
Abstract
Syndecans (SDC, SYND) comprise a group of four structurally related type 1 transmembrane heparan sulfate proteoglycans (HSPGs) that play important roles in tumorigenic processes. SDCs exert signaling via their protein cores and their conserved transmembrane and cytoplasmic domains or by forming complexes with growth factors (GFs). In classical Hodgkin’s lymphoma (cHL), a lymphoid neoplasm of predominantly B cell origin, SDC1 and SDC4 are the active SDCs, and a number of GF (vascular endothelial growth factor, fibroblast growth factor, etc.) signaling pathways have been studied. However, despite extensive pre-clinical and clinical research on SDC-mediated GF signaling in many cancer types, there is very limited data for this interaction in cHL. Thus, this review highlights the relevant literature focusing on the potential interactions of SDCs and GFs in cHL pathogenesis. Also discussed are the pre-clinical and clinical studies targeting signaling through these pathways.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The syndecans (SDC, SYND) comprise a family of highly conserved type I transmembrane proteins that are present on the surface of adherent cells and cells of the hematological system. In humans, there are four members, designated SDC1, SDC2, SDC3, which are tissue-specific, and SDC4, which is cell type-specific [1]. Each of these heparan sulfate proteoglycans (HSPGs) consists of an extracellular domain (ectodomain) carrying glycosaminoglycan (GAG) side chains, a transmembrane domain and a short cytoplasmic domain [2]. SDCs have been implicated in a wide range of cellular processes including differentiation, cell adhesion [3, 4], cytoskeletal organization, cell spreading and migration [5–7], infiltration, and angiogenesis [8, 9]. These processes are partly facilitated by the interaction of the heparan sulfate (HS) chains of SDCs, with other factors, including heparin-binding growth factors (GFs) such as fibroblast growth factors (FGFs), vascular endothelial growth factors (VEGFs), transforming growth factor-β (TGF-β), platelet-derived growth factors (PDGFs), and cytokines. These interactions play important roles in cancer development and progression [10]. In the tumor microenvironment (TM) of classical Hodgkin’s lymphoma (cHL), a lymphoid malignancy of predominantly B cell origin, a number of these GFs and cytokines are produced by the mosaic of different cell types including the neoplastic giant multinuclear Reed-Sternberg and mononuclear Hodgkin’s (HRS) cells. In addition, high levels of SDC1 expression have been detected in HRS cells [11]. Also, increased levels SDC1 in the sera of HL patients suggest shedding of its ectodomain [12]. This review highlights the relevant literature that provides insights into the role(s) of SDC-GF interactions in the pathogenesis and clinical course of cHL.
Expression of SDCs in different cellular subtypes
Expression of SDCs by normal B cells
In the hematopoietic system, SDC1 and SDC4 appear to be the most active syndecans in B cell lineage. SDC1 is expressed at distinct stages of differentiation of normal lymphoid cells (pre-B) and post-germinal center B cells, including mature plasma B cells and immunoblasts [13–19] (Fig. 1). In solid lymphatic tissue, SDC1 virtually controls the binding of B lymphocytes to the interstitial matrix. SDC4 is expressed in all stages of B cell development, except in the stem cell stage [20] (Fig. 1).

Schematics showing the formation of SDC1+ HRS cells. In this interpretation, upon leaving the GC, post-GC B cells differentiate into plasma B cells which express SDC1 and BCR. The lineage that gives rise to HRS cells suffered crippling mutation of their IgG, lost most of their B cell programming (not represented), and escaped immune detection, but some of them retain the SDC1 signature. Except in the B stem cells, SDC4 is expressed by all stages of B cell differentiation and also by the HRS cells
SDCs in Hodgkin’s lymphoma
Although increasing evidence implicates SDCs in tumor development and progression, only SDC1 and SDC4 showed any association with HL. Elevated serum levels of SDC1 (sSDC1) has been reported for HL but without a prognostic significance, perhaps due to the small number of patients in the study [12]. Variable levels of SDC1 expression have been detected in subsets of primary HRS cells in some patients’ biopsies [16, 21, 22]. A more recent immunohistochemical study showed that SDC1 is overexpressed by primary HRS cells of poor outcome and good outcome patients [11]. SDC1 is also transcriptionally upregulated by most of the cHL cell lines [11]. Stromal expression of SDC1 has been observed in 11/16 cHL cases [16]. SDC4 is expressed by most HL cell lines and by primary HRS cells [23, 24].
SDCs and plasma B Cell ancestry of HRS cells
SDC1 plays an important role in B cell differentiation and, by extension, the development of some B cell malignancies. As mentioned, SDC1 is expressed during the pre-B cell stages and by post-germinal center B cells. The HRS cells of HL typically lack B cell program due to epigenetic silencing. However, evidence suggests that these tumor cells are predominantly descendants of plasma B cells. Buettner et al. showed that several proteins that are important for the differentiation of B cells are also expressed by HRS cells of different subsets of HL patients, and in the samples examined, the tumor cells were SDC1-negative (SDC1−) [25]. Pax-5 [25], a transcription factor that limits the lymphoid progenitor cells to the B-lineage development by activating B-lineage-specific genes and suppressing non-B-lineage-specific genes, and IRF4 [25], which regulates germinal center B cell formation [26] and plasma cell differentiation [27], were consistently expressed by HRS cells [25]. Buettner and colleagues also showed that Bcl-6 [25], a transcription factor expressed by germinal center B cells, was detected in HRS cells of approximately 25 % of cHL cases [25]. Moreover, B lymphocyte-induced maturation protein-1 (Blimp-1), a key regulator of plasma cell differentiation, was expressed by a small proportion of HRS cells in 23 % of cHL cases [25]. The authors concluded that plasma cell differentiation might be initiated in a small subset of HRS cells but remains abortive.
A study was conducted to determine epigenetic similarities between cells of cHL and plasma cell myeloma since both share loss of gene expression program of mature B cells [28]. However, only a limited number of genes (e.g., IRF4/MUM1 and RYBP) were found to be jointly hyperacetylated and expressed in cHL and plasma cell myeloma cell lines [28]. Both the study by Buetner et al. [25] and Seitz et al. [28] suggest that the tumor cells of cHL are characterized by an abortive plasma cell phenotype with downregulation of B cell lineage-specific proteins.
Although the plasma cell phenotype is eradicated in HRS cells for most part, a subset of these cells do express SDC1 as well as other post-germinal center markers. In an immunohistochemical analysis of 101 cases of cHL, Bai et al. recognized three bcl6/CD10/MUM1/SDC1 immunophenotypes [29]. The late germinal center (GC)/early post-GC B cell-like immunophenotype were bcl6−/CD10−/MUM1+/SDC1− and occurred in 59/101 cases (59 %) [29]. Post-GC B cell-like immunophenotype showed a bcl6−/CD10−/MUM1+/SDC1+ immunophenotype in 24/101 cases (24 %) [29]. Indeterminate immunophenotype, which consisted of bcl6+/CD10−/MUM1+/SDC1+ in 14 cases and bcl6+/CD10−/MUM1+/SDC1+ in four cases, occurred in 18/101 cases (18 %) [29].
Studies in mice revealed that SDC1 expression by plasma B cells correlates with the onset of immunoglobulin release by B cells [30]. In HL, HRS cells do not produce functional immunoglobulin (IgG), a consequence of crippling mutations in immunoglobulin genes and a phenotype detected mostly in EBV-positive tumor cells [31]. Therefore, it is likely that some HRS cells continue to express SDC1 although they do not express IgG, unlike the myeloma neoplastic plasma cells that express both SDC1 and functional IgG.
In contrast to SDC1, SDC4 is expressed in all stages of B-lineage [20] and continues to be expressed by HRS cells [24]. Figure 1 summarizes the putative relationship between plasma B cell and SDC1/SDC4 HRS cells.
Shedding of SDC in Hodgkins’ lymphoma
Shedding of SDC ectodomain in HL
The detection of soluble SDC1 in the sera of cancer patients suggests shedding of its ectodomain. Soluble SDC1 was detected in the sera of HL patients [12]. Shedding of SDC1 ectodomain is largely a consequence of the proteolytic activities of heparanase [32] and sheddases including metalloproteinases (MMPs), membrane-bound metalloproteinases (MT-MMP1) [33], and a disintegrin and metalloproteases (ADAM10, ADAM17) [34–36]. Shedding of SDC1 may further be accelerated by Rab-5 [37] and GFs [38]. Deregulated expression of these molecules by different cell types in the TM has been reported for HL [39–41]. In multiple myeloma (MM), the shedding is partly a consequence of heparanase-mediated activation of ERK signaling, which leads to the increased expression of matrix metalloproteinase-9 (MMP-9) [42]. In HL, ERK signaling is aberrantly active and it is shared with multiple signaling pathways (CD30, CD40, and RANK) that regulate cell proliferation and survival [43], but its role in SDC1 shedding remains unclear, in this neoplasm. The proteoglycan (PG) and glycosamineglycans (GAGS) of SDC1 are known to bind GFs and shuttle these cargoes to the nucleus [44, 45] although this has not been demonstrated in HL. However, given a molecular milieu that includes sheddases, SDC1, and GFs in the TM of HL, it is likely that these molecules constitute paracrine and autocrine networks that contribute to proteolytic cleavage of SDC1.
Shedding of SDC1 may be accelerated by inflammatory cytokines
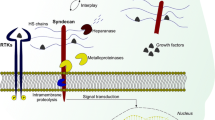
Several inflammatory mediators, including inflammatory cytokines, enhance SDC1 shedding in vitro [38, 46–49], thereby mediating the potent pathophysiologic role(s) of SDC1 in cancer. HL is notorious for number of inflammatory cytokines (IL-1, 2, 5, 6, 7, 8, 13, 17, TGF-β, RANTES, TNF-a, CCL28, TARC), which are produced by either the HRS cells or by reactive cells in the TM. An in vitro study showed that RANTES (CCL5) accelerated shedding of SDC1 ectodomain in CCR5 (receptor for RANTES)-expressing Hela cells, an interaction that involves activation of MAP kinase signaling [49]. The same study showed that RANTES forms GAG-dependent complexes with the sSDC1 as well as with those of CD44. Intriguingly, SDC1 expression is also regulated by inflammatory cytokines [50]. Additionally, lymphoma (including HL) patients with progressive disease showed elevated levels of serum CD44 before and after treatment [51] and the expression of CD44 splice variant v10 by HRS cells in HL is associated with aggressive behavior and high risk of relapse (within 2–3 years) [52]. In HL, inflammatory cytokines may accelerate shedding of SDC1 and potentiate its putative tumorigenic roles in this malignancy (Fig. 2a).

