
Abstract
Glucose-regulated protein 78 (GRP78) is a key chaperone and stress response protein. Previous studies have demonstrated that high GRP78 expression may be correlated with cancer progression and therapeutic response. However, the role of GRP78 in the metastasis of colon cancer is unclear. In this study, we used small interfering RNA (siRNA) to knock down GRP78 expression in colon cancer cells (HT-29 and DLD-1 cells). In wound-healing migration assays, we found that GRP78-knockdown (GRP78KD) cells showed better wound-healing ability than control cells. We also found that GRP78KD cells displayed a better migratory ability than control cells in migration and invasion assays. As we further dissected the underlying molecular mechanism, we found that silencing GRP78 may cause an increase in vimentin expression and a decrease in the E-cadherin level, which was correlated with the increase in migratory ability. In addition, we found that GRP78KD may activate the NRF-2/HO-1 pathway, and this activation was also correlated with the increase in cell invasiveness. Furthermore, we examined GRP78 expression in a tissue array and found that the GRP78 expression in metastatic adenocarcinoma in lymph nodes tended to be weaker than that in primary colonic adenocarcinoma. In conclusion, a low level of GRP78 may cause an increase in metastasis ability in colon cancer cells by altering E-cadherin and vimentin expression and activating the NRF-2/HO-1 signaling pathway. Our study demonstrates that low expression of GRP78 may correlate with a high risk of metastasis in colon cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glucose-regulated protein 78 (GRP78), referred to as BiP, is an immunoglobulin heavy chain-binding protein and stress protein [1]. It shares amino acid sequence similarity with heat-shock protein 70 (HSP70); therefore, it is also regarded as a member of the heat-shock protein 70 (HSP70) family [2]. GRP78 resides primarily in the endoplasmic reticulum (ER). When unfolded proteins accumulate in the lumen of the ER, the expression of GRP78 protein will be up-regulated to restore ER homeostasis. In terms of physiological processes, GRP78 plays essential roles in early embryonic development, and the GRP78 expression level is decreased during aging [3, 4].
During cancer progression, cancer cells are often under ER stress in the tumor microenvironment, which is characterized by glucose deprivation, acidosis, and severe hypoxia [5]. Increased expression of the ER stress protein GRP78 has been found in several types of cancer, including breast cancer, lung cancer, esophageal cancer, gastric cancer, colon cancer, and endometrial cancer [6–11]. However, the role of GRP78 expression in the prognosis of various cancer types is controversial. The GRP78 expression level in esophageal cancer is higher in early-stage, highly differentiated adenocarcinomas, and patients with higher GRP78 levels tend to have better survival rates [8]. In contrast, in gastric cancer, GRP78 expression is up-regulated, and patients with higher GRP78 expression have a poor prognosis [9]. The GRP78 expression level in breast cancer cells also serves as a prognostic marker for responsiveness to hormonal therapy based on estrogen starvation, and combination therapy targeting GRP78 may enhance the efficacy of and reduce the resistance to hormone therapy [6].
In general, GRP78 had an anti-apoptotic function and conferred drug resistance in cancer cells such as gastric cancer, colon cancer, breast cancer, and hepatocellular carcinoma (HCC) cells [6, 12–14]. Suppression of GRP78 enhanced drug efficacy in both proliferative and dormant cancer cells, which implied that GRP78-mediated drug resistance is not limited to proliferating cancer cells but also occurs in dormant cancer cells [15].
A previous study revealed that GRP78 was overexpressed in colon cancers and might be an important biomarker for malignant transformation [10]. GRP78 silencing enhanced epirubicin-induced apoptosis in colon cancer cells and suppressed colon cancer growth through the down-regulation of the VEGF/VEGFR2 signaling pathway [16, 17]. Although overexpression of GRP78 was noted in colon cancers, its expression level was negatively correlated with lymphatic invasion [18]. In addition, colon cancers with higher surface GRP78 expression showed reduced tumor proliferation and growth, but their invasive ability increased with surface GRP78 expression [19, 20]. However, GRP78 localizes mainly to the endoplasmic reticulum, and only a small fraction is found on the cell surface [21]. The role of the overall GRP78 expression level on metastasis in colon cancer remains unclear. In this study, we demonstrate that the down-regulation of GRP78 expression increases the migration and invasion ability of colon cancer cells by inducing vimentin expression, decreasing the E-cadherin level and activating the NRF-2/HO-1 signaling pathway.
