
Abstract
Background
Neurodegenerative diseases show various phenotypes of molecular and cellular malfunction including mitochondrial dysfunction and neuroinflammation. These molecular dynamics are based on the epigenetic regulation of the gene expression in the cells, which are vulnerable to progressive neurodegeneration. Histone deacetylases (HDAC) are the enzymes that remove acetyl group from histones or non-histone proteins for the transcriptional control. Thus, HDAC inhibitors (HDACi) have been proposed as prominent drugs for neurodegenerative diseases.
Objectives
In this study, we explain the molecular targets of the HDACi in the processes of neurodegeneration and neuroprotection.
Results
Treatment with HDACi altered the expression of specific genes that are associated with mitochondrial bioenergetics and neuroinflammation.
Conclusions
Mitochondrial bioenergetics- and neuroinflammation-related molecular targets of HDACi may be the key to the use of HDACi therapy for neurodegenerative diseases.
Purpose of review
We aimed to discover molecular targets of HDACi in progressive neurodegeneration and to use these targets in potential therapeutics to induce neuroprotection.
Recent findings
HDACi reverse cellular pathology in a mechanism involving mitochondrial bioenergetics and neuroinflammation, and the result is alleviation of pathologic phenotypes of neurodegenerative diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) are progressively developed with multifactorial pathogenic mechanisms and complex molecular dynamics. The pathogenic factors include abnormal protein processing, mitochondrial dysfunction, and neuroinflammation; these are closely associated with aging (Wang et al. 2019; Yan et al. 2020b). None of the neurodegenerative diseases are currently curable, and the limited available treatments only manage the symptoms. Because neurodegenerative diseases impose significant personal burdens and social costs, it is important to discover the patho-physiological mechanisms and potential therapeutic targets of these diseases.
Recent studies have shown that neuroinflammation and bioenergetics are linked to the progressive pathology of neurodegenerative diseases (Lin and Beal 2006). Neuroinflammation is induced by activation of microglia and astrocytes by molecules released from damaged cells in the neurodegeneration process. Through neuroinflammation, proinflammatory cytokines and chemokines are released peripherally and into the brain. At the molecular level, neuroinflammation is mainly dependent on the redox status of glial cells; reactive oxygen species (ROS) are produced by microglia after microglial activation (Kierdorf and Prinz 2013). Abnormal activation of microglia also leads to the production of ROS, proinflammatory cytokines, and related functional proteins that lead to the neurodegeneration process. An abnormal increase of ROS production affects intracellular redox status and induction of the expression of proinflammatory cytokines (Rojo et al. 2014). ROS are generated as the result of mitochondrial action, and mitochondria respond to ROS induced by the other associated cellular mechanisms (Handy and Loscalzo 2012). Thus, mitochondrial bioenergetics has been suggested as an emerging therapeutic target in neurodegenerative diseases. Mitochondria generate more than 90% of the body’s energy in the form of ATP. This occurs by the electron transport process that utilizes mitochondrial respiratory complexes I–IV for oxidative phosphorylation (OXPHOS). Mitochondrial dysfunction is associated with alteration of neuro-metabolic coupling and neural functional networking. This explains the high susceptibility of neurons to the bioenergetic deficits observed in neurodegenerative diseases (Uittenbogaard and Chiaramello 2014). The entire pathological neurodegenerative process including neuroinflammation and mitochondrial dysfunction can be explained by failure of the proper regulation of specific genes in the degenerating vulnerable neurons. For example, Yang et al., suggested that controlling mitochondrial bioenergetics reduced ROS production by histone modification (Yang et al. 2019). In addition, previous study showed that mitochondrial homeostasis by histone modification improved neuronal function (Weyemi et al. 2019).
Histone modification regulates chromatin dynamics and gene accessibility by covalent attachment or detachment of certain histone tail functional groups. Histone acetylation by histone acetyltransferase (HAT) and histone deacetylation by histone deacetylase (HDAC) are important epigenetic regulation steps for directing specific gene expression (Fig. 1). HDAC changes the affinity of histones for DNA, increasing accessibility of transcription factors to the cis-regulatory elements of DNA (Icardi et al. 2012; Rousseaux and Khochbin 2015). Abnormal regulation of these processes is implicated in the cellular mechanisms of a variety of disease processes, and control of HDAC-associated gene expression by new neurodegenerative disease therapies is a focus of scientific investigation (Huang et al. 2023; Zhou et al. 2023). HDAC inhibitors (HDACi) are potential therapies for treatment of neurodegenerative diseases, and some HDACi that target multiple classes or specific types of HDAC have been reported to improve the pathological symptoms of neurological diseases (Chuang et al. 2009; Gupta et al. 2020).

