
Abstract
Recent evidence suggests that cancerous inhibitor of protein phosphatase 2A (CIP2A) is an oncoprotein that acts as a novel therapeutic target in a variety of tumors. In this study, we investigated the clinical significance of CIP2A and its function in our large collection of prostate samples. Between August 2000 and December 2013, 126 patients with histologically confirmed PCa and 92 with benign prostate hyperplasia (BPH) were recruited into the study. Quantitative RT-PCR, Western blot, and immunohistochemistry analyses were used to quantify CIP2A expression in PCa clinical samples and cell lines. The relationships between CIP2A expression and clinicopathological features were analyzed. The functional role of CIP2A in PCa cells was evaluated by small interfering RNA-mediated depletion of the protein followed by analyses of cell proliferation and invasion. High expression of CIP2A staining was 86.51 % (109/126) in 126 cases of PCa and 17.39 % (16/92) in 92 cases of BPH, and the difference of CIP2A expression between PCa and BPH was statistically significant. CIP2A was significantly elevated in all five PCa cell lines when compared to the RWPE-1 cells at both the messenger RNA (mRNA) and protein levels. Silencing of CIP2A inhibited the proliferation of DU-145 cells which have a relatively high level of CIP2A in a time- and concentration-dependent manner, and the invasion and migration of DU-145 cells were distinctly suppressed. Furthermore, CIP2A knockdown led to substantial reductions in c-Myc levels in PCa cell lines, but no significant change in phosphorylated Akt expression after CIP2A knockdown in DU-145 cells. Our data suggest that the pathogenesis of human PCa maybe mediated by CIP2A, and CIP2A inhibition treatment may provide a promising strategy for the antitumor therapy of PCa, and thus CIP2A could represent selective targets for the molecularly targeted treatments of PCa.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Prostate cancer represents the most common cancer and the leading cause of cancer-related death in men in the USA, accounting for 30 % of male cancer diagnoses [1, 2]. This high rate of mortality is primarily due to metastasis of the primary tumor. Early detection and treatment before the tumor metastasizes is critical for improving patient survival. Locally advanced prostate cancer almost always progressed to castration-resistant prostate cancer (CRPC) which is characterized by insensitivity to androgen deprivation (castrate resistance), increased tumor size, and metastasis [3–5]. The key mechanistic principles underlying the transition from androgen-dependent to castrate-resistant cancer remains unclear; therefore, it is important to investigate the molecular mechanisms underlying the progression of PCa.
Cancerous inhibitor of protein phosphatase 2A (CIP2A), also referred as KIAA1524 or p90 tumor-associated antigen, is a novel human oncoprotein that is known to suppress PP2A phosphatase activity via stabilizing the level of c-Myc in human malignancies [6]. Previous studies have reported that CIP2A is overexpressed in various human cancers, and its overexpression is related to poor prognosis [7]. Increasing evidence showed that CIP2A can promote proliferation and cell invasion. Downregulation of p90/CIP2A in cancer cells reduced cell proliferation, and tumor formation in vivo [8, 9]. The knockdown of p90/CIP2A decreased the protective role of AKT in drug treatment. Taken together, CIP2A protein plays an important role in human cancers. However, the expression pattern of CIP2A in PCa and its involvement in aggressiveness of PCa has not been studied so far.
Materials and methods
Patients and tissue specimens
This study included 126 patients who had undergone radical prostatectomy and bilateral lymphadenectomy at the Department of Urology, First Affiliated Hospital of Jinan University between December 2001 and August 2013 and for whom archival tissues were available. No patient was managed preoperatively with either hormonal or radiation therapy, and no secondary cancers were observed. Ninety-two cases of benign prostate hyperplasia (BPH) were obtained from men undergoing suprapubic prostatectomy or transurethral plasmakinetic enucleation of prostate. Twenty-four cases of normal prostate tissue were obtained from bladder cancer patients who underwent radical cystoprostatectomy. The stages of cancer for all patients were determined by the American Joint Committee on Cancer (AJCC) 2002 system. The specimens were examined by two staff pathologists who were blinded to the clinical outcome and follow-up data. The evaluation of the specimen was performed according to the guidelines of the College of American Pathologists. Formalin-fixed, paraffin-embedded tumor tissues from these patients were evaluated. Besides, freshly frozen tissue samples were available. Samples were snap-frozen in liquid nitrogen immediately after surgery and experiments were performed. This study was approved by the Ethics Committee of the First Affiliated Hospital of Jinan University. All patients provided informed consent.