Materials and methods
Tissue samples
Two sets of tissue microarrays of colonic adenocarcinoma were purchased from US Biomax (Rockville, MD, USA) and Shanghai Outdo Biotech Co. (Shanghai, China). The first microarray (US Biomax) contained 30 cases of primary colonic adenocarcinoma, five cases of distantly metastatic adenocarcinoma, 25 cases of metastatic adenocarcinoma in lymph nodes, and normal colonic tissue. The second microarray (Shanghai Outdo Biotech Co.) consisted of 63 cases of low-grade (well-differentiated to moderately differentiated) and 29 cases of high-grade (poorly differentiated) colonic adenocarcinomas. The pathologic diagnoses of these cases were reconfirmed using microscopy. Immunohistochemical staining was performed on a BenchMark XT autostainer using an iView DAB detection kit (Ventana, Tucson, AZ, USA). The sections were incubated with GRP78 antibody (1:300; Santa Cruz Biotechnology, Dallas, TX, USA) for 1 h at 37 °C. Appropriate positive and negative controls were included in these assays. Both the intensity and extent of GRP78 expression in carcinoma cells were evaluated and scored by the method previously described [22]. The intensity of cytoplasmic staining was scored semiquantitatively as follows: 0 point, negative; 1 point, weakly positive; 2 points, moderately positive; or 3 points, strongly positive. The percentage of positive tumor cells (0–100 %) was multiplied by the intensity of GRP78 staining; therefore, the overall score ranged from 0 to 300. We divided the GRP78 expression scores (0–300) into three groups: low expression (0–100), intermediate expression (101–200), and high expression (201–300). For statistical analyses, we used Fisher’s exact test to determine whether there were significant differences in GRP78 expression among primary colonic adenocarcinoma, distantly metastatic adenocarcinoma, and metastatic adenocarcinoma in regional lymph nodes. The expression levels of GRP78 in low- and high-grade colonic adenocarcinoma were also compared.
Human colon adenocarcinoma cell culture
Human colon adenocarcinoma HT-29 (HTB-38) and DLD-1 (CCL-221) cell lines were purchased from the American Type Culture Collection (ATCC, Rockville, MD) and were isolated from Dukes’ stage B and C human colon adenocarcinoma, respectively. The cells were cultured in RPMI 1640 with 10 % fetal calf serum (FCS), penicillin (100 U/mL), and streptomycin (100 μg/mL) in a humidified incubator (37 °C, 5 % CO2). The cells were either sub-cultured or used before they reached 80 % confluence.
Small interfering RNA (siRNA) preparation
The expression of GRP78 was knocked down in HT-29 and DLD-1 cells using siRNA, as previously described [23]. The target sequence for the human GRP78 mRNA was 5′-AAGGTTACCCATGCAGTTGTT-3′, and the scrambled siRNA sequence was 5′-AAGGTGGTTGTTTTGTTCACT-3′. The GRP78 siRNA and scrambled siRNA were inserted into pSUPERIOR vectors to generate the pSUPERIOR-GRP78-siRNA and pSUPERIOR-scramble-siRNA plasmids, respectively. The siRNAs were then transfected into cells. Briefly, 1.5 × 105 cells were washed twice with Phosphate buffered saline (PBS) and mixed with 0.5 μg of plasmid. We applied one pulse for 20 ms under a fixed voltage of 1.4 kV on a Neon pipette-type microporator (Invitrogen Life Technologies, Grand Island, NY) [24, 25]. Successfully transfected HT-29 and DLD-1 cells were selected based on their antibiotic resistance.
Transwell migration assay
In vitro cell migration was examined using the BD Falcon cell culture insert (BD Biosciences). Aliquots of 1 × 105 DLD-1 cells expressing scrambled control or GRP78KD were suspended in 500 μL of serum-free RPMI 1640 and were seeded into the upper compartments of each chamber. The lower compartments were filled with 1 mL of RPMI 1640 with 10 % fetal calf serum. After incubation for 24 h at 37 °C in 5 % CO2, the non-migrating cells were removed from the upper surface of the membrane by scrubbing. The cells on the reverse side were stained with 0.1 % crystal violet, and migrating cells were counted under a microscope at ×100 magnification.
Wound-healing assay
Scramble control or GRP78KD HT-29 or DLD-1 cells were seeded into a six-well plate and allowed to grow to 80 % confluence in RPMI 1640. Subsequently, the cell monolayers were wounded using a 10-μL pipette tip. The wounded monolayers were washed four times with phosphate-buffered saline and incubated in RPMI 1640 for the indicated time periods.