Model of putative SDC1-mediated signaling of growth factor networks in HL. a The ectodomain of SDC1 can be shed by the activities of sheddases (such as MMPs), inflammatory cytokines, or heparanase. b WT1 upregulates endothelial expression of SDC1 and VEGF and promote angiogenesis. Also, sSDC1 (from either the epithelium or shed from HRS cells) may bind VEGF and stimulate angiogenesis. c Shed SDC1 mediate binding of FGF2 to FGFR3, resulting in phosphorylation of ERK5 which inhibit BMI1, resulting in HOXB9 upregulation. d Shed SDC1 mediated binding of HGF to c-MET, resulting in phosphorylation of MET (p-MET) and its downstream targets Akt (p-AKT) and Erk1/2 (p-ERK). e Shed SDC1 mediate the binding of IGF1 to its receptor IGF1R, also resulting in phosphorylation of Akt and Erk1/2. f Shed SDC1 mediated-of PDGF to PDGFR, resulting in phosphorylation at tyrosine residues 680 and 681 (Y680 and Y681, respectively). g Shed SDC1 also mediates the binding of VEGF to VEGFR. Activation of these signaling pathways lead to cell growth and proliferation, cell invasion, metastasis, angiogenesis, and cell survival. These pathways are also active in HL and may play a role in the behavior of HRS cells
Inhibition of SDC1 shedding by sphingosine-1-phosphate (S1P)
Sphingosine-1-phosphate (S1P) is a plasma-borne lipid, generated from the phosphorylation of sphingosine by isoenzymes sphingosine phosphate kinases 1 and 2 (Sphk1 and Sphk2). In the immune system, S1P is recognized as a major regulator of trafficking of T and B cells. S1P receptor, sphingosine-1-phosphate receptor (S1PR1), and Sphk1 are overexpressed by primary HRS cells and HL cell lines [11, 53, 54], which also express SDC1 [11]. S1P inhibits shedding of SDC1 from the surface of endothelial cells [55], a process that may reduce cell migration and metastasis in cancers where loss of SDC1 ectodomain is associated with increase metastatic potential [56], at least in an in vivo setting. In prostate cancer where SDC1 is overexpressed [57, 58], lower levels of circulating S1P, a consequence of downregulation of erythrocyte SphK1, strongly correlate with lymph node metastasis [59]. However, in vitro experiments showed that S1P potently stimulate migration HL cell lines SUPHD1 and KM-H2 [53] although the study did not indicate the level of SDC1 expression by these cell lines. In DLBCL, a B lymphoma in which SDC1 overexpression by primary tumor cells is a poor prognostic factor [60], sphingosine (Sph) (not S1P) induced cell death and blocked cell growth presumably independent of S1P receptors in different cell lines [61].
SDCs interact with APRIL to promote growth and proliferation of HRS cells
Studies showed that APRIL (a proliferation-inducing ligand) signaling via TACI (cyclophylin ligand interactor) and BCMA (B cell maturation antigen) receptors is reinforced by HSPGs anchored on the cell membrane or associated with the extracellular matrix [62–64]. GAG side chains of cell-anchored and extracellular HSPGs bind a basic QKQKKQ amino acid sequence proximal to the amino terminus of APRIL, an interaction that promotes tumor growth of T cell origin [63, 64]. The resulting oligomerization of APRIL enhances TACI and BCMA signaling by promoting the formation of a highly efficient signaling network. HRS cells express APRIL and BAFF, TACI, and BCMA, but not BAFF-R. In the presence of BAFF or APRIL, TACI and BCMA provide cell-survival and growth-inducing signals to HRS cells. Chiu and colleagues showed that treatment of HRS cell lines that expressed SDC1 and SDC4, with heparinitase and heparinase, reduced binding of APRIL to the HRS cells [23]. The binding of APRIL to HRS cells was abrogated by heparin, a compound known to mimic extracellular HSPGs [23]. The authors also showed that APRIL-induced HRS cell proliferation can be increased in the presence of heparin but attenuated by heparinitase and heparinase [23]. This study highlights the importance of membrane-anchored and matrix-associated HSPGs on the proliferation of HRS cells via interaction with APRIL.
Angiogenesis
Evidence suggests that SDC1 may also mediate angiogenic signaling. In myeloma, shedding of SDC1 from the surface of the neoplastic plasma B cells is facilitated by heparanase, resulting in endothelial invasion and angiogenesis [8]. Studies also showed that levels of sSDC1 correlate positively with levels of hepatocyte growth factor (HGF) expression and regulate its signaling in myeloma [65, 66], a mechanism implicated in angiogensis. Also in myeloma, heparanase facilitates the upregulation of HGF and VEGF and SDC1 ectodomains bound to VEGF and presented VEGF to endothelial cells, initiating angiogenesis [8, 42, 67]. The pro-angiogenic role of SDC1 also appears to be dependent on its ectodomain that binds to αvβ3 and αvβ5 integrins. In vitro studies identify a short peptide that mimics the SDC1 ectodomain core protein (synstatin) that inhibit SDC1 interactions with both integrins, resulting in reduced endothelial cell invasion and slowing of tumor growth [8, 9].
Although evidence that support angiogenesis in HL is slowly emerging, elevated levels of pro-angiogenic molecules (namely HGF, VEGF, TNF-alpha, and angiogenin) (including those that depend on SDC1 for enhanced binding and signaling) were detected in the sera of patients, before standard therapy, and subsequently decreased post treatment [68]. In the same study, HGF and VEGF correlated with IL-6 levels [68], another cytokine that can interact with SDC1 and also itself a negative prognostic factor in HL [69–71]. As mentioned, separate studies detected either sSDC1 or FGF2 in the sera of HL patients [12, 72]. In addition, the injection of HL cell lines KMH2, L428, and HDLM2 in conjunction with bone marrow mast cells into NOD/SCID mouse resulted in increase tumor vascularity [73]. However, the expression of a very few of the pro-angiogenic cytokines correlate with increase microvascular density (MVD). Immunohistochemical study showed strong expression of VEGF-D by HRS cells, and this correlated with a high number of tumor microvessels, suggesting a role for this GF in HL angiogenesis [74]. Vascular endothelial growth factor receptor-1 (VEGFR-1) and VEGFR-2, two important but highly related tyrosine kinase receptors, bind VEGF-A and promote survival of endothelial cells through the Raf-MEK-MAP kinase pathway [75, 76]. A retrospective study was conducted to evaluate the expression patterns of VEGF-A, VEGFR-1, and VEGFR-2 in cHL and NLPHL in a total of 194 cases [77]. The authors showed that HRS cells expressed VEGF-A, VEGFR-1, and VEGFR-2 in 90.3, 97.2, and 94.1 % of cases, respectively [77]. Importantly, there was a significant correlation between these markers and vessel branching [77]. Interestingly, morphometric data showed that increased angiogenesis, as evident by increased microvascularization, is a negative independent prognostic factor in HL [78]. Because sSDC1 can bind multiple pro-angiogenic cytokines, several of which are implicated in HL, and enhance their signaling, it is likely that the soluble form of this HSPG may have a pro-angiogenic role in HL (Fig. 2b).
SDC1-mediated angiogenesis in HL may further be potentiated by Wilm’s tumor protein—WT1 (Fig. 2b). SDC1 is transcriptionally upregulated by WT1 [79], and it is expressed predominantly by epithelial cells [16] where it modulates neovasculization and cellular differentiation. In malignant lymph nodes of HL, WT1 is overexpressed by endothelial cells [80] where SDC1 is also likely to be expressed [81, 82]. As indicated, sSDC1 is known to bind to pro-angiogenic molecules such as FGF and VEGF (both of which are overexpressed in TM of HL) and activate them, thereby promoting endothelial cell invasion and angiogenesis. In addition, WT1 directly upregulates VEGF, resulting in increase angiogenesis [83, 84].
Exosome biogenesis
SDC1 is important in the biogenesis of exosomes, small 30–120-nm microvesicles released by cells into body fluids. The cargo of these microvessicles may consist of nucleic acids (RNAs, miRNAs) and proteins, which can be shuttled between tumor cells and host cells. Also, exosomes have been implicated in disease metastasis and relapse. HL cell lines have been shown to release exosomes containing CD30, which stimulates granulocytes to secrete IL-8, a pro-angiogenic cytokine [85]. Of interest, soluble CD30 (sCD30) is an independent poor prognostic marker in HL [86], and SDC1 binds to IL-8 and prolongs its biological functions [87]. Elevated levels of IL-8 in the sera of HL patients is associated with B symptoms [88]. Formation of exosomes is a consequence of the interaction of the SDC1 cytoplasmic domain with both syntenin and ALIX to form a complex that facilitates the budding of intraluminal vesicles within endosomal membranes [89]. In addition, SDC1 has been found in exosomes derived from cells of colorectal cancer [90], bladder cancer [91], and prostate cancer [92]. A search conducted for the expression of follicular dendritic cell markers by HRS cells turned up molecules associated with exosome physiologies. Syntenin was expressed by subsets of HRS cells [93]. And although no evidence of ALIX activities in HL has been reported to date, exosomes derived from aggressive B cell lymphoma were ALIX-positive [94]. These observations suggest that SDC1 may contribute to the synthesis of exosomes involved in the release of CD30 and perhaps contribute to the poor prognosis associated with sCD30. A possible mechanism may involve SDC1 mediating the synthesis and release of CD30+ exosomes, which then stimulate reactive cells to release IL-8, and proteolytically cleaved (either by heparanase or MMPs) SDC1 may bind to IL-8 thereby potentiating angiogenesis. The Epstein-Barr virus latent membrane protein 1 (LMP1), which has been localized to HRS cells [95, 96], may play a role in the exosomal release of FGF2 [97] into the biofluids of HL patients where significantly higher concentrations of it (FGF2) are associated with systemic symptoms and an erythrocyte sedimentation rate [72].
Putative SDC1-mediating signaling mechanisms
FGF2-FGFR3
FGF2-FGFR3 signaling has been demonstrated in hematological malignancies, including MM, lymphoma, and leukemia. Studies indicated that binding of FGFs to HSPG is required for signaling transmission through FGF high-affinity receptor [98, 99]. SDC1 forms ternary complexes with FGF2 ligand and its receptor (HSGAG:FGF:FGFR), a complex that stabilizes the receptor dimerization and promotes FGFR trans-phosphorylation. In MM, SDC1 [100], FGF2 [101], and FGFR3 [102] are overexpressed by the neoplastic plasma cells, signatures associated with a poor outcome [101–103]. This setting provides an autocrine mechanism for FGFR3 signaling. This mechanism may also be enhanced by a paracrine mode since SDC1 and FGF2 are elevated in serum of MM patients [104]. The binding of FGF2 appears to be facilitated by heparanase. In melanoma cells, heparanase stimulates FGF2 signaling by degrading the cell surface heparan sulfate chains [105], a modification that enhances the binding of FGF2 to cell surfaces and leads to stimulation of ERK and focal adhesion kinase phosphorylation (FAK) [105]. Signaling pathways that involve the activation of ERK are also important regulators of cell growth and invasion and are altered during melanoma progression to metastatic phenotype [106, 107]. In MM, ERK is activated by insulin receptor signaling which is enhanced by heparanase [108]. And in HL, ERK signaling is activated by CD30, CD40, RANK [43], and FGF2 [109].
In HL, SDC1 [16, 21, 22, 110], FGF2 [111], and FGFR3 [111, 112] are overexpressed by HRS cells and other cell types in the TM, suggesting autocrine and paracrine mechanisms (Fig. 3). The oncogenic role(s) of FGFR3 is due to either its translocation or by activated mutation in different cancer types. In HL cell lines, multiple copies of FGFR3 have been reported [112], and it is constitutively active in primary HRS cells [111]. SDC1 [12] and FGF2 [72] are elevated in the sera of HL patients. These observations suggest an active role of SDC1-FGF2-FGRF3 signaling in HL pathogenesis. In HL, signaling arising from FGF2 occurred via ERK-5 pathway [109]. Treatment of the HL cell line KM-H2 with either an inhibitory antibody against the FGF2 protein or with genistein (an inhibitor of tyrosine kinase known to inhibit growth factor receptor signaling) resulted in significant decreases of nuclear phospho-ERK5 (p-ERK5) and HOXB9 mRNA expression [109] (Table 1, Figs. 2 and 4). Overexpression of HOXB9 promotes metastasis, tumor cell growth, and angiogenesis in cancers with a very poor clinical outcome [113, 114]. In addition, the expression and release of FGF2 by HL cell lines may be dependent on LMP1 expression [115].