Immunohistochemistry
Specimens were fixed in 10 % neutral buffered formalin, embedded in paraffin, and cutted into serial sections at a thickness of 3 μm. Paraffin-embedded tissues were dewaxed in xylene, rehydrated by serial concentrations of ethanol, and then rinsed in phosphate buffer solution (PBS) followed by treated with 3 % H2O2 to refrain endogenous peroxidase. After being heated in a microwave at 750 W for 15 min to repair the tissue antigen, the sections were incubated with 10 % normal goat serum at room temperature for 10 min to block nonspecific reactions. This was followed by a PBS wash and incubation with rabbit polyclonal antibody against human CIP2A (Novus Biologicals, Littleton, CO, USA, dilution 1:400) for 12 h at 4 °C, and biotinylated goat anti-rabbit serum IgG was used as secondary antibody. After a PBS wash, the sections were developed in diaminobenzidine (DAB) substrate. The sections were then counter-stained in hematoxylin for 2 min and then dehydrated in ethanol and xylene before being mounted. Sections were re-prepared by EnVision immunohistochemical staining. The staining results of colon cancer tissue sections which CIP2A positive had already known were regarded as positive control, PBS instead of primary antibodies was as negative control.
Evaluation of immunohistochemical results
Digital images of each tissue microarray were manually scored and displayed according to staining intensity and morphology. Positive CIP2A staining was characterized by brown-yellow granules located diffusely in the cell cytoplasm. Lack of any obvious purple-brown or brown-red pigmentation in the cytoplasm of tumor cell was considered negative. For quantitative analyses of expression, five visual fields were randomized selected per section under high power microscope (×400), and 200 cells were counted in each high power field. The proportion of positively stained tumor cells was graded as follows: 0 (no positive tumor cells), 1 (<10 % positive tumor cells), 2 (10–50 % positive tumor cells), and (>50 % positive tumor cells). The cells at each intensity of staining were recorded on a scale of 0 (no staining), 1 (weak staining, light yellow), 2 (moderate staining, yellow brown), and 3 (strong staining, brown). The staining index (SI) was calculated as follows: SI = staining intensity proportion of positively stained tumor cells. Using this method of assessment, we evaluated the expression of CIP2A in PCa by SI (scored as 0, 1, 2, 3, 4, 6, or 9). The SI score of 6 or above was used to define as high expression and SI of 4 or less as low expression of CIP2A. The results were scored by two independent pathologists who were blinded to the subtype of the tumors.
Cell culture
Human prostate normal epithelial cell line RWPE-1 and prostate cancer cell lines LNCaP-AI, LNCaP-AD, DU145, PC3, and 22RV1 were obtained directly from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai Institute of Cell Biology, Chinese Academy of Sciences, China) for fewer than 6 months. LNCaP-AD, PC3, and DU145 were maintained in Roswell Park Memorial Institute (RPMI) medium 1640 (GIBCO, Carlsbad, CA, USA) supplemented with 10 % fetal bovine serum (FBS), 2 mmol/L l-glutamine and 25 mmol/L HEPES. LNCaP-AI and 22RV1 cells were maintained in phenol red-free RPMI medium 1640 (GIBCO) supplemented with 10 % charcoal-stripped FBS, 300 mg/L l-glutamine, 2000 mg/L glucose, and 2000 mg/L NaHCO3. RWPE-1 cells were maintained in Keratinocyte-SFM (10724, GIBCO), supplemented with 5 mg/mL human recombinant epidermal growth factor (rEGF) and 0.05 mg/mL bovine pituitary extract l-glutamine. All cell lines were maintained in a humidified incubator at 5 % CO2 and 37 °C.