Invasion assay
The BD BioCoat™ Matrigel Invasion Chamber (BD Biosciences) was utilized to determine the invasiveness of cells. DLD-1 cells (1 × 105) expressing scrambled control or GRP78KD suspended in 500 μL of serum-free RPMI 1640 were seeded into the upper compartments of each chamber, and the lower compartments were filled with 1 mL of RPMI 1640 containing 10 % fetal calf serum. After incubation for 24 h at 37 °C in 5 % CO2, the non-migrating cells were removed from the upper surface of the membrane by scrubbing. The cells on the reverse side were stained with 0.1 % crystal violet, and invading cells were counted under a microscope at ×100 magnification.
Evaluation of cell migration using the x’CELLigence biosensor system
Experiments were carried out using the RTCA DP instrument (Roche Diagnostics GmbH, Germany), which was placed in a humidified incubator maintained at 5 % CO2 at 37 °C. Cell migration was assessed using specifically designed 16-well plates (CIM-plate 16; Roche Diagnostics GmbH) with 8 μm pores. These plates are similar to conventional Transwell plates but with microelectrodes located on the underside of the membrane of the upper chamber. FCS medium (10 %) was added in the lower chamber, and cells were seeded into the upper chamber at 20,000 cells/well in serum-free medium. The CIM-plate 16 was monitored every 10 s for 40 min and then once every hour. Data analysis was carried out using RTCA v. 1.2 software, which was supplied with the instrument.
Protein extraction and western blot analysis
The method for total protein extraction was described previously [14, 23]. The cells were lysed using cell lysis buffer containing protease inhibitors (Complete Protease Inhibitor Tablets; Boehringer Mannheim, Indianapolis, IN) [14, 23]. The proteins (25 μg) in each sample were separated using 10 % SDS–PAGE under reducing conditions and electrotransferred onto polyvinylidene difluoride membranes (GE Healthcare). The membranes were incubated overnight at 4 °C with a primary antibody against a specific target and subsequently probed with a horseradish peroxidase-conjugated secondary antibody (1:5,000). The products were visualized with an enhanced chemiluminescence reagent (GE Healthcare) and detected using a VersaDoc 5000 (Bio-Rad Laboratories, Hercules, CA) [26].
Statistical analysis
All experiments were repeated a minimum of three times. All data collected from real-time RT–PCR and cell proliferation experiments are expressed as the means ± SD. The data presented in some figures are derived from a representative experiment that was quantitatively similar to the replicate experiments. When two groups of datasets were compared, statistical significance was determined using a two-tailed Student’s t test. Asterisks in the figures indicate significant differences between the indicated experimental groups and the corresponding control conditions (p < 0.05, see figure legends).
Results
Silencing of GRP78 enhanced the migratory ability of DLD-1 cells
To confirm the role of GRP78 in cancer metastasis, we used small interfering RNA (siRNA) to suppress the expression of GRP78 in HT-29 and DLD-1 colon cancer cells. As shown in Fig. 1a, the levels of GRP78 were suppressed by over 80 % in GRP78-siRNA (GRP78KD) DLD-1 cells compared with scrambled control DLD-1 cells. To further evaluate the influence of GRP78 on colon cancer cell migration, Transwell migration, wound-healing, and invasion assays were performed. As shown in Fig. 1, we found that migratory cells were more prevalent among GRP78KD DLD-1 cells than among scrambled control DLD-1 cells (Fig. 1b, c). We further confirmed the migration assay results using the x’CELLigence biosensor system and found that knockdown of GRP78 enhanced the cell migration of DLD-1 cells (Fig. 2a); the difference was statistically significant (Fig. 2b).

Silencing GRP78 expression enhanced cell migratory ability in a Transwell migration assay system. GRP78 expression was suppressed using small interfering RNA (siRNA). GRP78-siRNA and scrambled control RNA were transfected into DLD-1 cells, and transfected cells were selected with antibiotics. a The levels of GRP78 in scrambled control and GRP78-knockdown (GRP78KD) cells were determined by western blotting. b The migratory ability of scrambled control and GRP78-knockdown (GRP78KD) cells was determined using a Transwell migration assay as described in the “Materials and methods” section. The migrated cells were observed and counted under a microscope

Silencing GRP78 expression was found to enhance the migratory ability of cells using the x’CELLigence biosensor system. The x’CELLigence biosensor system was used to evaluate cell migration ability in DLD-1 cells transfected with scrambled control and GRP78KD. Cell migration was assessed using specially designed 16-well plates (CIM-plate 16; Roche Diagnostics, GmbH) with 8-μm pores and carried out using the RTCA DP instrument (Roche Diagnostics GmbH, Germany). The CIM-plate 16 was monitored every 10 s for 40 min and then once every hour. Data analysis was carried out using RTCA software 1.2, which was supplied with the instrument. The Y-axis represents the cell index
GRP78 silencing enhanced the migratory ability of colon cancer cells in a wound-healing assay
We also used a wound-healing assay to observe the influence of GRP78 on the migration ability of colon cancer cells. The lower migration ability in HT-29 cells compared with DLD-1 cells was also observed (Fig. 3). After 24 h, the gap between the wound edges was smaller in GRP78KD DLD-1 cells than in scrambled control DLD-1 cells (21 vs. 57 %) (Fig. 3b). This phenomenon was also observed in HT-29 colon cancer cells (Fig. 3a). These results indicate that silencing GRP78 may enhance the wound-healing migration ability of colon cancer cells.