Functions of histone acetylation and deacetylation in transcriptional regulation
Identifying the molecular targets of the HDACi to understand the cellular processes that overcome disease symptoms is a particularly important area of research. In this review, we address the main targets of small molecule inhibitors of HDACs in neurodegeneration and neuroprotection, focusing on neuroinflammation and bioenergetics.
Histone deacetylases (HDACs) as epigenetic controllers in neurodegeneration and neuroprotection
The neurodegenerative disease process is progressive; neuronal function declines over time and neurons ultimately die (Lamptey et al. 2022). Emerging research has uncovered that epigenetic alterations are a potential molecular target for treatment of neurodegenerative diseases (Berson et al. 2018; Coppede 2014). HDAC is an important epigenetic regulation step for the specific gene expression pattern for neural function (Fig. 1). HDACs remove acetyl groups from histone and non-histone proteins (Seto and Yoshida 2014), leading to changes in gene expression by triggering the chromatin de-condensation process. Deacetylation of histone or non-histone proteins have been studied in many cellular processes (Hubbert et al. 2002).
Eighteen types of HDACs have been identified in mammals. These are classified into four categories depending on primary sequence homology and structural similarities to yeast proteins (Fig. 2) (Yang and Seto 2008). Class I HDACs (HDAC1, 2, 3, and 8) show a similar sequence to the yeast transcription regulator Rpd3, and class II HDACs have similar sequences to the yeast Hda1 protein. Class I HDACs are found in ubiquitously in human tissues and mainly reside in the nucleus. Class I, II, and IV HDACs have a variety of histone deacetylase functions, activities, and substrate specificities. Class I HDACs have an entirely conserved deacetylase domain compared to the other classes. Class II HDACs are subdivided into two subclasses, Class IIa (HDAC4, 5, 7, and 9), and class IIb (HDAC6 and 10), depending on their domain compositions. Class IIa HDACs have unique N-terminal domains that regulate binding affinity and locations. Class IIb HDACs contain characteristically long c-terminal domains that consist of a second deacetylase domain and a zinc finger ubiquitin binding domain (HDAC6) or leucine-rich repeat domain (HDAC10). HDAC11, which has similar homology to both class I and class II HDACs, is in a separate class, class IV. Class I, II, and IV HDACs are the classical HDACs and have common zinc-dependent catalytic mechanisms that can be the targets of their inhibitors (Zhang et al. 2018a). In contrast, class III HDACs have nicotinamide adenine dinucleotide (NAD+)-dependent sites with similar sequences to the yeast Sir2 protein also known as Sir2-like protein (sirtuins); SIRT1–SIRT7 are included in this class.

Classification of histone deacetylases (HDAC) with their molecular structures and inhibitors
HDAC inhibitors (HDACi): structure and function
HDAC inhibitors (HDACi) are small natural or synthetic chemical compounds that inhibit the enzyme activities of HDACs. HDACi usually consist of a metal-binding moiety or functional group, a capping group and a linker. Metal-binding moieties promote the binding of catalytic metals to the active site of the HDAC enzymes. The capping group interacts with the amino acid of the lysine binding site, and a linker with a structural similarity interacts with the carbon chain in the acetyl-lysine substrate that connects the metal-binding moiety and capping groups (Shukla and Tekwani 2020). HDACi have different chemical structures, including hydroxamic acids, cyclic peptides, electrophilic ketones, short-chain fatty acids, benzamides, boronic acid-based compounds, benzofuranone- and sulfonamide-containing molecules, and α/β peptide structures. The hydroxamic acid class of HDACi generally have common structural characteristics like an opposing capping group; zinc-binding moiety (ZBM) in the catalytic pocket; and straight-chain alkyl, vinyl, or aryl linkers (Marks 2010).
The potential of HDACi as an effective treatment for neurodegenerative diseases by causing in hyperacetylation of histones and activation and/or inhibition of the expression of various gene subsets has been discussed (Choudhary et al. 2009; Lin et al. 2006). SAHA (vorinostat) is an HDACi that structurally belongs to the group of hydroxamic acids that potently inhibit class I HDACs (HDAC 1, 2, and 3), class II HDACs (HDAC6 and 7) and the class IV HDAC (HDAC11). CI-994 (tacedinaline) is a benzamide that inhibits class I HDACs (Zhang et al. 2018b). Short-chain fatty acids HDACi, sodium butyrate (SB) inhibits class I and IIa HDACs and readily cross the BBB (Park and Sohrabji 2016). RGFP109 is a specific HDAC1 and 3 (class I) inhibitor that also penetrates the BBB with potential neuroprotective effects (Paraskevopoulou et al. 2021). ACY-738 is potent, selective HDAC 6 inhibitor that also has the ability to penetrate the BBB (Burg et al. 2021). Tubacin selectively targets HDAC6 and deacetylation of α-tubulin. MS-275 (entinostat) is a potent benzamide HDACi for class I HDACs (Fig. 2).