Total RNA extraction and cDNA synthesis
Upon collection, the prostate tissues were snap frozen in liquid nitrogen and subsequently kept at −80 °C until required. The tissues were pulverized and total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. The purity and concentration of RNA were determined by spectrophotometric methods. Three micrograms of total RNA were reverse-transcribed into first-strand complementary DNA (cDNA) using reverse transcription system kit (Promega, Madison, WI, USA) according to the following protocol with the reaction kit. Briefly, samples were preincubated at 70 °C for 10 min; cooled on ice then added to a reaction mixture of 10 mmol/L dNTP mixture, 25 mmol/L MgCl2, 15 U of AMV reverse transcriptase, reverse transcription 10× buffer, 0.5 U of RNasin, and 0.5 μg oligo-(dT)15 primer; and scaled up to a final volume of 20 L. The reaction mixture was sequentially incubated at 44 °C for 15 min, 99 °C for 5 min and 4 °C for 5 min. The cDNA was stored at 20 °C before use.
Quantitative real-time polymerase chain reaction
Quantitative RT-PCR was performed using SYBR Master Mix (Takara) on an ABI Prism 7900 HT (Applied Biosystems). A human GAPDH gene was used as an endogenous control for sample normalization. Results were presented as the fold expression relative to that of GAPDH. PCR primers were as follows: for human GAPDH, forward 5′-GAGTCAACGGATTTGGTCGT-3′ and reverse 5′-GACAAGCTTCCCGTTCTCAG-3′; for CIP2A forward 5′-GGGAATTCCCTGATTCCTCTTCA-3′ and reverse 5′-CCCTCGAGCTAGAAGCTTACTTCCAT-3′.
Western blot
Western immunoblot analyses were performed with protein lysates obtained from snap-frozen tissue samples. Protein levels were determined using the BCA Protein Assay Kit (Pierce, USA).Thirty micrograms of the respective tissue protein were separated by SDS-PAGE (using 10 % gels) and transferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5 % nonfat milk and then incubated with primary antibodies against CIP2A (Novus Biologicals, Littleton, CO, USA, dilution 1:400), c-MYC (1:1000; Abcam, Cambridge, MA, USA), and β-actin (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4 °C for 12 h. Membranes were washed three times for 10 min each with Tris-buffered saline (50 mM Tris, pH 7.4, 0.9 % NaCl) containing 0.05 % Tween-20 (TBS-T) and incubated with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotech. Inc., Santa Cruz, CA, USA). Membranes were then washed again three times for 10 min each with TBS-T. Target protein bands were visualized using the enhanced chemiluminescence method. The intensity of the bands was quantified using the Tanon GIS system (Tanon, Shanghai, China), and the data were normalized to the ACTIN loading controls. All Western immunoblot analyses were performed three times.
Small interfering RNA transfection
The PCa cells were seeded onto the six-well plates at 2 × 105 cells per well before transfection. Cells were cultured for 24 h until cell density was around 50 %. Double-stranded small interfering RNA targeting CIP2A (50 nM, Gene Pharma; siCIP2A 1 5′-CUGUGGUUGUGUUUGCACUTT-3′; siCIP2A 2 5′-ACCAUUGAUAUCCUUAGAATT-3′) or scrambled control small interfering RNA (siRNA) (5′-UAACAAUGAGAGCACGGCTT-3′) were transfected into PCa cell line using Lipofectamine 2000 reagent (Invitrogen). About 48 h after transfection, total RNA was extracted with the TRIzol reagent and then added it to a reverse transcription reaction to generate cDNA.
Cell survival assay
The effects of CIP2A on PCa cells survival were determined by MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazoliumbromide) assay. Four groups of cells were seeded into 96-well plates (5 × 103 cells/well) and cultured for 120 h. After treatments, cells were incubated with MTT (Sigma-Aldrich, St. Louis, MO, USA, 20 μL/well) at 37 °C for 4 h, and then 200 μL DMSO was added into each well. Cells were subjected to absorbance reading at 570 nm using a 96-well microplate reader. Percentage of residual cell viability was determined as [(OD of experiment group − OD of blank group) / (OD of negative group−OD of blank group)] × 100 %. Assays were performed three times.
Cell migration assay
Motility capabilities in vitro were measured with transwell chambers (Corning, Corning, NY, USA). Four groups of cells (5 × 105) were seeded on the upper wells with serum-free medium. Medium with 20 % FBS was plated in the bottom wells as chemoattractants. After 48-h incubation, cells were fixed with methanol and stained with 1 % crystal violet for 30 min at 37 °C. Cells staying on the upper side of the membranes were wiped, while those on the lower side were counted and photographed with microscope.