GRP78 mediated migration of HT-29 and DLD-1 cells in a wound-healing migration assay. GRP78 expression was suppressed using small interfering RNA (siRNA) in HT-29 and DLD-1 cells. A wound-healing migration assay was performed to compare the migratory ability between scrambled control and GRP78-knockdown (GRP78KD) cells in a HT-29 and b DLD-1 cells. DLD-1 or HT-29 cells transfected with scrambled control or GRP78 KD were seeded into a six-well plate and allowed to grow to 80 % confluence in RPMI 1640. Then, the cell monolayers were wounded using a 10-μL pipette tip. The wounded monolayers were washed four times with phosphate-buffered saline and incubated in RPMI 1640 for the indicated time periods
Suppression of GRP78 expression enhanced cells’ invasive ability
The process of metastasis involves several different stages. To further confirm the role of GRP78 in cancer metastasis, an invasion assay was performed. As shown in Fig. 4, the invasive ability was increased dramatically in GRP78KD DLD-1 cells compared with scrambled control cells. These results indicate that suppression of GRP78 may enhance cancer invasiveness. Taken together, our results demonstrate that silencing GRP78 enhances the invasiveness of colon cancer cells.

Knockdown of GRP78 expression enhanced the cells’ invasive ability. The BD BioCoat™ Matrigel Invasion Chamber (BD Biosciences) was applied to determine the invasiveness of cells. Scrambled control or GRP78 KD DLD-1 cells (1 × 105 cells each) were seeded into the upper compartments of each chamber. After incubation for 24 h, the non-migrating cells were removed from the upper surface of the membrane by scrubbing. Cells on the reverse side were stained with 0.1 % crystal violet, and migrated cells were counted under a microscope at ×100 magnification
Knockdown of GRP78 resulted in a change in epithelial-to-mesenchymal transition (EMT) marker expression and led to activation of the NRF-2/HO-1 pathway
To dissect the mechanism underlying the phenomena described above, epithelial-to-mesenchymal transition (EMT) markers were evaluated. After the successful suppression of GRP78 expression, E-cadherin expression was down-regulated without a change in N-cadherin levels. The expression of vimentin, a mesenchymal marker, was markedly up-regulated (Fig. 5a). A previous study demonstrated that silencing of GRP78 may cause an increase in NRF-2 expression, which is correlated with sensitivity to epirubicin [16]. It is known that NRF-2 plays a role in mediating cell migration ability [27]. We further evaluated the expression levels of NRF-2 and HO-1 in colon cancer cells. As shown in Fig. 5b, we found that the level of NRF-2 was increased dramatically after silencing GRP78. The expression of the downstream molecule HO-1 was also up-regulated after GRP78 knockdown in colon cancer cells. These results indicate that the knockdown of GRP78 enhanced colon cancer metastasis through alterations in EMT biomarker expression and the activation of the NRF-2/HO-1 pathway.

Silencing of GRP78 altered EMT biomarker expression and activated the NRF-2/HO-1 pathway. a The levels of E-cadherin and vimentin in scrambled control and GRP78-knockdown (GRP78KD) cells were determined by western blotting. GAPDH was used as the control. b The levels of NRF-2 and HO-1 in scrambled control and GRP78-knockdown (GRP78KD) cells were determined by western blotting. GAPDH was used as the control
GRP78 expression levels in primary and metastatic colonic adenocarcinomas
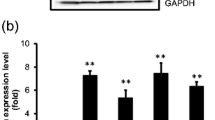
To better understand the role of GRP78 in colon cancers, we first compared the expression levels of GRP78 in normal colonic epithelia and primary colonic adenocarcinomas. Normal colonic glandular epithelial cells usually showed low or intermediate GRP78 expression scores (Fig. 6a). Primary colonic adenocarcinoma showed stronger GRP78 immunostaining compared to normal colonic epithelium (Fig. 6b). To clarify the role of GRP78 in colon cancer metastasis, GRP78 expression levels were compared between primary colon adenocarcinomas and lymph node metastases. GRP78 expression in metastatic adenocarcinoma in lymph nodes tended to be weaker than that in primary colonic adenocarcinoma (p < 0.01, Table 1, Fig. 6b, c). We further evaluated whether GRP78 expression levels were associated with differentiation of carcinoma cells. In the microarrays comprising low- and high-grade colonic adenocarcinomas, significantly stronger GRP78 expression was observed in low-grade adenocarcinomas (p < 0.01, Fig. 6d, e).