Functional recovery by HDACi under a neurodegeneration
Previous studies have shown that HDACi administration improved neural function. SAHA administration improved spatial reference memory in aged mice and APP/PS1-21 mice compared to the vehicle group (Benito et al. 2015). ACY-738 treatment restored histone acetylation and attenuated the ALS disease phenotype in wild-type FUS mice, which presented with hypoacetylation of histones (Rossaert et al. 2019). CI-994, a class I HDACi, enhanced synaptic plasticity, increased transcription of some learning-related genes in the hippocampus, and maintained long-term memory retention in contextual fear conditioning (CFC) (Burns et al. 2022). Administration of SB alleviated the neurodegenerative phenotype of presenilin-1 and -2 conditional double knockout (cDKO) mice, improved contextual memory deficits, enhanced neonatal neuronal differentiation in the cortex of cDKO mice, and significantly reduced phosphorylated tau. MS-275, an HDAC1 inhibitor, improved cognitive decline of AD mice. Valproic acid (VPA) administration significantly improved PD-like motor behaviors in LRRK2R1441G transgenic mice (Kim et al. 2019).
Sung et al. demonstrated that W2, a mercaptoacetamide-based class II HDACi, significantly decreased mRNA levels of γ-secretase components (Psen1, Psen2, and Nicastrin) and increased Mmp2 expression that is involved in Aβ degradation in an AD model of primary cortical neurons. I2, a hydroxyamide-based class I and II HDACi, significantly repressed expression of β-secretase (BACE1) and γ-secretase components (Nicastrin and Psen2) and upregulated Aβ degradation-related expression of genes such as Nep, Ece1, Mmp2, Ctsd, and Ctsb (Sung et al. 2013).
Molecular targets of HDACi in neuroinflammation
Neuroinflammation is a key pathological feature of neurological disorders and is mediated by central nervous system (CNS) immune cells, microglia and astrocytes. AD model 5xFAD mice showed decreased protein levels of proinflammatory cytokines (TNF-α, IL-6, and IL-1β) and decreased phosphorylated NF-κB level (Jiang et al. 2021). Previous studies suggested the functions and molecular targets of HDACi in the cellular mechanism of neuroinflammation. For example, SB, an HDACi for class I and IIa HDACs, alleviated the abnormal expression of genes related to immune and inflammatory responses in cDKO mice. SB also decreased the expression of GFAP, a marker of inflammation in the cortex and hippocampus of cDKO mice. Also, SB treatment rescued dysregulation of acetylated histone 3 (H3Ac) expression in cDKO mice (Cao et al. 2018) and reduced proinflammatory cytokine production, including production of TNF-a, IL-6, and IL-1β, in 5xFAD AD mice (Jiang et al. 2021). In another study, SB inhibited the expression of S100a9 and Ccl4 in cDKO mice (Cao et al. 2018). In microarray and RT-PCR analysis, the expression level of chemokine receptor 6 (Ccr6), which is critical in trafficking of T helper 17 (Th17) cells to the CNS, was decreased with HD and increased with HDACi 4b NKL22 treatment, a benzamide-type HDAC inhibitor that targets HDAC1 and HDAC3 (Thomas et al. 2008). MS-275, an HDAC1 inhibitor, induced cognitive decline of AD APP/PS1 mice, and reduced the mRNA expression levels of IL-1β, iNOS, and TNFα in LPS-induced, activated RAW 264.7 cells (Zhang and Schluesener 2013). Benito et al. (2015) used a genome-wide approach to investigate the molecular basis of HDACi therapy and found that CLK1 (CDC-like kinase1) is downregulated in an aged mouse model and is fully restored after SAHA administration. CLK1 is a protein kinase essential for phosphorylation of serine/arginine-rich (SR) proteins that are involved in gene splicing regulation. The four CLK family members CLK1/STY, CLK2, CLK3, and CLK4 are commonly used as a target for diseases caused by gene mis-splicing. Cao et al. (2018) have found that SB treatment inhibited the upregulation of inflammation-related genes such as S100a9 (S100 calcium binding protein A9), GFAP, and Ccl4 in the brains of cDKO mice. Ccl4, inflammatory chemokine (C–C motif) ligand 4, is a gene that encodes a mitogen-inducible monokine. Those encoded proteins are involved in inflammation/immune processes and cytokine-cytokine receptor interactions. Compound 106, a derivative of the HDACi 4b inhibitor (NKL22), induced negative regulation of Rnf35, a member of the tripartite motif (TRIM) protein family, in ataxia model KIKI mice. Compound 106 also decreased antigen-binding and immunoglobulin receptor binding activity with Igh-6, an immunoglobulin heavy locus and positive regulator of B cell proliferation (Rai et al. 2008). In 6-OHDA mice and 6-OHDA-treated neuroblastoma SH-SY5Y cells, tubastatin A, an HDAC6 inhibitor, decreased the inflammatory cytokines and inflammasome related gene expression including that of NLRP3, IL-18, IL-1β and IL-6 (Yan et al. 2020a) (Fig. 3).