Illustration of autocrine and paracrine (dashed arrows) interactions of growth factors (ligands for receptor tyrosine kinase—RTKs) and SDC1, the main HSPG, in the tumor microenvironment of HL. In this model, SDC1 mediate the binding of growth factors to their receptors on the surface of HRS cells, activating these pathways and thereby leading to cell growth, proliferation, and survival. Accumulation of sSDC1 may exaggerate the binding and produce more aggressive HRS cells

Structures of receptor tyrosine kinase inhibitors used in Hodgkin’s lymphoma. a Inhibitor of FGF2 signaling. b c-Met inhibitor used in blocking HGF-mediated signaling. c Inhibitor to IGF1-R signaling. d RTK inhibitors for PDGFR pathway. e Anti-VEGF and anti-angiogenesis molecules
VEGF-VEGFR
As indicated, SDC1-VEGF interaction appears important for angiogenesis and cellular invasion. In MM, this process is dependent on heparanse. Purushothaman et al. demonstrated that heparanase upregulated SDC1 shedding, via heparanase stimulation of ERK phosphorylation, with resulting upregulation of MMP-9, which then acted as a sheddase of SDC1 [42]. Interestingly, heparanase also mediates the upregulation of VEGF. VEGF likely binds to sSDC1 to stimulate endothelial cell invasion and angiogenesis. These roles may be mediated by the VEGFR signaling. Lamorte showed that SDC1 is involved in angiogenic phenotype of MM epithelial cells (MMECs) by promoting EC proliferation, survival, and modulating VEGF-VEGFR-2 signaling [116]. Rapraeger et al. showed that insulin-like growth factor 1 (IGF1R) coupling to SDC1 and αVβ3 integrin comprises a core activation mechanism activated by VE-cadherin that is necessary for VEGFR2 and integrin activation in the initial stages of endothelial cell dissemination during angiogenesis [117]. The TM of HL is populated with these molecular players. VEGF is expressed by HRS cells and by activated macrophages [118], and variants of VEGF proteins and their cognate receptors are upregulated in the sera of newly diagnosed cHL patients [119]. In addition, HRS cells expressed VEGF-A, VEGFR-1, and VEGFR-2 in 90.3, 97.2, and 94.1 % of cases, respectively, and this pattern correlated statistically with ramifications of blood vessels [77]. Since IGF1R is expressed by large subsets of HRS cells [120], it may be involved in enhancing the role of SDC1 in activation of VEGRF signaling [117], although details of αVβ3 integrin involvement is not known, in HL.
Several studies targeted the angiogenic roles of VEGF signaling in HL (Table 1). Evaluation of thalidomide, an inhibitor of VEGF [121] in combination with cyclophosphamide and dexamethasone [122] and vinblastine [123] in relapsed HL, showed clinical activities to this drug (thalidomide—Fig. 4). However, thalidomide was not further evaluated. Lenalidomide, a potent analog of thalidomide, has been investigated in HL (Fig. 4, Table 1). Treatment of a cohort of 38 heavily pretreated relapsed or refractory cHL patients with lenalidomide alone resulted in objective and cytostatic responses with modest toxicity [124]. In another study of lenalidomide as a single agent in heavily pretreated HL patients, Boll et al. reported 50 % overall response rate and low toxicity in the 12 subjects enrolled [125]. In addition, a recent study by Rueda et al. reported that lenalidomide combined with cyclophosphamide resulted in 38 % overall response rate (one complete remission and five partial responses), and clinical benefits to 62 %, in refractory and relapsing cHL patients who received autologous stem cell transplant [126]. Upon median follow-up of 19 months, 3-year progression-free and overall survival were 6 and 31 %, respectively [126].
The anti-tumor effects of bevacizumab, an anti-VEGF monoclonal antibody, were investigated in human HL xenografts in SCID mouse and in five patients with refractory/relapsed HL. In the animal model, bevacizumab treatment resulted in significant delay in tumor growth, whereas three of the five human subjects treated with bevacizumab and gemcitabine showed complete response [127].
HGF-c-MET
In MM, overexpression of SDC1 is an independent negative prognostic factor [100] and elevated HGF levels predict a poor prognosis, short-term responses to therapies, and early relapses [66, 128, 129]. Also in MM, SDC1 promotes HGF-induced signaling through its receptor MET and downstream activation of Ras/MAPK and PI3/Akt signaling pathways, resulting in enhanced cell proliferation and survival [130]. In addition, high levels of nuclear heparanase resulted in upregulation of HGF and SDC1 shedding, enhancing the HGF signaling [67]. Nuclear heparanase also regulates the expression of SDC1 in MM [131]. These observations suggest a heparanase-SDC1-HGF regulatory loop in MM. In diffuse large B cell lymphoma (DLBCL), SDC1 overexpression by the tumor cells is associated with poor prognosis [60] and HGF induces MEK-dependent activation of ERK and PI3K-dependent phosphorylation of PKB, GSK3, and FOXO3a in MET+ tumor cells [132], suggesting a possible role of SDC1 in HGF signaling in B lymphomas. Also in DLBCL, HGF induces PI3K-dependent α4β1 integrin-mediated adhesion to VCAM-1 and fibronectin [132]. In HL, c-MET is expressed only by EBV+ cases [133]. However, only five out of six HL cell lines were c-MET+ although all six were HGF+ [134]. Teofili et al. reported the frequent expression of c-MET by HRS cells, surrounded by CD21+ follicular dendritic cells that express HGF [135] (Fig. 3), suggesting a paracrine mechanism. In addition, elevated serum levels of HGF were detected in HL patients and this was associated with B symptoms [135]. The c-MET+ HRS cells express α4β1 and α5β1 integrins [135], indicating the possible existence of HGF signaling pathway between HRS cells and the reactive cellular background. In vitro studies showed that although treatment of the c-MET+ HL cell line L428 with HGF resulted in increased phosphorylation of MET (p-MET), and upregulation of p-AKT and p-ERK1/2, there was no effect on cell proliferation [136], suggesting that SDC1-mediated binding of HGF to its receptor may be important for stimulating cell growth [65]. However, the c-MET inhibitor SU11274 (Fig. 4) suppressed cell growth by inducing G2/M cell cycle arrest [136]. Additionally, changes in plasma heparanase levels correlated with the response to treatment in HL pediatric patients [137]. Although P3-kinase/protein kinase B and RAS/mitogen-activated protein kinase networks are active in HL [138, 139], to date, there is no data to support their activation by HGF in this neoplasm, as occurred in MM [65]. However, the activities of heparanase, SDC1, HGF, and MET in the TM of HL suggest that these molecules constitute an active network that contribute to HL pathogenesis.
IGG1-IGF1R
In vitro studies showed that overexpression of SDC1 leads to the activation of IGF1R (and increased expression of Ets), resulting in increased proliferation of HT-1080 cells [140]. Beauvis et al. (2010) showed that SDC1 couples IGF1R to activate integrin signaling, promoting tumorigenic activities [141]. Although these mechanisms are independent of IGF1, they are enhanced by this ligand [141]. Interestingly, IGF1R coupling to SDC1 and αVβ3 integrin appears to be a core a mechanism activated by VE-cadherin that is necessary for VEGFR2 and integrin activation in the initial stages of endothelial cell dissemination during angiogenesis [117]. Of significance to lymphomas, Huang et al. showed that EBV infection increases αv, β3, and β5 integrin subunit mRNAs as well as upregulates the expression of the αv β3 integrin protein on human B lymphocytes [142] and about 40–50 % of cHL are EBV+. In HL, IGF1R is overexpressed by HRS cells in an almost all-or-none-fashion in 55 % of cHL patients, and this is associated with a favorable 5-year progression-free survival [120]. In this same study, the authors showed that addition of IGF1 to cell cultures resulted in increased phosphorylation of IGF1R and its downstream targets Akt (p-AKT) and Erk1/2 (p-ERK), producing an increase in cell proliferation [120] (Fig. 2e). In addition, inhibition of IGF1R with cyclolignan picropodophyllin (PPP, a highly selective IGF1R inhibitor) resulted in decreased cell growth and induced a G2/M cell cycle arrest, as indicated by decrease in pCcd2 and an increase in CyclinB1 levels [120]. Although there is no report on in vivo levels of IGF1 in HL, these observations suggest IGF1-IGFR1 may be an autocrine signaling which may be mediated or enhanced by SDC1. The cell lines used in this study express SDC1 [120, 143].
PDGF-PDGFR
PDGF (as well as other growth factors including IGF) appears to regulate the levels of HSPGs and variations in the expression of HSPGs correlate with the GF signaling activation by an auto-regulatory loop mechanism [143]. PDGF-BB treatment resulted in increased SDC1 mRNA expression [144]. Treatment of aggressive breast cancer cell lines exposed to PDGF-BB with imatinib reduces the cell surface expression of HSPGs (namely SDC2 and SDC4), thereby inhibiting cell proliferation, invasion, and migration [145], suggesting that HSPGs are involved in PDGF-mediated signaling. In HL, signaling by PDGF and its cognate receptor, platelet-derived growth factor receptor (PDGFR) may also be influenced by HSPGs (namely SDC1 and SDC4 as these are the only ones active in HL). Both PDGF [146, 147] and its receptor PDGFRA [148, 149], as well as EphrinB1 (another ligand for PDGFRA), are expressed by primary HRS cells in a proportion of HL patients. Intriguingly, cytoplasmic and nuclear PDGF expression by primary HRS cells increase with disease progression in some cases [146] and shed SDC1 is known to bind and translocate GFs to the nucleus [44]. Also of interest, ephrins (namely EphrinB2) are known to upregulate the levels of SDC1 [150].
Activation of PDGFRA resulted in phosphorylation of specific intracellular tyrosines (mainly 680 and 681) of TRKA and of TRKB in HRS cells [148] (Fig. 2f). Of clinical relevance, high levels of PDGF have been detected in the sera of HL patients, but decreased after treatment [151]. Another study revealed that serum levels of PDGF in cHL patients remained elevated after treatment with ABVD and mediastinal radiation in stage IIA patients [152], indicating a possible role in resistance to radiation and chemotherapy. The expression of PDGF and PDGFR by the same HRS cells suggest an autocrine stimulation of tumor cell growth (Fig. 3). Inhibition of PDGFRA signaling with imatinib affected cell survival and proliferation of HL cell lines [148]. Sorafenib and lestaurtinib (Fig. 4), both already being used in clinical trials, inhibited proliferation of HRS cell lines, via deactivation of PDGF signaling, at concentrations achievable in patients [153]. In MM, PDGF-AB stimulates tumor growth and angiogenesis as evident by increased microvascular density (MVD) [154], which is also a negative independent prognostic factor in HL [78]. Given the affinity of HSPGs to bind to growth factors such as PDGFA [155], it is likely that SDC1 plays a role in tumorigenic signaling of PDGF-PDGFR in HL.