GRP78 expression in normal colonic mucosa, primary colonic adenocarcinoma, and metastatic colonic adenocarcinoma in lymph nodes. a Intermediate GRP78 expression in normal colonic epithelium. b High GRP78 expression in primary colonic adenocarcinoma. c Intermediate GRP78 expression in metastatic colonic adenocarcinoma in a lymph node. High-grade colonic adenocarcinoma (e) tended to have weaker GRP78 expression than low-grade colonic adenocarcinoma (d). Original magnification ×100
Discussion
Colorectal cancer is the third most common cancer among both men and women in the United States and is the second leading cause of cancer-related death [28]. According to statistical data from the Department of Health, colorectal cancer has become the most common cancer in Taiwan. In addition, it is also the third most commonly diagnosed malignancy in the world [29]. Colorectal cancer can be surgically cured when it is confined to a primary site, and metastasis is the main cause of death in colorectal cancer patients. In this study, we demonstrated that metastatic colon cancer cells had lower GRP78 expression than colon cancer cells at the primary site. With the down-regulation of GRP78 expression, colon cancer cells gained migration ability through EMT (Fig. 7).

A summary schematic diagram illustrating the role of GRP78 in the migration of colon cancer cells. Suppression of GRP78 expression may influence the EMT program (via a decrease in E-cadherin expression and increase in vimentin expression). In addition, the NRF-2/NO-1 pathway may be activated after GRP78 silencing. The migratory ability increases due to the enhancement of the EMT program and the activation of the NRF-2/NO-1 pathway after GRP78 suppression
A previous study found that GRP78 caused resistance to chemotherapy-induced apoptosis in colon cancer [16]. GRP78 down-regulators caused cell death in both glucose-deprived and etoposide-resistant HT-29 colon cancer cells, and it was found that the down-regulation of GRP78 suppressed colon cancer growth through decreased angiogenesis [13, 17, 30]. Although GRP78 typically localizes to the endoplasmic reticulum, cell surface-localized GRP78 is regarded as a possible anticancer treatment target [31]. GRP78 overexpression was noted in colon cancers, but the role of cell surface GRP78 in colon cancer metastasis has remained controversial [19, 20]. Interestingly, GRP78 expression was negatively correlated with lymphatic invasion in a study of human colorectal cancer specimens [18]. In this study, we found that the suppression of GRP78 in colon cancer cells increased their migration ability, increased vimentin expression, and decreased E-cadherin expression.
The epithelial-to-mesenchymal transition is a hallmark of cancer metastasis. E-cadherin is the major epithelial marker and is a key player in cell polarity and organization of the epithelium [32]; it is often decreased during the progression from adenoma to carcinoma and the development of tumor metastasis [33]. Down-regulation of E-cadherin was observed to accompany an increase in migration ability in colon cancers [34–36]. In this study, suppression of GRP78 was associated with the down-regulation of E-cadherin, which may contribute to enhanced migration ability.
Vimentin is an intermediate filament protein. All intermediate filaments share a common tripartite structure consisting of a highly conserved central α-helical rod domain and variable N-terminal head and C-terminal tail domains [37]. Focal contacts participate in the movement of cells on a substratum. The architecture of focal contacts is disturbed in vimentin-deficient fibroblasts, and vimentin regulates focal contacts and helps to stabilize cell–extracellular matrix (ECM) adhesions in endothelial cells; all of these observations support a role for vimentin in regulating focal adhesions [38, 39]. Vimentin is one epithelial-to-mesenchymal transition marker, and it has recently been considered to be a prerequisite for EMT induction [40]. Although the majority of colorectal cancers (53–84 %) carry an aberrantly methylated vimentin gene, vimentin expression may be increased in colon cancers in certain situations [41, 42]. In this study, the suppression of GRP78 was associated with up-regulation of vimentin, which may contribute to enhanced migration ability in colon cancer cells. Calcium homeostasis and influx have been widely reported to be involved in the mesenchymal transition. Calcium signals play a pivotal role in cancer cell migration and invasion [43, 44]. It is reported when increasing the concentration of cytosolic calcium would promote the process of cancer malignant migration through calcium-related proteins, such as Orail and STIM1 [45]. Chelating intracellular calcium would abolish EGF or hypoxia-induced EMT in breast cancer cells [46]. Previous studies have indicated that EGF can induce calcium signaling via store-operated calcium channels along with subsequent regulation of cell proliferation and migration [47–50]. In addition, KCNN4 channels participate in the EMT induced by PRL-3 in colorectal cancer [51]. However, the correlation between GRP78 and calcium influx remains unclear. It will be worthwhile to further dissect this relationship.
