0472-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-017-0472-6