SDC4, expressed by HRS cells, may transduce signaling via PDGF-PDGFR pathway, which may also be related to changes in reactive oxygen species (ROS). SDC4 regulates generation of ROS through interactions with Nox1 [156]. Nox1 and its related homologs are NADPH oxidase family of enzymes responsible for the catalytic one-electron transfers of oxygen to generate superoxide or hydrogen peroxide. SDC4-mediated increases in ROS potentiate PDGF-mediated MAP kinase activation [156]. In addition, a syndecan(4)/PDGFR chimera resulted in increase MAPK activities [157]. In HL, although SDC4 [23, 24], PDGF [146, 147], PDGFR [148, 149], and MAP kinase activities [43] have been detected in the HRS cells, a SDC4-mediated increase in ROS may not potentiate PDGF-mediated MAP kinase activation, because CYBB (NOX2/gp91phox), a homolog of Nox1, is downregulated, a consequence of gene deletion in some cases, thereby contributing to impairment of ROS synthesis by HRS cells [158]. However, an SDC4-PDGF interaction my produce other consequences in HL.
Signaling via TGF-β binding
SDCs also bind TGF-β to cell surface, [159, 160] including the surface of tumor cells. A study by Yang et al. showed that TGF-β binds to SDCs expressed on the plasma membrane of cell lines and primary tumor cells of the B cell origin, a process that may be involved in the regulation of intratumoral T cell differentiation in B cell non-Hodgkin’s lymphoma [161]. It has been speculated that HS-mediated TGF-β binding is to protect TGF-β against proteolytic degradation [162]. The SDC-bound TGF-β in these instances still retains its active form, indicating a potent SDC-TGF-β interaction in tumorigenesis. In HL, various isoforms of TGF-β mRNA and proteins were detected by different types of cells including primary HRS cells [163], reactive T lymphocytes [164], and eosinophils [165], in the TM (Fig. 3). From a clinical standpoint, the presence of cytotoxic (TIA-1+ and granzyme B+) and regulatory T cells (FOXP3+) correlates with poor overall survival in HL [166–169]. In B cell non-Hodgkin’s lymphoma (NHL), soluble TGF-β promote regulatory T (Treg) cells by enhancing expression of Foxp3 in CD4+ T cells and suppressed effector helper T (TH) cells by inhibiting expression of IFN-c and IL-17 [161]. In HL, TGF-β (and IL-10) produced by Treg and HRS cells (Fig. 3) exerts inhibitory effects on T cell effector functions, especially on cytotoxic T lymphocytes (CTLs) [170–173]. A study conducted on mostly stage IIA HL patients showed no decrease in the serum concentration of TGF-β after ABVD and mediastinal radiation, suggesting a possible role of TGF-β in resistance to both radiotherapy and chemotherapy in HL. The studies of TGF-β in HL paint an unfavorable role of this GF in the pathology of this lymphoma. However, given high expression of SDC1 in HL [11] and its binding of TGF-β to the surface of lymphoma cells [161], it is possible that SDC1 mediate the pathological role(s) of TGF-β in this malignancy. One possible role is the suppression of T cell functions. Additionally, separate studies showed that SDC1 and TGF-β may mediate expression levels of each other. RNA interference studies showed that changes in SDC1 resulted in altered levels of TGF-β [174]. In contrast, SDC1 expression is induced by TGF-β through the PKA-dependent pathway [175].
An SDC1-TGF-β signaling may also play a role in the generation of fibrosis in HL. TGF-β is known to induce fibrosis [176, 177] and it stimulates collagen synthesis [178], characteristics of some proportions of nodular sclerosing cHL subtype where there is strong expression of TGF-β [173]. Intriguingly, SDC1 is also a marker for fibrosis [179], and sSDC1 increases fibroblast proliferation and the release of TGF-β [180].
SDC1 and IL6
IL-6 is a pro-inflammatory cytokine produced by HRS cells and its overexpression is associated with poor prognosis and anemia [69, 70]. Studies indicated that IL-6 might have differential roles on the expression of SDC1. In B lymphoid cells, IL-6 regulates the expression of SDC1 at the post-transcriptional level. Exogenous application of IL-6 in growth medium of cultured murine B lymphoid cells resulted in decreased SDC1 expression, in a dose- and time-dependent manner, an effect that is reversible after IL-6 withdrawal [181]. In contrast, the overexpression of SDC1 resulted in a ten-fold increase of IL-6 expression in malignant mesothelioma cells [182]. In breast cancer, SDC1 modulates the functions of β-integrin-dependent and IL-6-dependent functions in cell adhesion, migration, and resistance to irradiation [183]. In MM, the neoplastic plasma cells are SDC1+ and they also produce IL-6. In fact, large proportions of SDC1+/IL-6+ malignant cells detected in MM patients were associated with resistant relapse or primary refractory disease [184] and high blood levels of sSDC1 and IL-6 (and HGF) predict shorter survival [185]. These observation suggest that SDC1 and IL6 may involved a feedback loop, although no single study has demonstrated such, to date. Interestingly, HRS cells in HL tumor biopsies express either SDC1 [11] or IL-6 [186], and either of these proteins are elevated in the sera of HL patients [12, 187]; however, IL-6 levels correlate with poor prognosis [187]. In addition, the expression of IL-6 and its receptor by HRS cells suggest an autocrine role in the proliferation of these cells [186]. It is quite possible that the pathophysiological contributions of IL-6 in HL may be potentiated by SDC1.
Conclusion
Ample evidence suggests that SDCs mediate cancer development and progression by enhancing the binding of growth factors and cytokines to their cognate receptors, activating signaling pathways that give rise to angiogensis, cell growth and proliferation, and cellular invasion and metastasis. In HL, heparanase, inflammatory cytokines, and sheddases may cause shedding of SDC1 ectodomain. The shed SDC1 will then bind to and mediate the signaling of GFs such as FGF2, HGF, IGF1, PDGF, and VEGF, via autocrine and paracrine mechanisms. Pre-clinical and clinical studies in HL showed that inhibition a number of these pathways lead to decrease cell growth and proliferation (Table 1, Fig. 4). Perhaps these drug-inhibitory effects can be enhanced with greater understanding of the contribution of HSPGs (namely syndecans) to HL pathology. It is hopeful that the evidence discussed here will encourage future active research in the molecular function(s) of SDCs in the pathology of HL.
References
Tkachenko E, Rhodes JM, Simons M. Syndecans: new kids on the signaling block. Circ Res. 2005;96(5):488–500. doi:10.1161/01.RES.0000159708.71142.c8.
Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–77. doi:10.1146/annurev.biochem.68.1.729.
Liu W, Litwack ED, Stanley MJ, Langford JK, Lander AD, Sanderson RD. Heparan sulfate proteoglycans as adhesive and anti-invasive molecules. Syndecans and glypican have distinct functions. J Biol Chem. 1998;273(35):22825–32.
Beauvais DM, Burbach BJ, Rapraeger AC. The syndecan-1 ectodomain regulates alphavbeta3 integrin activity in human mammary carcinoma cells. J Cell Biol. 2004;167(1):171–81. doi:10.1083/jcb.200404171.
Beauvais DM, Rapraeger AC. Syndecan-1-mediated cell spreading requires signaling by alphavbeta3 integrins in human breast carcinoma cells. Exp Cell Res. 2003;286(2):219–32.
Lee H, Kim Y, Choi Y, Choi S, Hong E, Oh ES. Syndecan-2 cytoplasmic domain regulates colon cancer cell migration via interaction with syntenin-1. Biochem Biophys Res Commun. 2011;409(1):148–53. doi:10.1016/j.bbrc.2011.04.135.
Lee JH, Park H, Chung H, Choi S, Kim Y, Yoo H, et al. Syndecan-2 regulates the migratory potential of melanoma cells. J Biol Chem. 2009;284(40):27167–75. doi:10.1074/jbc.M109.034678.
Purushothaman A, Uyama T, Kobayashi F, Yamada S, Sugahara K, Rapraeger AC, et al. Heparanase-enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood. 2010;115(12):2449–57. doi:10.1182/blood-2009-07-234757.
Beauvais DM, Ell BJ, McWhorter AR, Rapraeger AC. Syndecan-1 regulates alphavbeta3 and alphavbeta5 integrin activation during angiogenesis and is blocked by synstatin, a novel peptide inhibitor. J Exp Med. 2009;206(3):691–705. doi:10.1084/jem.20081278.
Gharbaran R. Advances in the molecular functions of syndecan-1 (SDC1/CD138) in the pathogenesis of malignancies. Crit Rev Oncol Hematol. 2015;94(1):1–17. doi:10.1016/j.critrevonc.2014.12.003.
Gharbaran R, Goy A, Tanaka T, Park J, Kim C, Hasan N, et al. Fibroblast growth factor-2 (FGF2) and syndecan-1 (SDC1) are potential biomarkers for putative circulating CD15+/CD30+ cells in poor outcome Hodgkin lymphoma patients. J Hematol Oncol. 2013;6:62. doi:10.1186/1756-8722-6-62.
Vassilakopoulos TP, Kyrtsonis MC, Papadogiannis A, Nadali G, Angelopoulou MK, Tzenou T, et al. Serum levels of soluble syndecan-1 in Hodgkin’s lymphoma. Anticancer Res. 2005;25(6C):4743–6.
Rapraeger A, Jalkanen M, Bernfield M. Cell surface proteoglycan associates with the cytoskeleton at the basolateral cell surface of mouse mammary epithelial cells. J Cell Biol. 1986;103(6 Pt 2):2683–96.
Hayashi K, Hayashi M, Jalkanen M, Firestone JH, Trelstad RL, Bernfield M. Immunocytochemistry of cell surface heparan sulfate proteoglycan in mouse tissues. A light and electron microscopic study. J Histochem Cytochem. 1987;35(10):1079–88.
Sanderson RD, Lalor P, Bernfield M. B lymphocytes express and lose syndecan at specific stages of differentiation. Cell Regul. 1989;1(1):27–35.
O’Connell FP, Pinkus JL, Pinkus GS. CD138 (syndecan-1), a plasma cell marker immunohistochemical profile in hematopoietic and nonhematopoietic neoplasms. Am J Clin Pathol. 2004;121(2):254–63. doi:10.1309/617D-WB5G-NFWX-HW4L.
Wijdenes J, Vooijs WC, Clement C, Post J, Morard F, Vita N, et al. A plasmocyte selective monoclonal antibody (B-B4) recognizes syndecan-1. Br J Haematol. 1996;94(2):318–23.
Costes V, Magen V, Legouffe E, Durand L, Baldet P, Rossi JF, et al. The Mi15 monoclonal antibody (anti-syndecan-1) is a reliable marker for quantifying plasma cells in paraffin-embedded bone marrow biopsy specimens. Hum Pathol. 1999;30(12):1405–11.
Chilosi M, Adami F, Lestani M, Montagna L, Cimarosto L, Semenzato G, et al. CD138/syndecan-1: a useful immunohistochemical marker of normal and neoplastic plasma cells on routine trephine bone marrow biopsies. Mod Pathol. 1999;12(12):1101–6.
Yamashita Y, Oritani K, Miyoshi EK, Wall R, Bernfield M, Kincade PW. Syndecan-4 is expressed by B lineage lymphocytes and can transmit a signal for formation of dendritic processes. J Immunol. 1999;162(10):5940–8.
Carbone A, Gloghini A, Gaidano G, Franceschi S, Capello D, Drexler HG, et al. Expression status of BCL-6 and syndecan-1 identifies distinct histogenetic subtypes of Hodgkin’s disease. Blood. 1998;92(7):2220–8.
Carbone A, Gloghini A, Gattei V, Degan M, Improta S, Aldinucci D, et al. Reed-Sternberg cells of classical Hodgkin’s disease react with the plasma cell-specific monoclonal antibody B-B4 and express human syndecan-1. Blood. 1997;89(10):3787–94.
Chiu A, Xu W, He B, Dillon SR, Gross JA, Sievers E, et al. Hodgkin lymphoma cells express TACI and BCMA receptors and generate survival and proliferation signals in response to BAFF and APRIL. Blood. 2007;109(2):729–39. doi:10.1182/blood-2006-04-015958.