Heme oxygenase (HO) regulates the intracellular heme level by cleaving heme into carbon monoxide (CO), biliverdin, and free iron [52], and there are three HO isoforms: HO-1, HO-2, and HO-3. HO-1, as a heat shock protein-32, controls the rate-limiting step in the degradation of heme, and it is an inducible enzyme with relatively low expression in most tissues. HO-1 also serves as a defense mechanism against oxidative stress. Furthermore, HO-1 regulates cell proliferation, modulates the inflammatory response, and facilitates angiogenesis. Previous studies have revealed that HO-1 expression is increased in colon cancer tissue [53, 54]. HO-1 is a target gene of the basic leucine zipper (bZIP) transcription factor NRF-2, which regulates the adaptive response to oxidative stress in multicellular organisms. Under normal conditions, NRF-2 remains transcriptionally inactive in the cytoplasm, and its protein level is kept low through its degradation by the proteasome [55]. Previous reports demonstrated that increased NRF-2 expression is associated with poor prognosis in various cancers [56–58] and that NRF-2 expression increases chemoresistance in colon cancer [16, 59]. A recent study also found that colon cancers with higher levels of NRF-2 expression had an increased incidence of lymph node and distant metastases [60]. In this study, we demonstrate that the suppression of GRP78 increases NRF-2 and HO-1 protein levels in colon cancer cells (Fig. 7), which may contribute to their increased capacity for metastasis.
In conclusion, our work indicates that GRP78 may suppress colon cancer cell migration. GRP78 silencing can enhance the expression of vimentin and decrease the expression of E-cadherin in colon cancers. In addition, the expression levels of NRF-2 and HO-1 expression are elevated after the suppression of GRP78 expression. Our results provide further information about the mechanisms associated with the progression and regulation of colon cancers.
References
Munro S, Pelham HR. An Hsp70-like protein in the ER: identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell. 1986;46:291–300.
Haas IG. Bip (grp78), an essential hsp70 resident protein in the endoplasmic reticulum. Experientia. 1994;50:1012–20.
Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–51.
Pfaffenbach KT, Lee AS. The critical role of grp78 in physiologic and pathologic stress. Curr Opin Cell Biol. 2011;23:150–6.
Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006;6:45–54.
Fu Y, Li J, Lee AS. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007;67:3734–40.
Wang Q, He Z, Zhang J, Wang Y, Wang T, Tong S, et al. Overexpression of endoplasmic reticulum molecular chaperone grp94 and grp78 in human lung cancer tissues and its significance. Cancer Detect Prev. 2005;29:544–51.
Langer R, Feith M, Siewert JR, Wester HJ, Hoefler H. Expression and clinical significance of Glucose Regulated Proteins GRP78 (BiP) and GRP94 (GP96) in human adenocarcinomas of the esophagus. BMC Cancer. 2008;8:70.
Zheng HC, Takahashi H, Li XH, Hara T, Masuda S, Guan YF, et al. Overexpression of GRP78 and GRP94 are markers for aggressive behavior and poor prognosis in gastric carcinomas. Hum Pathol. 2008;39:1042–9.
Xing X, Lai M, Wang Y, Xu E, Huang Q. Overexpression of glucose-regulated protein 78 in colon cancer. Clin Chim Acta. 2006;364:308–15.
Bifulco G, Miele C, Di Jeso B, Beguinot F, Nappi C, Di Carlo C, et al. Endoplasmic reticulum stress is activated in endometrial adenocarcinoma. Gynecol Oncol. 2012;125:220–5.
Song MS, Park YK, Lee JH, Park K. Induction of glucose-regulated protein 78 by chronic hypoxia in human gastric tumor cells through a protein kinase c-epsilon/erk/ap-1 signaling cascade. Cancer Res. 2001;61:8322–30.
Park HR, Ryoo IJ, Choo SJ, Hwang JH, Kim JY, Cha MR, et al. Glucose-deprived HT-29 human colon carcinoma cells are sensitive to verrucosidin as a GRP78 down-regulator. Toxicology. 2007;229:253–61.