Statistical analysis
Data analyses were performed using SPSS statistical package 15.0 (SPSS Inc, USA). Patient characteristics were expressed as the mean ± SD for continuous variables, and as the count and percent for discrete variables. Data were analyzed using Pearson’s chi-square test and Fisher’s exact test. Statistical significance was taken at the P < 0.05 level.
Results
CIP2A expression is correlated with clinicopathological features of PCa
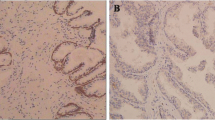
CIP2A expression was examined by immunohistochemistry, Western blotting, and quantitative real-time PCR in tissue samples from a total of 218 patients, including 92 BPH, 22 prostate intraepithelial neoplasia (PIN), 74 clinically localized PCa, and 30 metastatic cases. The results showed that the positive rate of CIP2A staining was 86.51 % (109/126) in 126 cases of prostatic carcinoma and 17.39 % (16/92) in 92 cases of BPH, and the difference of CIP2A expression between PCa and BPH was statistically significant (P < 0.001). Representative staining of CIP2A in PCa tissue is shown in Fig. 1, and positive staining of CIP2A was mainly observed in the cytoplasm. In addition, CIP2A staining was stronger in cells of prostatic carcinoma with metastasis than in those of prostatic carcinoma without metastasis. These data suggest that CIP2A expression is closely related with the occurrence and development of PCa.

a High expression of CIP2A in the PCa (magnification ×40); b low expression of CIP2A in the PCa (magnification ×40)
Furthermore, the expression of CIP2A mRNA in prostate cancer tissues normalized to GAPDH mRNA was detected by quantitative real-time PCR (Fig. 2a). The expression level of CIP2A mRNA was significantly increased in PCa tissues compared with that in BPH tissues and normal prostate tissue (P < 0.01). The expression of CIP2A mRNA exhibited different expression patterns in terms of localization depending on pathological category of PCa (Fig. 2b). The results were confirmed by Western blot analyses, and we also found the protein expression levels of CIP2A in PCa was higher than in BPH tissues and normal prostate tissue, and the difference among these groups had statistic significance (P < 0.01) (Fig. 3).

Quantitative real-time PCR showing expression level of CIP2A mRNA. a CIP2A mRNA expression in PCa tissues, BPH tissues, and normal prostate tissue (*P < 0.05;**P < 0.01); b CIP2A mRNA expression in PIN localized PCa and metastatic PCa (*P < 0.05;**P < 0.01)

Western blots showing expression of CIP2A in PCa tissues, BPH tissues, and normal prostate tissue
High expression of CIP2A in PCa cell lines
Quantitative RT-PCR and Western blot analyses were used to determine the levels of CIP2A mRNA and protein in five PCa cell lines and the prostate normal epithelial cell line RWPE-1. CIP2A was significantly elevated in all five PCa cell lines when compared to the RWPE-1 cells at both the mRNA and protein levels (Fig. 4a–b).

a Quantitative real-time PCR showing expression level of CIP2A mRNA in PCa cells and normal control cell line; b Western blots showing expression of CIP2A mRNA in PCa cells and normal control cell line
Effects of CIP2A depletion on cell proliferation and MYC expression in PCa
To explore the effects of CIP2A suppression on the proliferation of human PCa cells, we specifically knocked down the CIP2A expression in DU-145 cells using RNA interference. The DU-145 cell line was chosen because of its high abundance of CIP2A. The efficacy of CIP2A siRNA for knockdown of CIP2A mRNA and protein was confirmed by quantitative real-time polymerase chain reaction (qRT-PCR) and Western blot analysis, respectively. We observed that CIP2A mRNA and protein levels were significantly reduced in cells transfected with specific siRNA for CIP2A compared with those transfected with control siRNA. Thus, the CIP2A siRNA could effectively knockdown CIP2A expression at both transcriptional and translational levels.
We next studied the impact of CIP2A silencing on cell proliferation. The results of the MTT assay showed that CIP2A siRNA significantly reduced the proliferation rate of DU-145 cells compared with the negative control cells (P < 0.01) (Fig. 5a). Furthermore, CIP2A knockdown led to substantial reductions in MYC levels in DU-145 cells, but no significant change in phosphorylated Akt expression after CIP2A knockdown in DU-145 cells (Fig. 5b).