Ehlers A, Oker E, Bentink S, Lenze D, Stein H, Hummel M. Histone acetylation and DNA demethylation of B cells result in a Hodgkin-like phenotype. Leukemia. 2008;22(4):835–41. doi:10.1038/leu.2008.12.
Buettner M, Greiner A, Avramidou A, Jack HM, Niedobitek G. Evidence of abortive plasma cell differentiation in Hodgkin and Reed-Sternberg cells of classical Hodgkin lymphoma. Hematol Oncol. 2005;23(3–4):127–32. doi:10.1002/hon.764.
Willis SN, Good-Jacobson KL, Curtis J, Light A, Tellier J, Shi W, et al. Transcription factor IRF4 regulates germinal center cell formation through a B cell-intrinsic mechanism. J Immunol. 2014;192(7):3200–6. doi:10.4049/jimmunol.1303216.
Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol. 2006;7(7):773–82. doi:10.1038/ni1357.
Seitz V, Thomas PE, Zimmermann K, Paul U, Ehlers A, Joosten M, et al. Classical Hodgkin’s lymphoma shows epigenetic features of abortive plasma cell differentiation. Haematologica. 2011;96(6):863–70. doi:10.3324/haematol.2010.031138.
Bai M, Panoulas V, Papoudou-Bai A, Horianopoulos N, Kitsoulis P, Stefanaki K, et al. B-cell differentiation immunophenotypes in classical Hodgkin lymphomas. Leuk Lymphoma. 2006;47(3):495–501. doi:10.1080/10428190500306784.
Lalor PA, Nossal GJ, Sanderson RD, McHeyzer-Williams MG. Functional and molecular characterization of single, (4-hydroxy-3-nitrophenyl)acetyl (NP)-specific, IgG1+ B cells from antibody-secreting and memory B cell pathways in the C57BL/6 immune response to NP. Eur J Immunol. 1992;22(11):3001–11. doi:10.1002/eji.1830221136.
Kuppers R, Rajewsky K, Zhao M, Simons G, Laumann R, Fischer R, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A. 1994;91(23):10962–6.
Yang Y, Macleod V, Bendre M, Huang Y, Theus AM, Miao HQ, et al. Heparanase promotes the spontaneous metastasis of myeloma cells to bone. Blood. 2005;105(3):1303–9. doi:10.1182/blood-2004-06-2141.
Manon-Jensen T, Multhaupt HA, Couchman JR. Mapping of matrix metalloproteinase cleavage sites on syndecan-1 and syndecan-4 ectodomains. FEBS J. 2013;280(10):2320–31. doi:10.1111/febs.12174.
Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111(5):635–46.
Endo K, Takino T, Miyamori H, Kinsen H, Yoshizaki T, Furukawa M, et al. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J Biol Chem. 2003;278(42):40764–70. doi:10.1074/jbc.M306736200.
Pruessmeyer J, Martin C, Hess FM, Schwarz N, Schmidt S, Kogel T, et al. A disintegrin and metalloproteinase 17 (ADAM17) mediates inflammation-induced shedding of syndecan-1 and -4 by lung epithelial cells. J Biol Chem. 2010;285(1):555–64. doi:10.1074/jbc.M109.059394.
Hayashida K, Stahl PD, Park PW. Syndecan-1 ectodomain shedding is regulated by the small GTPase Rab5. J Biol Chem. 2008;283(51):35435–44. doi:10.1074/jbc.M804172200.
Subramanian SV, Fitzgerald ML, Bernfield M. Regulated shedding of syndecan-1 and -4 ectodomains by thrombin and growth factor receptor activation. J Biol Chem. 1997;272(23):14713–20.
Thorns C, Bernd HW, Hatton D, Merz H, Feller AC, Lange K. Matrix-metalloproteinases in Hodgkin lymphoma. Anticancer Res. 2003;23(2B):1555–8.
Campos AH, Vassallo J, Soares FA. Matrix metalloproteinase-9 expression by Hodgkin-Reed-Sternberg cells is associated with reduced overall survival in young adult patients with classical Hodgkin lymphoma. PLoS One. 2013;8(9):e74793. doi:10.1371/journal.pone.0074793.
Ebsen H, Schroder A, Kabelitz D, Janssen O. Differential surface expression of ADAM10 and ADAM17 on human T lymphocytes and tumor cells. PLoS One. 2013;8(10):e76853. doi:10.1371/journal.pone.0076853.
Purushothaman A, Chen L, Yang Y, Sanderson RD. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J Biol Chem. 2008;283(47):32628–36. doi:10.1074/jbc.M806266200.
Zheng B, Fiumara P, Li YV, Georgakis G, Snell V, Younes M, et al. MEK/ERK pathway is aberrantly active in Hodgkin disease: a signaling pathway shared by CD30, CD40, and RANK that regulates cell proliferation and survival. Blood. 2003;102(3):1019–27. doi:10.1182/blood-2002-11-3507.
Stewart MD, Ramani VC, Sanderson RD. Shed syndecan-1 translocates to the nucleus of cells delivering growth factors and inhibiting histone acetylation: a novel mechanism of tumor-host cross-talk. J Biol Chem. 2015;290(2):941–9. doi:10.1074/jbc.M114.608455.
Hsia E, Richardson TP, Nugent MA. Nuclear localization of basic fibroblast growth factor is mediated by heparan sulfate proteoglycans through protein kinase C signaling. J Cell Biochem. 2003;88(6):1214–25. doi:10.1002/jcb.10470.
Fitzgerald ML, Wang Z, Park PW, Murphy G, Bernfield M. Shedding of syndecan-1 and -4 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J Cell Biol. 2000;148(4):811–24.
Marshall LJ, Ramdin LS, Brooks T, DPhil PC, Shute JK. Plasminogen activator inhibitor-1 supports IL-8-mediated neutrophil transendothelial migration by inhibition of the constitutive shedding of endothelial IL-8/heparan sulfate/syndecan-1 complexes. J Immunol. 2003;171(4):2057–65.
Park PW, Foster TJ, Nishi E, Duncan SJ, Klagsbrun M, Chen Y. Activation of syndecan-1 ectodomain shedding by Staphylococcus aureus alpha-toxin and beta-toxin. J Biol Chem. 2004;279(1):251–8. doi:10.1074/jbc.M308537200.
Charnaux N, Brule S, Chaigneau T, Saffar L, Sutton A, Hamon M, et al. RANTES (CCL5) induces a CCR5-dependent accelerated shedding of syndecan-1 (CD138) and syndecan-4 from HeLa cells and forms complexes with the shed ectodomains of these proteoglycans as well as with those of CD44. Glycobiology. 2005;15(2):119–30. doi:10.1093/glycob/cwh148.
Day RM, Mitchell TJ, Knight SC, Forbes A. Regulation of epithelial syndecan-1 expression by inflammatory cytokines. Cytokine. 2003;21(5):224–33.
Ristamaki R, Joensuu H, Salmi M, Jalkanen S. Serum CD44 in malignant lymphoma: an association with treatment response. Blood. 1994;84(1):238–43.
Beham-Schmid C, Heider KH, Hoefler G, Zatloukal K. Expression of CD44 splice variant v10 in Hodgkin’s disease is associated with aggressive behaviour and high risk of relapse. J Pathol. 1998;186(4):383–9. doi:10.1002/(SICI)1096-9896(199812)186:4<383::AID-PATH202>3.0.CO;2-A.
Kluk MJ, Ryan KP, Wang B, Zhang G, Rodig SJ, Sanchez T. Sphingosine-1-phosphate receptor 1 in classical Hodgkin lymphoma: assessment of expression and role in cell migration. Laboratory investigation; a journal of technical methods and pathology. 2013;93(4):462–71. doi:10.1038/labinvest.2013.7.
Doctor of Philosophy: Expression of lipid signaling molecules in epithelial and lymphoid malignancies [database on the Internet]. University of Birmingham. 2014. Available from: http://etheses.bham.ac.uk/5190/2/Abdullah14PhD.pdf. Accessed:
Zeng Y, Adamson RH, Curry FR, Tarbell JM. Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am J Physiol Heart Circ Physiol. 2014;306(3):H363–72. doi:10.1152/ajpheart.00687.2013.
Nikolova V, Koo CY, Ibrahim SA, Wang Z, Spillmann D, Dreier R, et al. Differential roles for membrane-bound and soluble syndecan-1 (CD138) in breast cancer progression. Carcinogenesis. 2009;30(3):397–407. doi:10.1093/carcin/bgp001.
Shimada K, Anai S, Fujii T, Tanaka N, Fujimoto K, Konishi N. Syndecan-1 (CD138) contributes to prostate cancer progression by stabilizing tumour-initiating cells. J Pathol. 2013;231(4):495–504. doi:10.1002/path.4271.
Chen D, Adenekan B, Chen L, Vaughan ED, Gerald W, Feng Z, et al. Syndecan-1 expression in locally invasive and metastatic prostate cancer. Urology. 2004;63(2):402–7. doi:10.1016/j.urology.2003.08.036.
Nunes J, Naymark M, Sauer L, Muhammad A, Keun H, Sturge J, et al. Circulating sphingosine-1-phosphate and erythrocyte sphingosine kinase-1 activity as novel biomarkers for early prostate cancer detection. Br J Cancer. 2012;106(5):909–15. doi:10.1038/bjc.2012.14.
Bodoor K, Matalka I, Hayajneh R, Haddad Y, Gharaibeh W. Evaluation of BCL-6, CD10, CD138 and MUM-1 expression in diffuse large B-cell lymphoma patients: CD138 is a marker of poor prognosis. Asian Pac J Cancer Prev. 2012;13(7):3037–46.
Bode C, Berlin M, Rostel F, Teichmann B, Graler MH. Evaluating sphingosine and its analogues as potential alternatives for aggressive lymphoma treatment. Cell Physiol Biochem. 2014;34(5):1686–700. doi:10.1159/000366370.
Dillon SR, Gross JA, Ansell SM, Novak AJ. An APRIL to remember: novel TNF ligands as therapeutic targets. Nat Rev Drug Discov. 2006;5(3):235–46. doi:10.1038/nrd1982.
Ingold K, Zumsteg A, Tardivel A, Huard B, Steiner QG, Cachero TG, et al. Identification of proteoglycans as the APRIL-specific binding partners. J Exp Med. 2005;201(9):1375–83. doi:10.1084/jem.20042309.
Hendriks J, Planelles L, de Jong-Odding J, Hardenberg G, Pals ST, Hahne M, et al. Heparan sulfate proteoglycan binding promotes APRIL-induced tumor cell proliferation. Cell Death Differ. 2005;12(6):637–48. doi:10.1038/sj.cdd.4401647.
Derksen PW, Keehnen RM, Evers LM, van Oers MH, Spaargaren M, Pals ST. Cell surface proteoglycan syndecan-1 mediates hepatocyte growth factor binding and promotes Met signaling in multiple myeloma. Blood. 2002;99(4):1405–10.
Seidel C, Borset M, Hjertner O, Cao D, Abildgaard N, Hjorth-Hansen H, et al. High levels of soluble syndecan-1 in myeloma-derived bone marrow: modulation of hepatocyte growth factor activity. Blood. 2000;96(9):3139–46.
Ramani VC, Yang Y, Ren Y, Nan L, Sanderson RD. Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing HGF expression and activity. J Biol Chem. 2011;286(8):6490–9. doi:10.1074/jbc.M110.183277.