Chiou JF, Tai CJ, Huang MT, Wei PL, Wang YH, An J, et al. Glucose-regulated protein 78 is a novel contributor to acquisition of resistance to sorafenib in hepatocellular carcinoma. Ann Surg Oncol. 2010;17:603–12.
Lee AS. Grp78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 2007;67:3496–9.
Chang YJ, Huang YP, Li ZL, Chen CH. GRP78 knockdown enhances apoptosis via the down-regulation of oxidative stress and Akt pathway after epirubicin treatment in colon cancer DLD-1 cells. PLoS ONE. 2012;7:e35123.
Kuo LJ, Hung CS, Chen WY, Chang YJ, Wei PL. Glucose-regulated protein 78 silencing down-regulates vascular endothelial growth factor/vascular endothelial growth factor receptor 2 pathway to suppress human colon cancer tumor growth. J Surg Res 2013
Takahashi H, Wang JP, Zheng HC, Masuda S, Takano Y. Overexpression of GRP78 and GRP94 is involved in colorectal carcinogenesis. Histol Histopathol. 2011;26:663–71.
Hardy B, Raiter A, Yakimov M, Vilkin A, Niv Y. Colon cancer cells expressing cell surface GRP78 as a marker for reduced tumorigenicity. Cell Oncol (Dordr). 2012;35:345–54.
Li Z, Zhang L, Zhao Y, Li H, Xiao H, Fu R, et al. Cell-surface GRP78 facilitates colorectal cancer cell migration and invasion. Int J Biochem Cell Biol. 2013;45:987–94.
Wang M, Wey S, Zhang Y, Ye R, Lee AS. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid Redox Signal. 2009;11:2307–16.
Hirsch FR, Varella-Garcia M, Bunn Jr PA, Di Maria MV, Veve R, Bremmes RM, et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol. 2003;21:3798–807.
Chang YJ, Chiu CC, Wu CH, An J, Wu CC, Liu TZ, et al. Glucose-regulated protein 78 (GRP78) silencing enhances cell migration but does not influence cell proliferation in hepatocellular carcinoma. Ann Surg Oncol. 2010;17:1703–9.
Wei PL, Kuo LJ, Huang MT, Ting WC, Ho YS, Wang W, et al. Nicotine enhances colon cancer cell migration by induction of fibronectin. Ann Surg Oncol 2011
Lien YC, Wang W, Kuo LJ, Liu JJ, Wei PL, Ho YS, et al. Nicotine promotes cell migration through alpha7 nicotinic acetylcholine receptor in gastric cancer cells. Ann Surg Oncol 2011
Wei PL, Kuo LJ, Wang W, Lin FY, Liu HH, How T, et al. Silencing of glucose-regulated protein 78 (GRP78) enhances cell migration through the upregulation of vimentin in hepatocellular carcinoma cells. Ann Surg Oncol. 2012;19 Suppl 3:S572–9.
Pan H, Wang H, Zhu L, Mao L, Qiao L, Su X. The role of Nrf2 in migration and invasion of human glioma cell U251. World Neurosurg. 2013;80:363–70.
Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66.
Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108.
Hwang JH, Kim JY, Cha MR, Ryoo IJ, Choo SJ, Cho SM, et al. Etoposide-resistant HT-29 human colon carcinoma cells during glucose deprivation are sensitive to piericidin A, a GRP78 down-regulator. J Cell Physiol. 2008;215:243–50.
Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013;32:805–18.
Perez-Moreno M, Jamora C, Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. Cell. 2003;112:535–48.
Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for e-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190–3.
Wei PL, Chang YJ, Ho YS, Lee CH, Yang YY, An J, et al. Tobacco-specific carcinogen enhances colon cancer cell migration through alpha7-nicotinic acetylcholine receptor. Ann Surg. 2009;249:978–85.
Hur K, Toiyama Y, Takahashi M, Balaguer F, Nagasaka T, Koike J, et al. MicroRNA-200c modulates epithelial-to-mesenchymal transition (EMT) in human colorectal cancer metastasis. Gut 2012
Lu MH, Huang CC, Pan MR, Chen HH, Hung WC. Prospero homeobox 1 promotes epithelial–mesenchymal transition in colon cancer cells by inhibiting E-cadherin via miR-9. Clin Cancer Res. 2012;18:6416–25.
Eriksson JE, Dechat T, Grin B, Helfand B, Mendez M, Pallari HM, et al. Introducing intermediate filaments: from discovery to disease. J Clin Invest. 2009;119:1763–71.