a CIP2A knockdown inhibited cell proliferation of DU-145 cells. Cell number was measured by MTT assay; b Western blots showing that siRNA treatment of CIP2A markedly decreased CIP2A levels and MYC levels in DU-145 cells
Silencing of CIP2A is associated with decreased migration and invasiveness of PCa cells
We used the transwell assay to verify the effect of CIP2A deletion on migration and invasion of PCa in vitro. The results of DU-145 cells showed that both in invasion assay and migration assay, the number of DU-145 cells that penetrated through the membrane in the si-CIP2A-treated group passed out less cells than the control siRNA-transfected cells group (P < 0.01) (Fig. 6).

Inhibition of invasion and migration of DU-145 cells by siRNA treatment of CIP2A
Discussion
PCa is the most frequent cancer among men over 50 years old in industrialized countries. As the second cause for cancer-related death, it is a global public health problem. Recurrent or metastatic PCa progression usually is androgen-dependent, and most PCa are responsive to the available hormone therapies, so androgen deprivation therapy is generally considered first-line therapy at this time point [10]. Unfortunately, castration-resistant prostate cancer in virtually develop within a median of 18–24 months after castration in these patients, and current anticancer treatments are not effective [11]. The incomplete understanding of molecular features of PCa might be one of the reasons for this unsatisfied situation, although recent gene expression studies have significantly improved our knowledge. Therefore, it is important to investigate the molecular mechanisms and identify new biomarkers responsible for PCa progression to provide effective strategies for the prevention and therapy of this disease.
CIP2A is a novel human oncoprotein that can promote tumor transformation and maintain the malignant phenotype. Recent studies suggest that CIP2A modulates cell proliferation and lineage development and is implicated in a number of tumor types [12–14]. Studies in cell culture and mouse models of cancer have indicated that CIP2A also could promote various biological activities; however, the functional role of CIP2A in PCa progression and metastasis remains elusive. The aim of our study was to examine the impact on the oncogenetic process through investigating the expression and function of CIP2A in PCa.
In this study, we examined the expression of CIP2A in clinical PCa tissues by immunohistochemistry, Western blotting, and qRT-PCR. The immunohistochemistry analysis showed that the positive rate of CIP2A staining was 86.51 % (109/126) in 126 cases of prostatic carcinoma and 17.39 % (16/92) in 92 cases of BPH, and the difference of CIP2A expression between PCa and BPH was statistically significant (P < 0.001). Furthermore, CIP2A staining was stronger in prostatic carcinoma with metastasis than in prostatic carcinoma without metastasis. The results of qRT-PCR analysis showed that CIP2A mRNA level in PCa tissues revealed more than twofold increases compared with that in the BPH tissues. It suggested that CIP2A might play a role in the tumorigenesis of PCa.
Additionally, qRT-PCR and Western blot analyses were used to determine the levels of CIP2A mRNA and protein in five PCa cell lines and the prostate normal epithelial cell line RWPE-1. Results show that CIP2A was significantly elevated in all five PCa cell lines when compared to the RWPE-1 cells at both the mRNA and protein levels. To extend our clinical studies and investigate its biological function, we employed siRNA to knockdown CIP2A expression in PCa cell line DU-145. The DU-145 cell line was chosen because of its high abundance of CIP2A. Depletion of endogenous CIP2A attenuated proliferation of DU-145 cells in vitro, MTT cell proliferation assay showed that CIP2A siRNA significantly reduced the proliferation rate of DU-145 cells compared with the control siRNA-transfected cells, and the data presented dose-dependency. Furthermore, we used the transwell assay to verify the effect of CIP2A on migration of PCa cells in vitro. The results showed depletion of CIP2A could inhibit cell migration and invasion in vitro, suggesting that CIP2A expression can significantly promoted PCa cell proliferation, migration, and invasion. Collectively, these data strongly suggest that CIP2A served as a potential therapeutic target in PCa, and the CIP2A pathway is a promising target for rational cancer therapy.
Previous studies indicated that CIP2A inhibits PP2A activity toward the oncogenic transcription factor c-MYC Ser62 and thereby stabilizes the c-MYC protein by preventing its proteolytic degradation [15, 16]. Consistently, our data showed that the depletion of CIP2A expression in PCa cell line DU-145 resulted in reduced c-MYC protein levels. CIP2A knockdown led to substantial reductions in c-Myc levels in PCa cell lines, but no significant change in phosphorylated Akt expression after CIP2A knockdown in DU-145 cells.