Passam FH, Alexandrakis MG, Moschandrea J, Sfiridaki A, Roussou PA, Siafakas NM. Angiogenic molecules in Hodgkin’s disease: results from sequential serum analysis. Int J Immunopathol Pharmacol. 2006;19(1):161–70.
Reynolds GM, Billingham LJ, Gray LJ, Flavell JR, Najafipour S, Crocker J, et al. Interleukin 6 expression by Hodgkin/Reed-Sternberg cells is associated with the presence of ‘B’ symptoms and failure to achieve complete remission in patients with advanced Hodgkin’s disease. Br J Haematol. 2002;118(1):195–201.
Hohaus S, Massini G, Giachelia M, Vannata B, Bozzoli V, Cuccaro A, et al. Anemia in Hodgkin’s lymphoma: the role of interleukin-6 and hepcidin. J Clin Oncol. 2010;28(15):2538–43. doi:10.1200/JCO.2009.27.6873.
Casasnovas RO, Mounier N, Brice P, Divine M, Morschhauser F, Gabarre J, et al. Plasma cytokine and soluble receptor signature predicts outcome of patients with classical Hodgkin’s lymphoma: a study from the Groupe d’Etude des Lymphomes de l’Adulte. J Clin Oncol. 2007;25(13):1732–40. doi:10.1200/JCO.2006.08.1331.
Kowalska M, Kamińska, Janina., Fuksiewicz, Małgorzata., Kotowicz, Beata., Siedlecka, Alicja., Tajer, Joanna., Walewski, Jan. Serum VEGF and bFGF levels in patients with Hodgkin’s lymphoma. NOWOTWORY Journal of Oncology. 2007;57(4):179e–82e.
Mizuno H, Nakayama T, Miyata Y, Saito S, Nishiwaki S, Nakao N, et al. Mast cells promote the growth of Hodgkin’s lymphoma cell tumor by modifying the tumor microenvironment that can be perturbed by bortezomib. Leukemia. 2012;26(10):2269–76. doi:10.1038/leu.2012.81.
Bardelli M, Leucci E, Schurfeld K, Bellan C, Passiatore G, Rocchigiani M, et al. VEGF-D is expressed in activated lymphoid cells and in tumors of hematopoietic and lymphoid tissues. Leuk Lymphoma. 2007;48(10):2014–21. doi:10.1080/10428190701540975.
Gille H, Kowalski J, Li B, LeCouter J, Moffat B, Zioncheck TF, et al. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem. 2001;276(5):3222–30. doi:10.1074/jbc.M002016200.
Takahashi T, Yamaguchi S, Chida K, Shibuya M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 2001;20(11):2768–78. doi:10.1093/emboj/20.11.2768.
Dimtsas GS, Georgiadi EC, Karakitsos P, Vassilakopoulos TP, Thymara I, Korkolopoulou P, et al. Prognostic significance of immunohistochemical expression of the angiogenic molecules vascular endothelial growth factor-A, vascular endothelial growth factor receptor-1 and vascular endothelial growth factor receptor-2 in patients with classical Hodgkin lymphoma. Leuk Lymphoma. 2014;55(3):558–64. doi:10.3109/10428194.2013.813629.
Korkolopoulou P, Thymara I, Kavantzas N, Vassilakopoulos TP, Angelopoulou MK, Kokoris SI, et al. Angiogenesis in Hodgkin’s lymphoma: a morphometric approach in 286 patients with prognostic implications. Leukemia. 2005;19(6):894–900. doi:10.1038/sj.leu.2403690.
Cook DM, Hinkes MT, Bernfield M, Rauscher 3rd FJ. Transcriptional activation of the syndecan-1 promoter by the Wilms’ tumor protein WT1. Oncogene. 1996;13(8):1789–99.
Vadasz Z, Shasha-Lavsky H, Nov Y, Bejar J, Lurie M, Tadmor T, et al. Wilms’ tumor gene 1: a possible new proangiogenic factor in Hodgkin lymphoma. Appl Immunohistochem Mol Morphol. 2013;21(2):177–80. doi:10.1097/PAI.0b013e318259852a.
Sebestyen A, Berczi L, Mihalik R, Paku S, Matolcsy A, Kopper L. Syndecan-1 (CD138) expression in human non-Hodgkin lymphomas. Br J Haematol. 1999;104(2):412–9.
Alexopoulou AN, Multhaupt HA, Couchman JR. Syndecans in wound healing, inflammation and vascular biology. Int J Biochem Cell Biol. 2007;39(3):505–28. doi:10.1016/j.biocel.2006.10.014.
McCarty G, Awad O, Loeb DM. WT1 protein directly regulates expression of vascular endothelial growth factor and is a mediator of tumor response to hypoxia. J Biol Chem. 2011;286(51):43634–43. doi:10.1074/jbc.M111.310128.
Katuri V, Gerber S, Qiu X, McCarty G, Goldstein SD, Hammers H, et al. WT1 regulates angiogenesis in Ewing sarcoma. Oncotarget. 2014;5(9):2436–49.
Hansen HP, Reiners KS, Simhadri V, Böll B, Engert A, Pogge von Strandmann E. CD30-containing exosomes are released from Hodgkin lymphoma cell lines and trigger normal bystander cells to release angiogenic interleukin-8. In: Immunologie IV. Montag. 2007. http://registration.akm.ch/einsicht.php?XNABSTRACT_ID=51536&XNSPRACHE_ID=1&XNKONGRESS_ID=62&XNMASKEN_ID=900
Zanotti R, Trolese A, Ambrosetti A, Nadali G, Visco C, Ricetti MM, et al. Serum levels of soluble CD30 improve International Prognostic Score in predicting the outcome of advanced Hodgkin’s lymphoma. Ann Oncol. 2002;13(12):1908–14.
Goger B, Halden Y, Rek A, Mosl R, Pye D, Gallagher J, et al. Different affinities of glycosaminoglycan oligosaccharides for monomeric and dimeric interleukin-8: a model for chemokine regulation at inflammatory sites. Biochemistry. 2002;41(5):1640–6.
Trumper L, Jung W, Dahl G, Diehl V, Gause A, Pfreundschuh M. Interleukin-7, interleukin-8, soluble TNF receptor, and p53 protein levels are elevated in the serum of patients with Hodgkin’s disease. Ann Oncol. 1994;5 Suppl 1:93–6.
Baietti MF, Zhang Z, Mortier E, Melchior A, Degeest G, Geeraerts A, et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat Cell Biol. 2012;14(7):677–85. doi:10.1038/ncb2502.
Choi DS, Lee JM, Park GW, Lim HW, Bang JY, Kim YK, et al. Proteomic analysis of microvesicles derived from human colorectal cancer cells. J Proteome Res. 2007;6(12):4646–55. doi:10.1021/pr070192y.
Welton JL, Khanna S, Giles PJ, Brennan P, Brewis IA, Staffurth J, et al. Proteomics analysis of bladder cancer exosomes. Mol Cell Proteomics. 2010;9(6):1324–38. doi:10.1074/mcp.M000063-MCP201.
Soekmadji C, Russell PJ, Nelson CC. Exosomes in prostate cancer: putting together the pieces of a puzzle. Cancers (Basel). 2013;5(4):1522–44. doi:10.3390/cancers5041522.
Kim SH, Choe JY, Jeon Y, Huh J, Jung HR, Choi YD, et al. Frequent expression of follicular dendritic cell markers in Hodgkin lymphoma and anaplastic large cell lymphoma. J Clin Pathol. 2013;66(7):589–96. doi:10.1136/jclinpath-2012-201425.
Aung T, Chapuy B, Vogel D, Wenzel D, Oppermann M, Lahmann M, et al. Exosomal evasion of humoral immunotherapy in aggressive B-cell lymphoma modulated by ATP-binding cassette transporter A3. Proc Natl Acad Sci U S A. 2011;108(37):15336–41. doi:10.1073/pnas.1102855108.
Lee IS, Shin YK, Chung DH, Park SH. LMP1-induced downregulation of CD99 molecules in Hodgkin and Reed-Sternberg cells. Leuk Lymphoma. 2001;42(4):587–94. doi:10.3109/10428190109099318.
Knecht H, Sawan B, Lichtensztejn Z, Lichtensztejn D, Mai S. 3D Telomere FISH defines LMP1-expressing Reed-Sternberg cells as end-stage cells with telomere-poor ‘ghost’ nuclei and very short telomeres. Lab Invest. 2010;90(4):611–9. doi:10.1038/labinvest.2010.2.
Ceccarelli S, Visco V, Raffa S, Wakisaka N, Pagano JS, Torrisi MR. Epstein-Barr virus latent membrane protein 1 promotes concentration in multivesicular bodies of fibroblast growth factor 2 and its release through exosomes. Int J Cancer. 2007;121(7):1494–506. doi:10.1002/ijc.22844.
Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64(4):841–8.
Rapraeger AC, Krufka A, Olwin BB. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 1991;252(5013):1705–8.
Seidel C, Sundan A, Hjorth M, Turesson I, Dahl IM, Abildgaard N, et al. Serum syndecan-1: a new independent prognostic marker in multiple myeloma. Blood. 2000;95(2):388–92.
Sato N, Hattori Y, Wenlin D, Yamada T, Kamata T, Kakimoto T, et al. Elevated level of plasma basic fibroblast growth factor in multiple myeloma correlates with increased disease activity. Jpn J Cancer Res. 2002;93(4):459–66.
Chang H, Stewart AK, Qi XY, Li ZH, Yi QL, Trudel S. Immunohistochemistry accurately predicts FGFR3 aberrant expression and t(4;14) in multiple myeloma. Blood. 2005;106(1):353–5. doi:10.1182/blood-2005-01-0033.
Chandesris MO, Soulier J, Labaume S, Crinquette A, Repellini L, Chemin K, et al. Detection and follow-up of fibroblast growth factor receptor 3 expression on bone marrow and circulating plasma cells by flow cytometry in patients with t(4;14) multiple myeloma. Br J Haematol. 2007;136(4):609–14. doi:10.1111/j.1365-2141.2006.06479.x.
Kyrtsonis MC, Vassilakopoulos TP, Siakantaris MP, Kokoris SI, Gribabis DA, Dimopoulou MN, et al. Serum syndecan-1, basic fibroblast growth factor and osteoprotegerin in myeloma patients at diagnosis and during the course of the disease. Eur J Haematol. 2004;72(4):252–8. doi:10.1046/j.0902-4441.2003.00205.x.
Reiland J, Kempf D, Roy M, Denkins Y, Marchetti D. FGF2 binding, signaling, and angiogenesis are modulated by heparanase in metastatic melanoma cells. Neoplasia. 2006;8(7):596–606. doi:10.1593/neo.06244.
Easty DJ, Bennett DC. Protein tyrosine kinases in malignant melanoma. Melanoma Res. 2000;10(5):401–11.
Smalley KS. A pivotal role for ERK in the oncogenic behaviour of malignant melanoma? Int J Cancer. 2003;104(5):527–32. doi:10.1002/ijc.10978.
Purushothaman A, Babitz SK, Sanderson RD. Heparanase enhances the insulin receptor signaling pathway to activate extracellular signal-regulated kinase in multiple myeloma. J Biol Chem. 2012;287(49):41288–96. doi:10.1074/jbc.M112.391417.
Nagel S, Burek C, Venturini L, Scherr M, Quentmeier H, Meyer C, et al. Comprehensive analysis of homeobox genes in Hodgkin lymphoma cell lines identifies dysregulated expression of HOXB9 mediated via ERK5 signaling and BMI1. Blood. 2007;109(7):3015–23. doi:10.1182/blood-2006-08-044347.