Eckes B, Dogic D, Colucci-Guyon E, Wang N, Maniotis A, Ingber D, et al. Impaired mechanical stability, migration and contractile capacity in vimentin-deficient fibroblasts. J Cell Sci. 1998;111(Pt 13):1897–907.
Tsuruta D, Jones JC. The vimentin cytoskeleton regulates focal contact size and adhesion of endothelial cells subjected to shear stress. J Cell Sci. 2003;116:4977–84.
Ivaska J. Vimentin: Central hub in EMT induction? Small GTPases. 2011;2:51–3.
Forsyth CB, Tang Y, Shaikh M, Zhang L, Keshavarzian A. Alcohol stimulates activation of snail, epidermal growth factor receptor signaling, and biomarkers of epithelial–mesenchymal transition in colon and breast cancer cells. Alcohol Clin Exp Res. 2010;34:19–31.
Zhou J, Yang J, Li K, Mo P, Feng B, Wang X, et al. RhoE is associated with relapse and prognosis of patients with colorectal cancer. Ann Surg Oncol. 2013;20:175–82.
Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer. 2011;11:609–18.
Monet M, Lehen’kyi V, Gackiere F, Firlej V, Vandenberghe M, Roudbaraki M, et al. Role of cationic channel TRPV2 in promoting prostate cancer migration and progression to androgen resistance. Cancer Res. 2010;70:1225–35.
Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15:124–34.
Davis FM, Azimi I, Faville RA, Peters AA, Jalink K, Putney Jr JW, et al. Induction of epithelial–mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene. 2014;33:2307–16.
Ma R, Sansom SC. Epidermal growth factor activates store-operated calcium channels in human glomerular mesangial cells. Journal of the American Society of Nephrology : JASN. 2001;12:47–53.
Li WP, Tsiokas L, Sansom SC, Ma R. Epidermal growth factor activates store-operated Ca2+ channels through an inositol 1,4,5-trisphosphate-independent pathway in human glomerular mesangial cells. J Biol Chem. 2004;279:4570–7.
Chen YF, Chiu WT, Chen YT, Lin PY, Huang HJ, Chou CY, et al. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc Natl Acad Sci U S A. 2011;108:15225–30.
Yang IH, Tsai YT, Chiu SJ, Liu LT, Lee HH, Hou MF, et al. Involvement of STIM1 and Orai1 in EGF-mediated cell growth in retinal pigment epithelial cells. J Biomed Sci. 2013;20:41.
Lai W, Liu L, Zeng Y, Wu H, Xu H, Chen S, et al. KCNN4 channels participate in the EMT induced by PRL-3 in colorectal cancer. Med Oncol. 2013;30:566.
Maines MD. The heme oxygenase system: past, present, and future. Antioxid Redox Signal. 2004;6:797–801.
Becker JC, Fukui H, Imai Y, Sekikawa A, Kimura T, Yamagishi H, et al. Colonic expression of heme oxygenase-1 is associated with a better long-term survival in patients with colorectal cancer. Scand J Gastroenterol. 2007;42:852–8.
Kang KA, Maeng YH, Zhang R, Yang YR, Piao MJ, Kim KC, et al. Involvement of heme oxygenase-1 in Korean colon cancer. Tumour Biol. 2012;33:1031–8.
Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116.
Ma Q, He X. Molecular basis of electrophilic and oxidative defense: promises and perils of nrf2. Pharmacol Rev. 2012;64:1055–81.
Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1–NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420.
Shibata T, Kokubu A, Gotoh M, Ojima H, Ohta T, Yamamoto M, et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology 2008;135:1358–68, 1368 e1351-1354.
Akhdar H, Loyer P, Rauch C, Corlu A, Guillouzo A, Morel F. Involvement of Nrf2 activation in resistance to 5-fluorouracil in human colon cancer HT-29 cells. Eur J Cancer. 2009;45:2219–27.
Hu T, Yao Y, Yu S, Guo H, Han L, Wang W, et al. Clinicopathologic significance of CXCR4 and Nrf2 in colorectal cancer. J Biomed Res. 2013;27:283–90.
Acknowledgments
This study was supported by grants from the National Science Council (NSC101-2314-B-038-029-MY3 and NSC101-2314-B-038-016-MY3) and MOHW103-TDU-B-212-113001.
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chang, YJ., Chen, WY., Huang, CY. et al. Glucose-regulated protein 78 (GRP78) regulates colon cancer metastasis through EMT biomarkers and the NRF-2/HO-1 pathway. Tumor Biol. 36, 1859–1869 (2015). https://doi.org/10.1007/s13277-014-2788-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-014-2788-x