Molecular mechanisms of HDACi functions in neuroinflammation and bioenergetics
Molecular targets of HDACi in bioenergetic metabolism
Accumulating evidence indicates that dysregulated lipid metabolism and altered cholesterol homeostasis have strong associations with protein aggregation and progression of neurodegeneration (Estes et al. 2021). After treatment with SB, VPA, and TSA, the CYP46A1 gene involved in cerebral cholesterol catabolism was significantly upregulated (Nunes et al. 2010). The CYP46A1 gene encodes cholesterol 24-hydroxylase that is involved in a major mechanism of conversion of cholesterol into 24S-hydroxycholesterol to achieve cholesterol clearance in neurons (Lund et al. 1999). Hudry et al. (2010) demonstrated that overexpression of the CYP46A1 gene before or after the onset of amyloid plaques significantly reduced Aβ pathology in a mouse model of AD. TSA treatment also altered cholesterol metabolism in neuronal cells by upregulating the expression of ABCA1 genes, that involved cholesterol efflux and by downregulating the expression of HMGCS, HMGCR, MVK and LDLR that are involved in cholesterol synthesis and uptake (Nunes et al. 2013).
The effects of HDAC inhibition on lipid metabolism were also studied using non-targeted liposome analysis combined with transcriptomic analysis in the spinal cords of FUS mice. Abnormal levels of glycerophospholipids, sphingolipids, and cholesterol esters, were found in FUS mouse model compared with normal mice. Abnormal alterations in these lipids were completely or partially rescued by ACY-738 in FUS mice. ACY-738 treatment significantly restored transcription factors that are associated with lipid metabolism in FUS mice (Burg et al. 2021). ACY-738 treatment partially restored reduced levels of genes associated with fatty acid and cholesterol biosynthesis (7-Dhcr, Sqle) and β-oxidation (Acsl6, Acad11), and upregulated mRNA expression levels of genes associated with glycolysis, the pentose phosphate pathway (PPP) (Hk2, Pdk4), and lipid transport (Apod, Apoe, Fabp4) (Burg et al. 2021). ACY-738 treatment significantly induced downregulation of Pla1ax, Pla2g3, Pla2g4e, and Pnpla7, and upregulation of Pla2g4a transcription factors in FUS mice. The elevated levels of cholesterol esters and dysregulation of phospholipids is observed in ALS FUS mouse model. Pla1ax, Pla2g3, Pla2g4a, Pla2g4e, and Pnpla7 are genes that encode phospholipases that function in lipid metabolism. ACY-738 administration partially restored the expression of metabolic genes and substantially saved metabolic disorders in FUS mice. (Rossaert et al. 2019). ACY-738 also significantly induced downregulation of the transcription factor expression of Ppara, Pparg, Rxra, Srebf1, and Tfap2b and upregulation of the expression of Srebf2, another transcription factor; these transcription factors are all involved in lipid metabolism (Burg et al. 2021). Peroxisome proliferator-activated receptor (PPAR) isoforms including PPARG, PPARA, and PPARD belong to a nuclear receptor family. PPARG has the highest expression in the central nervous system and functions as a lipid sensor throughout the body. Each member of the PPAR family regulates a unique subset of genes responsible for lipid and carbohydrate energy metabolism and also regulates a variety of cellular processes such as Aβ degradation and mitochondrial activation (Khan et al. 2019; Rudko et al. 2020). ACY-738 also significantly repressed the expression of Kruppel-like factor 5 (KLF5) and nuclear respiratory factor 1 (NRF1). KLF transcriptional factors regulate various biological processes, including cell proliferation and differentiation and axon growth of CNS neurons in vitro and in vivo (Moore et al. 2009). Previous studies have shown that KLF5 has an important function in controlling metabolism at the cellular, tissue, and systemic levels (Oishi and Manabe 2018). The NRF1 gene encodes a homodimeric transcription factor that activates the expression of several key metabolic genes that regulate mitochondrial DNA transcription and replication (Fig. 3).