Tzankov A, Zimpfer A, Pehrs AC, Lugli A, Went P, Maurer R, et al. Expression of B-cell markers in classical Hodgkin lymphoma: a tissue microarray analysis of 330 cases. Mod Pathol. 2003;16(11):1141–7. doi:10.1097/01.MP.0000093627.51090.3F.
Khnykin D, Troen G, Berner JM, Delabie J. The expression of fibroblast growth factors and their receptors in Hodgkin’s lymphoma. J Pathol. 2006;208(3):431–8. doi:10.1002/path.1900.
Joos S, Granzow M, Holtgreve-Grez H, Siebert R, Harder L, Martin-Subero JI, et al. Hodgkin’s lymphoma cell lines are characterized by frequent aberrations on chromosomes 2p and 9p including REL and JAK2. Int J Cancer. 2003;103(4):489–95. doi:10.1002/ijc.10845.
Huang K, Yuan R, Wang K, Hu J, Huang Z, Yan C, et al. Overexpression of HOXB9 promotes metastasis and indicates poor prognosis in colon cancer. Chin J Cancer Res. 2014;26(1):72–80. doi:10.3978/j.issn.1000-9604.2014.01.11.
Seki H, Hayashida T, Jinno H, Hirose S, Sakata M, Takahashi M, et al. HOXB9 expression promoting tumor cell proliferation and angiogenesis is associated with clinical outcomes in breast cancer patients. Ann Surg Oncol. 2012;19(6):1831–40. doi:10.1245/s10434-012-2295-5.
Wakisaka N, Murono S, Yoshizaki T, Furukawa M, Pagano JS. Epstein-Barr virus latent membrane protein 1 induces and causes release of fibroblast growth factor-2. Cancer Res. 2002;62(21):6337–44.
Lamorte S, Ferrero S, Aschero S, Monitillo L, Bussolati B, Omede P, et al. Syndecan-1 promotes the angiogenic phenotype of multiple myeloma endothelial cells. Leukemia. 2012;26(5):1081–90. doi:10.1038/leu.2011.290.
Rapraeger AC, Ell BJ, Roy M, Li X, Morrison OR, Thomas GM, et al. Vascular endothelial-cadherin stimulates syndecan-1-coupled insulin-like growth factor-1 receptor and cross-talk between alphaVbeta3 integrin and vascular endothelial growth factor receptor 2 at the onset of endothelial cell dissemination during angiogenesis. FEBS J. 2013;280(10):2194–206. doi:10.1111/febs.12134.
Doussis-Anagnostopoulou IA, Talks KL, Turley H, Debnam P, Tan DC, Mariatos G, et al. Vascular endothelial growth factor (VEGF) is expressed by neoplastic Hodgkin-Reed-Sternberg cells in Hodgkin’s disease. J Pathol. 2002;197(5):677–83. doi:10.1002/path.1151.
Liu F, Song YQ, Zhang C, Fu ZY, Ping LY, Ying ZT, et al. Plasma levels of VEGF-C, VEGF-D, VEGFR-2 and VEGFR-3 in patients with newly diagnosed lymphomas. J Exp Hematol. 2011;19(5):1184–8.
Liang Z, Diepstra A, Xu C, van Imhoff G, Plattel W, Van Den Berg A, et al. Insulin-like growth factor 1 receptor is a prognostic factor in classical Hodgkin lymphoma. PLoS One. 2014;9(1), e87474. doi:10.1371/journal.pone.0087474.
Ribatti D, Vacca A. Therapeutic renaissance of thalidomide in the treatment of haematological malignancies. Leukemia. 2005;19(9):1525–31. doi:10.1038/sj.leu.2403852.
Garcia-Sanz R, Gonzalez-Lopez TJ, Vazquez L, Hermida G, Graciani IF, San Miguel JF. The combination of thalidomide, cyclophosphamide and dexamethasone is potentially useful in highly resistant Hodgkin’s lymphoma. Eur J Haematol. 2010;84(3):266–70. doi:10.1111/j.1600-0609.2009.01375.x.
Kuruvilla J, Song K, Mollee P, Panzarella T, McCrae J, Nagy T, et al. A phase II study of thalidomide and vinblastine for palliative patients with Hodgkin’s lymphoma. Hematology. 2006;11(1):25–9. doi:10.1080/10245330500276592.
Fehniger TA, Larson S, Trinkaus K, Siegel MJ, Cashen AF, Blum KA, et al. A phase 2 multicenter study of lenalidomide in relapsed or refractory classical Hodgkin lymphoma. Blood. 2011;118(19):5119–25. doi:10.1182/blood-2011-07-362475.
Boll B, Borchmann P, Topp MS, Hanel M, Reiners KS, Engert A, et al. Lenalidomide in patients with refractory or multiple relapsed Hodgkin lymphoma. Br J Haematol. 2010;148(3):480–2. doi:10.1111/j.1365-2141.2009.07963.x.
Rueda A, Garcia-Sanz R, Pastor M, Salar A, Labrador J, Quero-Blanco C, et al. A phase II study to evaluate lenalidomide in combination with metronomic-dose cyclophosphamide in patients with heavily pretreated classical Hodgkin lymphoma. Acta Oncol. 2015;54(6):933–8. doi:10.3109/0284186X.2015.1007212.
Reiners KS, Gossmann A, von Strandmann EP, Boll B, Engert A, Borchmann P. Effects of the anti-VEGF monoclonal antibody bevacizumab in a preclinical model and in patients with refractory and multiple relapsed Hodgkin lymphoma. J Immunother. 2009;32(5):508–12. doi:10.1097/CJI.0b013e3181a25daf.
Seidel C, Borset M, Turesson I, Abildgaard N, Sundan A, Waage A. Elevated serum concentrations of hepatocyte growth factor in patients with multiple myeloma. The Nordic Myeloma Study Group. Blood. 1998;91(3):806–12.
Seidel C, Lenhoff S, Brabrand S, Anderson G, Standal T, Lanng-Nielsen J, et al. Hepatocyte growth factor in myeloma patients treated with high-dose chemotherapy. Br J Haematol. 2002;119(3):672–6.
Derksen PW, de Gorter DJ, Meijer HP, Bende RJ, van Dijk M, Lokhorst HM, et al. The hepatocyte growth factor/Met pathway controls proliferation and apoptosis in multiple myeloma. Leukemia. 2003;17(4):764–74. doi:10.1038/sj.leu.2402875.
Chen L, Sanderson RD. Heparanase regulates levels of syndecan-1 in the nucleus. PLoS One. 2009;4(3), e4947. doi:10.1371/journal.pone.0004947.
Tjin EP, Groen RW, Vogelzang I, Derksen PW, Klok MD, Meijer HP, et al. Functional analysis of HGF/MET signaling and aberrant HGF-activator expression in diffuse large B-cell lymphoma. Blood. 2006;107(2):760–8. doi:10.1182/blood-2005-05-1929.
Weimar IS, de Jong D, Muller EJ, Nakamura T, van Gorp JM, de Gast GC, et al. Hepatocyte growth factor/scatter factor promotes adhesion of lymphoma cells to extracellular matrix molecules via alpha 4 beta 1 and alpha 5 beta 1 integrins. Blood. 1997;89(3):990–1000.
Pons E, Uphoff CC, Drexler HG. Expression of hepatocyte growth factor and its receptor c-met in human leukemia-lymphoma cell lines. Leuk Res. 1998;22(9):797–804.
Teofili L, Di Febo AL, Pierconti F, Maggiano N, Bendandi M, Rutella S, et al. Expression of the c-met proto-oncogene and its ligand, hepatocyte growth factor, in Hodgkin disease. Blood. 2001;97(4):1063–9.
Xu C, Plattel W, van den Berg A, Ruther N, Huang X, Wang M, et al. Expression of the c-Met oncogene by tumor cells predicts a favorable outcome in classical Hodgkin’s lymphoma. Haematologica. 2012;97(4):572–8. doi:10.3324/haematol.2011.056101.
Ben Arush MW, Shafat I, Ben Barak A, Shalom RB, Vlodavsky I, Ilan N. Plasma heparanase as a significant marker of treatment response in children with Hodgkin lymphoma: pilot study. Pediatr Hematol Oncol. 2009;26(4):157–64. doi:10.1080/08880010902754917.
Meadows SA, Vega F, Kashishian A, Johnson D, Diehl V, Miller LL, et al. PI3Kdelta inhibitor, GS-1101 (CAL-101), attenuates pathway signaling, induces apoptosis, and overcomes signals from the microenvironment in cellular models of Hodgkin lymphoma. Blood. 2012;119(8):1897–900. doi:10.1182/blood-2011-10-386763.
Steidl C, Connors JM, Gascoyne RD. Molecular pathogenesis of Hodgkin’s lymphoma: increasing evidence of the importance of the microenvironment. J Clin Oncol. 2011;29(14):1812–26. doi:10.1200/JCO.2010.32.8401.
Peterfia B, Fule T, Baghy K, Szabadkai K, Fullar A, Dobos K, et al. Syndecan-1 enhances proliferation, migration and metastasis of HT-1080 cells in cooperation with syndecan-2. PLoS One. 2012;7(6), e39474. doi:10.1371/journal.pone.0039474.
Beauvais DM, Rapraeger AC. Syndecan-1 couples the insulin-like growth factor-1 receptor to inside-out integrin activation. J Cell Sci. 2010;123(Pt 21):3796–807. doi:10.1242/jcs.067645.
Huang S, Stupack D, Liu A, Cheresh D, Nemerow GR. Cell growth and matrix invasion of EBV-immortalized human B lymphocytes is regulated by expression of alpha(v) integrins. Oncogene. 2000;19(15):1915–23. doi:10.1038/sj.onc.1203509.
Dobra K, Andang M, Syrokou A, Karamanos NK, Hjerpe A. Differentiation of mesothelioma cells is influenced by the expression of proteoglycans. Exp Cell Res. 2000;258(1):12–22. doi:10.1006/excr.2000.4915.
Worapamorn W, Tam SP, Li H, Haase HR, Bartold PM. Cytokine regulation of syndecan-1 and -2 gene expression in human periodontal fibroblasts and osteoblasts. J Periodontal Res. 2002;37(4):273–8.
Malavaki CJ, Roussidis AE, Gialeli C, Kletsas D, Tsegenidis T, Theocharis AD, et al. Imatinib as a key inhibitor of the platelet-derived growth factor receptor mediated expression of cell surface heparan sulfate proteoglycans and functional properties of breast cancer cells. FEBS J. 2013;280(10):2477–89. doi:10.1111/febs.12163.
Mainou-Fowler T, Angus B, Miller S, Proctor SJ, Taylor PR, Wood KM. Micro-vessel density and the expression of vascular endothelial growth factor (VEGF) and platelet-derived endothelial cell growth factor (PdEGF) in classical Hodgkin lymphoma (HL). Leuk Lymphoma. 2006;47(2):223–30. doi:10.1080/01674820500305838.
Passam FH, Alexandrakis MG, Kafousi M, Fotinou M, Darivianaki K, Tsirakis G, et al. Histological expression of angiogenic factors: VEGF, PDGFRalpha, and HIF-1alpha in Hodgkin lymphoma. Pathol Res Pract. 2009;205(1):11–20. doi:10.1016/j.prp.2008.07.007.