In conclusion, our data suggest that both CIP2A mRNA and protein were obviously expressed in a higher degree in PCa tissues and cell lines. CIP2A knockdown inhibited PCa cells proliferation, invasion, and migration in a time- and dose-dependent manner. Our research indicate that CIP2A may play a significant role in the regulation of aggressiveness in human PCa, and the CIP2A inhibition treatment may provide a promising strategy for the anti-tumor therapy of PCa. Though this mechanism is not completely clear, our study tries to reveal it in our further research.
References
Ferlay J, Parkin DM, Steliarova-Foucher E. Estimates of cancer incidence and mortality in Europe in 2008. Eur J Cancer. 2010;46:765–81.
Ren SC, Chen R, Sun YH. Prostate cancer research in China. Asian J Androl. 2013;15:350–3.
Heidenreich A, Bellmunt J, Bolla M, Joniau S, Mason M, et al. EAU guidelines on prostate cancer. Part 1: screening, diagnosis, and treatment of clinically localized disease. Eur Urol. 2011;59(1):61–71.
Saad F, Pantel K. The current role of circulating tumor cells in the diagnosis and management of bone metastases in advanced prostate cancer. Future Oncol. 2012;8(3):321–31.
Lassi K, Dawson NA. Emerging therapies in castrate-resistant prostate cancer. Curr Opin Oncol. 2009;21:260–5.
Li W, Ge Z, Liu C, Liu Z, Bjorkholm M, Jia J, et al. CIP2A is overexpressed in gastric cancer and its depletion leads to impaired clonogenicity, senescence, or differentiation of tumor cells. Clin Cancer Res. 2008;14:3722–8.
Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, Bottzauw T, et al. CIP2A inhibits PP2A in human malignancies. Cell. 2007;130:51–62.
Khanna A, Bockelman C, Hemmes A, Junttila MR, Wiksten JP, Lundin M, et al. MYC-dependent regulation and prognostic role of CIP2A in gastric cancer. J Natl Cancer Inst. 2009;101:793–805.
Come C, Laine A, Chanrion M, Edgren H, Mattila E, Liu X, et al. CIP2A is associated with human breast cancer aggressivity. Clin Cancer Res. 2009;15:5092–100.
Qu YY, Dai B, Kong YY, Ye DW, Yao XD, et al. Prognostic factors in Chinese patients with metastatic castration-resistant prostate cancer treated with docetaxel-based chemotherapy. Asian J Androl. 2013;15(1):110–5.
Petrylak DP, Tangen CM, Hussain MH, Lara Jr PN, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351(15):1513–20.
Wiegering A, Pfann C, Uthe FW, Otto C, Rycak L, Mäder U, et al. CIP2A influences survival in colon cancer and is critical for maintaining Myc expression. PLoS One. 2013;8(10):e75292.
Hemmes A, Leminen A, Westermarck J, Haglund C, Butzow R, et al. Prognostic role of CIP2A expression in serous ovarian cancer. Br J Cancer. 2011;105(7):989–95.
Ren J, Li W, Yan L, Jiao W, Tian S, et al. Expression of CIP2A in renal cell carcinomas correlates with tumour invasion, metastasis and patients survival. Br J Cancer. 2011;105(12):1905–11.
Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, et al. CIP2 inhibits PP2A in human malignancies. Cell. 2007;130:51–62.
Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, et al. A signaling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6:308–18.
Acknowledgments
This study was supported by the grants from the Science and Technology Research Projects of Guangzhou (No. 11A72070508, 2011Y-00003)
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Additional information
The Publisher and Editor retract this article in accordance with the recommendations of the Committee on Publication Ethics (COPE). After a thorough investigation we have strong reason to believe that the peer review process was compromised.
An erratum to this article is available at http://dx.doi.org/10.1007/s13277-017-5487-6.
About this article

Cite this article
Guo, Z., Liu, D. & Su, Z. RETRACTED ARTICLE: CIP2A mediates prostate cancer progression via the c-MYC signaling pathway. Tumor Biol. 36, 3583–3589 (2015). https://doi.org/10.1007/s13277-014-2995-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-014-2995-5