Molecular targets of HDACi in other mechanisms of neurodegeneration
HDAC inhibition has been reported to alter other pathological processes including the function of transcription factors. Using RNA-sequencing (RNA-seq) and chromatin immunoprecipitation-sequencing (ChIP-seq) technologies, previous studies reported that SAHA partially restored physiological gene expression and improved synaptic function and memory formation in aged mice. SAHA completely or partially restored the expression of genes associated with RNA processing that were downregulated in the aged CA1 region of APP/PS1-21 mice (Benito et al. 2015). SAHA rescued downregulation of genes involved in functional pathways related to synaptic plasticity in the CA1 region neurons (Hanson et al. 2013). Recently, Benito et al. used the GWAS approach to investigate the molecular basis of HDACi therapy and found that genes associated with RNA processing and splicing were affected in CA1 neurons of the aged mice. Downregulation of these genes is associated with decreased H4K12ac level in neuron. After SAHA administration, some of these genes such as CLK1, Srsf10, Srsf7, Tra2a, and Srsf3, were fully restored (Benito et al. 2015). CI-994, a selective inhibitor of HDACs 1, 2, and 3, enhances synaptic plasticity, increases transcription of some learning-related genes in the hippocampus, and maintains long-term memory retention. When comparing all enhancer acetylation and bulk and single-nucleus RNA sequencing transcriptional change datasets, the results suggested that the main epigenetic and transcriptional targets of HDACi therapy may be the genes involved in synaptic communication and MAPK signaling (Burns et al. 2022).
In transcriptomic analysis of HD and ataxia model mice, Scn11a, voltage-gated sodium channels, were upregulated in both diseases, and downregulated by HDACi (HDACi 4b and compound 106). The mRNA expression level of chloride intracellular channel 6 (Clic6) was decreased in HDACi 4b treated HD mice (Thomas et al. 2008). CI-994, a class I HDACi, combined with CFC functions as a molecular facilitator for the synaptic and intracellular communication including Kcna1 (voltage-gated potassium channels) (Burns et al. 2022). RGF-109, a selective inhibitor of HDAC1 and 3 improved motor control and reduced transcriptional dysregulation in HD mice including that of Neurod2 and Nr4a2 in R6/1 HD model mice (Hecklau et al. 2021). HDAC6 modulates amyloid beta-induced cognitive dysfunction in rats by regulating protein tyrosine kinase 2 beta (PTK2B), a tyrosine kinase involved in calcium-induced regulation of ion channels (Liu et al. 2020).
RGFP966, HDAC3 inhibitor, was administered to HD model HdhQ7/Q11 knock-in mice and decreased the H3K9ac in the hippocampus and striatum. Arc (activity-regulated cytoskeleton-associated protein) and Nr4a2 (nuclear receptor 4A2; Nurr1) expression levels in hippocampus were detected by RT-PCR and western blot. The transcriptional levels of Arc and Nr4a2 were decreased in knock-in mice, and these gene expression levels in the RGFP966-treated group were indistinguishable from those of wild-type mice (Suelves et al. 2017). HDAC inhibitor 4b increased the level of Mobp, an actin-binding and myelin-processing protein, that is decreased in HD mice. SB alleviated the neurodegenerative phenotype of cDKO mice, enhanced neonatal neuronal differentiation in the cortex, and significantly reduced the level of phosphorylated tau in cDKO mice (Cao et al. 2018). HDACi 4b inhibitor significantly increased frataxin (FXN) mRNA expression level in the cerebellum, cortex, and striatum of R6/2 HD mice (Herman et al. 2006; Lai et al. 2019; Thomas et al. 2008).
Conclusion
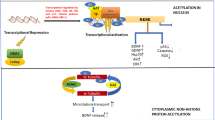
An understanding of the progressive pathology of neurodegenerative diseases requires an understanding of the epigenetic regulation of the gene expression in specific cells in the nervous system. HDACs remove acetyl groups from histones to provide access to DNA for transcription. In this review, we focused on the molecular targets of HDACs in disease development with the assumption that knowledge of these targets may be useful in the development of new neurodegenerative disease therapies. Many small molecules that regulate HDACs have been the focus of neurodegeneration and neuroprotection research. HDACi reverse cellular pathology by modulating mitochondrial bioenergetics and neuroinflammation, leading to alleviation of neurodegenerative diseases (Fig. 4). However, the functional mechanisms of these small molecules are not fully understood yet. For instance, the efficiency of blood–brain barrier (BBB) penetration of these small molecules cannot be fully explained for the neuroprotective effects in the progressive neurodegeneration. As previously reported, HDACi showed differential levels of BBB crossing with region-specific changes of rodent histone acetylation (Hooker et al. 2010). In conclusion, the molecular targets of HDACi modulate the neuroinflammation and bioenergetics including lipid metabolism. HDACi may be neuroprotective and may have potential as therapies for neurodegenerative disorders.