Renne C, Willenbrock K, Kuppers R, Hansmann ML, Brauninger A. Autocrine- and paracrine-activated receptor tyrosine kinases in classic Hodgkin lymphoma. Blood. 2005;105(10):4051–9. doi:10.1182/blood-2004-10-4008.
Brown RE, Nazmi RK. The Reed-Steinberg cell: molecular characterization by proteomic analysis with therapeutic implications. Ann Clin Lab Sci. 2002;32(4):339–51.
Yuan K, Hong TM, Chen JJ, Tsai WH, Lin MT. Syndecan-1 up-regulated by ephrinB2/EphB4 plays dual roles in inflammatory angiogenesis. Blood. 2004;104(4):1025–33. doi:10.1182/blood-2003-09-3334.
Guler N, Yilmaz S, Ayaz S, Yilmaz M, Aki Z, Dagdas S, et al. The platelet-derived growth factor level (PDGF) in Hodgkin’s disease and non-Hodgkin’s lymphoma and its relationship disease activation. Hematology. 2005;10(1):53–7. doi:10.1080/10245330400020405.
Villani F, Busia A, Villani M, Vismara C, Viviani S, Bonfante V. Serum cytokine in response to chemo-radiotherapy for Hodgkin’s disease. Tumori. 2008;94(6):803–8.
Holz MS, Janning A, Renne C, Gattenlohner S, Spieker T, Brauninger A. Induction of endoplasmic reticulum stress by sorafenib and activation of NF-kappaB by lestaurtinib as a novel resistance mechanism in Hodgkin lymphoma cell lines. Mol Cancer Ther. 2013;12(2):173–83. doi:10.1158/1535-7163.MCT-12-0532.
Tsirakis G, Pappa CA, Kanellou P, Stratinaki MA, Xekalou A, Psarakis FE, et al. Role of platelet-derived growth factor-AB in tumour growth and angiogenesis in relation with other angiogenic cytokines in multiple myeloma. Hematol Oncol. 2012;30(3):131–6. doi:10.1002/hon.1014.
Feyzi E, Lustig F, Fager G, Spillmann D, Lindahl U, Salmivirta M. Characterization of heparin and heparan sulfate domains binding to the long splice variant of platelet-derived growth factor A chain. J Biol Chem. 1997;272(9):5518–24.
Kim J, Lee JH, Park HS, Hwang J, Han IO, Bae YS, et al. Syndecan-4 regulates platelet-derived growth factor-mediated MAP kinase activation by altering intracellular reactive oxygen species. FEBS Lett. 2008;582(18):2725–30. doi:10.1016/j.febslet.2008.06.055.
Choi S, Lee E, Kwon S, Park H, Yi JY, Kim S, et al. Transmembrane domain-induced oligomerization is crucial for the functions of syndecan-2 and syndecan-4. J Biol Chem. 2005;280(52):42573–9. doi:10.1074/jbc.M509238200.
Giefing M, Winoto-Morbach S, Sosna J, Doring C, Klapper W, Kuppers R, et al. Hodgkin-Reed-Sternberg cells in classical Hodgkin lymphoma show alterations of genes encoding the NADPH oxidase complex and impaired reactive oxygen species synthesis capacity. PLoS One. 2013;8(12), e84928. doi:10.1371/journal.pone.0084928.
Lyon M, Rushton G, Gallagher JT. The interaction of the transforming growth factor-betas with heparin/heparan sulfate is isoform-specific. J Biol Chem. 1997;272(29):18000–6.
McCaffrey TA, Falcone DJ, Du B. Transforming growth factor-beta 1 is a heparin-binding protein: identification of putative heparin-binding regions and isolation of heparins with varying affinity for TGF-beta 1. J Cell Physiol. 1992;152(2):430–40. doi:10.1002/jcp.1041520226.
Yang ZZ, Grote DM, Ziesmer SC, Xiu B, Yates NR, Secreto FJ, et al. Soluble and membrane-bound TGF-beta-mediated regulation of intratumoral T cell differentiation and function in B-cell non-Hodgkin lymphoma. PLoS One. 2013;8(3), e59456. doi:10.1371/journal.pone.0059456.
McCaffrey TA, Falcone DJ, Vicente D, Du B, Consigli S, Borth W. Protection of transforming growth factor-beta 1 activity by heparin and fucoidan. J Cell Physiol. 1994;159(1):51–9. doi:10.1002/jcp.1041590108.
Newcom SR, Gu L. Transforming growth factor beta 1 messenger RNA in Reed-Sternberg cells in nodular sclerosing Hodgkin’s disease. J Clin Pathol. 1995;48(2):160–3.
Hsu SM, Lin J, Xie SS, Hsu PL, Rich S. Abundant expression of transforming growth factor-beta 1 and -beta 2 by Hodgkin’s Reed-Sternberg cells and by reactive T lymphocytes in Hodgkin’s disease. Hum Pathol. 1993;24(3):249–55.
Kadin M, Butmarc J, Elovic A, Wong D. Eosinophils are the major source of transforming growth factor-beta 1 in nodular sclerosing Hodgkin’s disease. Am J Pathol. 1993;142(1):11–6.
Koreishi AF, Saenz AJ, Persky DO, Cui H, Moskowitz A, Moskowitz CH, et al. The role of cytotoxic and regulatory T cells in relapsed/refractory Hodgkin lymphoma. Appl Immunohistochem Mol Morphol. 2010;18(3):206–11. doi:10.1097/PAI.0b013e3181c7138b.
Alvaro T, Lejeune M, Salvado MT, Bosch R, Garcia JF, Jaen J, et al. Outcome in Hodgkin’s lymphoma can be predicted from the presence of accompanying cytotoxic and regulatory T cells. Clin Cancer Res. 2005;11(4):1467–73. doi:10.1158/1078-0432.CCR-04-1869.
Asano N, Oshiro A, Matsuo K, Kagami Y, Ishida F, Suzuki R, et al. Prognostic significance of T-cell or cytotoxic molecules phenotype in classical Hodgkin’s lymphoma: a clinicopathologic study. J Clin Oncol. 2006;24(28):4626–33. doi:10.1200/JCO.2006.06.5342.
Kelley TW, Pohlman B, Elson P, Hsi ED. The ratio of FOXP3+ regulatory T cells to granzyme B+ cytotoxic T/NK cells predicts prognosis in classical Hodgkin lymphoma and is independent of bcl-2 and MAL expression. Am J Clin Pathol. 2007;128(6):958–65. doi:10.1309/NB3947K383DJ0LQ2.
Skinnider BF, Mak TW. The role of cytokines in classical Hodgkin lymphoma. Blood. 2002;99(12):4283–97. doi:10.1182/blood-2002-01-0099.
Herbst H, Foss HD, Samol J, Araujo I, Klotzbach H, Krause H, et al. Frequent expression of interleukin-10 by Epstein-Barr virus-harboring tumor cells of Hodgkin’s disease. Blood. 1996;87(7):2918–29.
Marshall NA, Christie LE, Munro LR, Culligan DJ, Johnston PW, Barker RN, et al. Immunosuppressive regulatory T cells are abundant in the reactive lymphocytes of Hodgkin lymphoma. Blood. 2004;103(5):1755–62. doi:10.1182/blood-2003-07-2594.
Kadin ME, Agnarsson BA, Ellingsworth LR, Newcom SR. Immunohistochemical evidence of a role for transforming growth factor beta in the pathogenesis of nodular sclerosing Hodgkin’s disease. Am J Pathol. 1990;136(6):1209–14.
Szatmari T, Dobra K. The role of syndecan-1 in cellular signaling and its effects on heparan sulfate biosynthesis in mesenchymal tumors. Front Oncol. 2013;3:310. doi:10.3389/fonc.2013.00310.
Hayashida K, Johnston DR, Goldberger O, Park PW. Syndecan-1 expression in epithelial cells is induced by transforming growth factor beta through a PKA-dependent pathway. J Biol Chem. 2006;281(34):24365–74. doi:10.1074/jbc.M509320200.
Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18(7):816–27. doi:10.1096/fj.03-1273rev.
Pohlers D, Brenmoehl J, Loffler I, Muller CK, Leipner C, Schultze-Mosgau S, et al. TGF-beta and fibrosis in different organs - molecular pathway imprints. Biochim Biophys Acta. 2009;1792(8):746–56. doi:10.1016/j.bbadis.2009.06.004.
Lijnen P, Petrov V. Transforming growth factor-beta 1-induced collagen production in cultures of cardiac fibroblasts is the result of the appearance of myofibroblasts. Methods Find Exp Clin Pharmacol. 2002;24(6):333–44.
Tromp J, van der Pol A, Klip IT, de Boer RA, Jaarsma T, van Gilst WH, et al. Fibrosis marker syndecan-1 and outcome in patients with heart failure with reduced and preserved ejection fraction. Circ Heart Fail. 2014;7(3):457–62. doi:10.1161/CIRCHEARTFAILURE.113.000846.
Kliment CR, Englert JM, Gochuico BR, Yu G, Kaminski N, Rosas I, et al. Oxidative stress alters syndecan-1 distribution in lungs with pulmonary fibrosis. J Biol Chem. 2009;284(6):3537–45. doi:10.1074/jbc.M807001200.
Sneed TB, Stanley DJ, Young LA, Sanderson RD. Interleukin-6 regulates expression of the syndecan-1 proteoglycan on B lymphoid cells. Cell Immunol. 1994;153(2):456–67. doi:10.1006/cimm.1994.1042.
Szatmari T, Mundt F, Heidari-Hamedani G, Zong F, Ferolla E, Alexeyenko A, et al. Novel genes and pathways modulated by syndecan-1: implications for the proliferation and cell-cycle regulation of malignant mesothelioma cells. PLoS One. 2012;7(10):e48091. doi:10.1371/journal.pone.0048091.
Hassan H, Greve B, Pavao MS, Kiesel L, Ibrahim SA, Gotte M. Syndecan-1 modulates beta-integrin-dependent and interleukin-6-dependent functions in breast cancer cell adhesion, migration, and resistance to irradiation. FEBS J. 2013;280(10):2216–27. doi:10.1111/febs.12111.
Frassanito MA, Cusmai A, Iodice G, Dammacco F. Autocrine interleukin-6 production and highly malignant multiple myeloma: relation with resistance to drug-induced apoptosis. Blood. 2001;97(2):483–9.
Andersen NF, Standal T, Nielsen JL, Heickendorff L, Borset M, Sorensen FB, et al. Syndecan-1 and angiogenic cytokines in multiple myeloma: correlation with bone marrow angiogenesis and survival. Br J Haematol. 2005;128(2):210–7. doi:10.1111/j.1365-2141.2004.05299.x.
Tesch H, Jucker M, Klein S, Abts H, Gunther A, Krueger GR, et al. Hodgkin and Reed-Sternberg cells express interleukin 6 and interleukin 6 receptors. Leuk Lymphoma. 1992;7(4):297–303. doi:10.3109/10428199209049781.
Kurzrock R, Redman J, Cabanillas F, Jones D, Rothberg J, Talpaz M. Serum interleukin 6 levels are elevated in lymphoma patients and correlate with survival in advanced Hodgkin’s disease and with B symptoms. Cancer Res. 1993;53(9):2118–22.
Acknowledgements
No financial support was provided for this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
None
Rights and permissions
About this article

Cite this article
Gharbaran, R. Insights into the molecular roles of heparan sulfate proteoglycans (HSPGs—syndecans) in autocrine and paracrine growth factor signaling in the pathogenesis of Hodgkin’s lymphoma. Tumor Biol. 37, 11573–11588 (2016). https://doi.org/10.1007/s13277-016-5118-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-016-5118-7