Understanding of the pathological mechanism of neurodegenerative diseases focusing on neuroinflammation and bioenergetic failure and the potential therapeutic candidates for neurodegeneration
Data availability
All data are available in the original article.
Abbreviations
- HDAC:
-
Histone deacetylase
- HDACi:
-
Histone deacetylase inhibitor
- AD:
-
Alzheimer’s disease
- PD:
-
Parkinson’s disease
- HD:
-
Huntington’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- ROS:
-
Reactive oxygen species
- ATP:
-
Adenosine triphosphate
- OXPHOS:
-
Oxidative phosphorylation
- HAT:
-
Histone acetyltransferase
- NAD+ :
-
Nicotinamide adenine dinucleotide
- ZBM:
-
Zinc-binding moiety
- SAHA:
-
Suberoylanilide hydroxamic acid (vorinostat)
- SB:
-
Sodium butyrate
- BBB:
-
Blood–brain barrier
- GWAS:
-
Genome-wide association study
- VPA:
-
Valproic acid
- CFC:
-
Contextual fear conditioning
- TSA:
-
Trichostatin A
- CNS:
-
Central nervous system
- ChIP-seq:
-
Chromatin immunoprecipitation-sequencing
- 6-OHDA:
-
6-Hydroxydopamine
References
Benito E et al (2015) HDAC inhibitor-dependent transcriptome and memory reinstatement in cognitive decline models. J Clin Investig 125:3572–3584. https://doi.org/10.1172/JCI79942
Berson A, Nativio R, Berger SL, Bonini NM (2018) Epigenetic regulation in neurodegenerative diseases. Trends Neurosci 41:587–598. https://doi.org/10.1016/j.tins.2018.05.005
Burg T et al (2021) Histone deacetylase inhibition regulates lipid homeostasis in a mouse model of amyotrophic lateral sclerosis. Int J Mol Sci 22:11224. https://doi.org/10.3390/ijms222011224
Burns AM et al (2022) The HDAC inhibitor CI-994 acts as a molecular memory aid by facilitating synaptic and intracellular communication after learning. Proc Natl Acad Sci USA 119:e2116797119. https://doi.org/10.1073/pnas.2116797119
Cao T et al (2018) Histone deacetylase inhibitor alleviates the neurodegenerative phenotypes and histone dysregulation in presenilins-deficient mice. Front Aging Neurosci 10:137. https://doi.org/10.3389/fnagi.2018.00137
Choudhary C et al (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325:834–840. https://doi.org/10.1126/science.1175371
Chuang DM et al (2009) Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci 32:591–601. https://doi.org/10.1016/j.tins.2009.06.002
Coppede F (2014) The potential of epigenetic therapies in neurodegenerative diseases. Front Genet 5:220. https://doi.org/10.3389/fgene.2014.00220
Estes RE, Lin B, Khera A, Davis MY (2021) Lipid metabolism influence on neurodegenerative disease progression: is the vehicle as important as the cargo? Front Mol Neurosci 14:788695. https://doi.org/10.3389/fnmol.2021.788695
Gupta R, Ambasta RK, Kumar P (2020) Pharmacological intervention of histone deacetylase enzymes in the neurodegenerative disorders. Life Sci 243:117278. https://doi.org/10.1016/j.lfs.2020.117278
Handy DE, Loscalzo J (2012) Redox regulation of mitochondrial function. Antioxid Redox Signal 16:1323–1367. https://doi.org/10.1089/ars.2011.4123
Hanson JE, La H, Plise E, Chen Y-H, Ding X, Hanania T, Sabath EV, Alexandrov V, Brunner D, Leahy E, Steiner P, Liu L, Scearce-Levie K, Zhou Q (2013) SAHA enhances synaptic function and plasticity in vitro but has limited brain availability in vivo and does not impact cognition. PLoS ONE 8(7):e69964. https://doi.org/10.1371/journal.pone.0069964
Hecklau K et al (2021) The effects of selective inhibition of histone deacetylase 1 and 3 in huntington’s disease mice. Front Mol Neurosci 14:616886. https://doi.org/10.3389/fnmol.2021.616886
Herman D et al (2006) Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat Chem Biol 2:551–558. https://doi.org/10.1038/nchembio815
Hooker JM et al (2010) Histone deacetylase inhibitor, MS-275, exhibits poor brain penetration: PK studies of [C]MS-275 using Positron Emission Tomography. ACS Chem Neurosci 1:65–73. https://doi.org/10.1021/cn9000268
Huang J, Chen X, Lin H, Chen X (2023) β-Hydroxybutyrate impairs nasopharyngeal carcinoma cell aggressiveness via histone deacetylase 4 inhibition. Mol Cell Toxicol. https://doi.org/10.1007/s13273-023-00378-7
Hubbert C et al (2002) HDAC6 is a microtubule-associated deacetylase. Nature 417:455–458. https://doi.org/10.1038/417455a
Hudry E et al (2010) Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer’s disease. Mol Ther 18:44–53. https://doi.org/10.1038/mt.2009.175
Icardi L, De Bosscher K, Tavernier J (2012) The HAT/HDAC interplay: multilevel control of STAT signaling. Cytokine Growth Factor Rev 23:283–291. https://doi.org/10.1016/j.cytogfr.2012.08.002
Jiang Y et al (2021) Sodium butyrate ameliorates the impairment of synaptic plasticity by inhibiting the neuroinflammation in 5XFAD mice. Chem Biol Interact 341:109452. https://doi.org/10.1016/j.cbi.2021.109452
Khan MA et al (2019) Current progress on peroxisome proliferator-activated receptor gamma agonist as an emerging therapeutic approach for the treatment of Alzheimer’s disease: an update. Curr Neuropharmacol 17:232–246. https://doi.org/10.2174/1570159x16666180828100002
Kierdorf K, Prinz M (2013) Factors regulating microglia activation. Front Cell Neurosci 7:44. https://doi.org/10.3389/fncel.2013.00044
Kim T et al (2019) HDAC inhibition by valproic acid induces neuroprotection and improvement of PD-like behaviors in LRRK2 R1441G transgenic mice. Exp Neurobiol 28:504–515. https://doi.org/10.5607/en.2019.28.4.504
Lai JI et al (2019) Transcriptional profiling of isogenic Friedreich ataxia neurons and effect of an HDAC inhibitor on disease signatures. J Biol Chem 294:1846–1859. https://doi.org/10.1074/jbc.RA118.006515
Lamptey RNL et al (2022) A review of the common neurodegenerative disorders: current therapeutic approaches and the potential role of nanotherapeutics. Int J Mol Sci. https://doi.org/10.3390/ijms23031851
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795. https://doi.org/10.1038/nature05292
Lin HY et al (2006) Targeting histone deacetylase in cancer therapy. Med Res Rev 26:397–413. https://doi.org/10.1002/med.20056
Liu Z, Hao KM, Wang HY, Qi WX (2020) Histone deacetylase-6 modulates amyloid beta-induced cognitive dysfunction rats by regulating PTK2B. NeuroReport 31:754–761. https://doi.org/10.1097/WNR.0000000000001481
Lund EG, Guileyardo JM, Russell DW (1999) cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci USA 96:7238–7243. https://doi.org/10.1073/pnas.96.13.7238
Marks PA (2010) The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs 19:1049–1066. https://doi.org/10.1517/13543784.2010.510514
Moore DL et al (2009) KLF family members regulate intrinsic axon regeneration ability. Science 326:298–301. https://doi.org/10.1126/science.1175737
Nunes MJ et al (2010) Sp proteins play a critical role in histone deacetylase inhibitor-mediated derepression of CYP46A1 gene transcription. J Neurochem 113:418–431. https://doi.org/10.1111/j.1471-4159.2010.06612.x
Nunes MJ et al (2013) Histone deacetylase inhibition decreases cholesterol levels in neuronal cells by modulating key genes in cholesterol synthesis, uptake and efflux. PLoS ONE 8:e53394. https://doi.org/10.1371/journal.pone.0053394
Oishi Y, Manabe I (2018) Krüppel-like factors in metabolic homeostasis and cardiometabolic disease. Front Cardiovasc Med 5:69. https://doi.org/10.3389/fcvm.2018.00069
Paraskevopoulou F et al (2021) Impaired inhibitory GABAergic synaptic transmission and transcription studied in single neurons by Patch-seq in Huntington’s disease. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.2020293118
Park MJ, Sohrabji F (2016) The histone deacetylase inhibitor, sodium butyrate, exhibits neuroprotective effects for ischemic stroke in middle-aged female rats. J Neuroinflamm 13:300. https://doi.org/10.1186/s12974-016-0765-6
Rai M et al (2008) HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS ONE 3:e1958. https://doi.org/10.1371/journal.pone.0001958
Rojo AI et al (2014) Redox control of microglial function: molecular mechanisms and functional significance. Antioxid Redox Signal 21:1766–1801. https://doi.org/10.1089/ars.2013.5745
Rossaert E et al (2019) Restoration of histone acetylation ameliorates disease and metabolic abnormalities in a FUS mouse model. Acta Neuropathol Commun 7:107. https://doi.org/10.1186/s40478-019-0750-2
Rousseaux S, Khochbin S (2015) Histone Acylation beyond acetylation: terra incognita in chromatin biology. Cell J 17:1–6. https://doi.org/10.22074/cellj.2015.506
Rudko OI, Tretiakov AV, Naumova EA, Klimov EA (2020) Role of PPARs in progression of anxiety: literature analysis and signaling pathways reconstruction. PPAR Res 2020:8859017. https://doi.org/10.1155/2020/8859017
Seto E, Yoshida M (2014) Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol 6:a018713. https://doi.org/10.1101/cshperspect.a018713
Shukla S, Tekwani BL (2020) Histone deacetylases inhibitors in neurodegenerative diseases, neuroprotection and neuronal differentiation. Front Pharmacol 11:537. https://doi.org/10.3389/fphar.2020.00537
Suelves N, Kirkham-McCarthy L, Lahue RS, Gines S (2017) A selective inhibitor of histone deacetylase 3 prevents cognitive deficits and suppresses striatal CAG repeat expansions in Huntington’s disease mice. Sci Rep 7:6082. https://doi.org/10.1038/s41598-017-05125-2
Sung YM et al (2013) Mercaptoacetamide-based class II HDAC inhibitor lowers Abeta levels and improves learning and memory in a mouse model of Alzheimer’s disease. Exp Neurol 239:192–201. https://doi.org/10.1016/j.expneurol.2012.10.005
Thomas EA et al (2008) The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci USA 105:15564–15569. https://doi.org/10.1073/pnas.0804249105
Uittenbogaard M, Chiaramello A (2014) Mitochondrial biogenesis: a therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr Pharm Des 20:5574–5593. https://doi.org/10.2174/1381612820666140305224906
Wang Y, Xu E, Musich PR, Lin F (2019) Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci Ther 25:816–824. https://doi.org/10.1111/cns.13116
Weyemi U et al (2019) Histone H2AX promotes neuronal health by controlling mitochondrial homeostasis. Proc Natl Acad Sci USA 116:7471–7476. https://doi.org/10.1073/pnas.1820245116
Yan S et al (2020a) Pharmacological inhibition of HDAC6 attenuates NLRP3 inflammatory response and protects dopaminergic neurons in experimental models of Parkinson’s disease. Front Aging Neurosci 12:78. https://doi.org/10.3389/fnagi.2020.00078
Yan X et al (2020b) Abnormal mitochondrial quality control in neurodegenerative diseases. Front Cell Neurosci 14:138. https://doi.org/10.3389/fncel.2020.00138
Yang XJ, Seto E (2008) The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 9:206–218. https://doi.org/10.1038/nrm2346
Yang J et al (2019) HDAC inhibition induces autophagy and mitochondrial biogenesis to maintain mitochondrial homeostasis during cardiac ischemia/reperfusion injury. J Mol Cell Cardiol 130:36–48. https://doi.org/10.1016/j.yjmcc.2019.03.008
Zhang ZY, Schluesener HJ (2013) Oral administration of histone deacetylase inhibitor MS-275 ameliorates neuroinflammation and cerebral amyloidosis and improves behavior in a mouse model. J Neuropathol Exp Neurol 72:178–185. https://doi.org/10.1097/NEN.0b013e318283114a
Zhang L et al (2018a) Zinc binding groups for histone deacetylase inhibitors. J Enzyme Inhib Med Chem 33:714–721. https://doi.org/10.1080/14756366.2017.1417274
Zhang S, Fujita Y, Matsuzaki R, Yamashita T (2018b) Class I histone deacetylase (HDAC) inhibitor CI-994 promotes functional recovery following spinal cord injury. Cell Death Dis 9:460. https://doi.org/10.1038/s41419-018-0543-8
Zhou J, Feng X, Wang D (2023) HDAC3 deacetylates H3K27ac and H3K9ac on the TrkC promoter to exacerbate sevoflurane-induced neurotoxicity. Mol Cell Toxicol. https://doi.org/10.1007/s13273-023-00394-7
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) funded by Korea Ministry of Science and ICT (MSIT), and Institute of Information & Communications Technology Planning & Evaluation (IITP) (NRF-2022R1A2C1011996, NRF-2022K1A3A1A20015190, RS-2023-00302751, 2020-0-01343).
Author information
Authors and Affiliations
Contributions
YP and HS designed the study. YP, SY, SYH, and HS wrote the paper. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare that they have no conflicts of interest. Yeongwon Park declares that she has no conflicts of interest. Shangfei Yu declares that she has no conflicts of interest. Seung Yong Hwang declares that he has no conflicts of interest. Hyemyung Seo declares that she has no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Park, Y., Yu, S., Hwang, S. et al. Molecular targets of histone deacetylase inhibitors in neurodegeneration and neuroprotection. Mol. Cell. Toxicol. (2024). https://doi.org/10.1007/s13273-024-00441-x
Accepted:
Published:
DOI: https://doi.org/10.1007/s13273-024-00441-x