
Abstract
Background
Sevoflurane (Sev) is a widely used general anesthetic that can cause neurotoxicity. Histone acetylation is a vital epigenetic mechanism responding to environmental stresses.
Objective
This study was conducted to analyze the role of histone deacetylase 3 (HDAC3) in Sev-induced neurotoxicity and provide a theoretical reference for the treatment of anesthetic neurotoxicity.
Results
HDAC3 was upregulated in Sev-treated HT-22 cells. Inhibition of HDAC3 upregulated cell viability and downregulated apoptosis, oxidative stress and secretion of inflammatory cytokines. HDAC3 reduced H3K27ac and H3K9ac levels on the TrkC promoter to inhibit TrkC expression. TrkC downregulation reversed the alleviative role of si-HDAC3 in Sev-induced neurotoxicity.
Conclusion
HDAC3 enhanced Sev-induced neurotoxicity by reducing H3K27ac and H3K9ac to repress TrkC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Volatile anesthetics are extensively used for surgical operations and considered to be clinically safe for most of the time. Sevoflurane (Sev) is a volatile general anesthetic tailored for inhalational induction to induce anesthesia both in pediatric and adult populations (De Hert and Moerman 2015). It owns its popularity due to rapid anesthetic induction, lack of airway irritation, and good pharmacokinetic performance (Apai et al. 2021). However, accumulating evidence has verified that Sev is neurotoxic to damage the function and structure of the brain, especially in infants (Sun et al. 2022). Sev has deleterious effects on neuronal plasticity, hippocampal neural survival, and neurocognitive performance dependent on exposure time and dose, which is documented in rats (Xiao et al. 2016; Zhou et al. 2016). Moreover, it has also been found to trigger apoptosis, oxidative stress (OS), and inflammation in human neuroglioma cells (Liu et al. 2020). However, the exact molecular pathway by which Sev induces neurotoxicity remains elusive and warrants further exploration.
Histone acetylation, one common form of epigenetic modifications, is mediated by histone acetyltransferases (HATs) and histone deacetylases (HDACs). Acetylated histones are often less compact and more accessible to RNA polymerase and the transcriptional machinery, thus activating the transcription of target genes, while histone deacetylation results in transcriptional repression because of chromatin compaction (Chelladurai et al. 2021). Essentially, histone acetylation and deacetylation by HATs and HDACs significantly involve the regulation of neurodevelopment, brain function, and neurodegenerative diseases (Gupta et al. 2021). For instance, ablation of HDAC2 and HDAC3 is correlated with increased mRNA levels of brain-derived neurotrophic factor (BDNF) and improved memory (Sartor et al. 2019); HDAC3 and HDAC6 are identified as possible regulators of neurotoxicity in the case of ischemic stroke (Chen et al. 2012). Especially, Sev exerts neurotoxicity by downregulating hippocampal HDAC2 and HDAC3, consequently impairing spatial learning and memory of pregnant rats (Yu et al. 2020). However, growing evidence has revealed that HDAC3 exerts both beneficial and damaging functions in the brain (D’Mello 2020). Small interfering RNAs (siRNAs) are a group of double-stranded RNA molecules, 20–25 base pairs in length, which take action within the RNAi pathway to interfere with the transcribed mRNA of a specific targeted gene and prevent its translation. Therefore, we used siRNAs to intervene HDAC3 expression and further observe cell behaviors and physiological and biochemical changes after gene intervention.
On another note, tyrosine kinase (Trk) receptors, including TrkA, TrkB and TrkC, have a high affinity with multiple neurotrophins, such as nerve growth factor, BDNF, and neurotrophin-3/4, thus regulating the homeostasis of the nervous system (Reichardt 2006). TrkB and TrkC are shown to promote the growth of cholinergic neurons to protect against Alzheimer’s disease (Gonzalez et al. 2022). TrkA and TrkB are identified to play an alleviative role in bupivacaine-induced neuronal apoptosis and neurite loss (Guo et al. 2017). In particular, TrkC overexpression protects against neurotoxic damage by Sev in human induced pluripotent stem cell-derived neurons (Zhang et al. 2020a, b). Moreover, TrkC can be elevated by HDAC inhibitor (Dedoni et al. 2021), suggesting that HDAC3 could deacetylate TrkC. In light of the aforementioned associations, we speculated that HDAC3 mediates histone deacetylation of TrkC to regulate Sev-induced neurotoxicity. The primary objective of our study is to decipher the molecular route of HDAC3 in Sev-induced neurotoxicity and provide a new rationale for the management of anesthetic neurotoxicity.
Materials and methods
Cell culture
Mouse hippocampal neurons HT-22 were supplied by Cell Bank of Chinese Academy of Sciences (Shanghai, China) and cultured in a T25 flask containing Dulbecco’s modified Eagle medium/F12 (Thermo Fisher Scientific, Waltham, MA, USA) incorporated with 10% fetal bovine serum (Thermo Fisher Scientific) and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA) under a condition of 5% CO2 and 37℃. The medium was replaced twice a week.
Cell treatment
Cells were seeded in the 6-well plates at a density of 2 × 104 cells/well. With regard to Sev treatment, HT-22 cells were placed in a gas-tight chamber and exposed to 1%, 2%, and 4% Sev for 6 h respectively, with the control cells cultured in the equivalent condition without Sev (Zhang et al. 2020a, b). Small interfering RNA (siRNA) negative control (si-NC) and siRNAs targeting HDAC3 and TrkC (si-HDAC3-1, si-HDAC3-2, si-TrkC-1, and si-TrkC-2) were purchased from Genepharma (Shanghai, China). Based on the manufacturer’s protocol, the above RNAs (80 pmol) were transfected into HT-22 cells using Lipofectamine 3000 (Invitrogen). After 48 h, transfection efficiency was tested, followed by Sev treatment. RGFP966 (inhibitor of HDAC3) was provided by MedChemExpress Co., Ltd. (Monmouth Junction, NJ, USA) and was added to the medium at a concentration of 10 μM (Louis Sam Titus et al. 2019) and was cultured with Sev exposure, with the equal volume of dimethylsulfoxide (DMSO) as the control. The sequence information of used siRNAs were as follow: si-NC, SS: CAAAGUGUUGACUAUGGGACA, AS: TGTCCCATAGTCAACACTTTGGGG; si-HDAC3-1, SS: CGGUGUUGGACAUAUGAAACA, AS: UUUCAUAUGUCCAACACCGGG; si-HDAC3-2, SS: GAUGAUGAUGUGUAUAAUAAA, AS: UAUUAUACACAUCAUCAUCUG; si-TrkC-1, SS: GGCUGAGUGCUACAAUCUAAG, AS: UAGAUUGUAGCACUCAGCCAG; si-TrkC-2, SS: GCUGAGUGCUACAAUCUAAGC, AS: UUAGAUUGUAGCACUCAGCCA.
Cell counting kit-8 (CCK-8) assay
Cell viability was tested by means of the CCK-8 assay (Beyotime, Jiangsu, China). HT-22 cells were seeded in the 96-well plates (5 × 103cells/well). After Sev exposure, 10 μL of CCK-8 reagent was incorporated and cultured at 37℃ for 2 h, upon which the optical density at a wavelength of 450 nm was measured using a microplate reader (Bio-Rad 680, Bio-Rad, Hercules, CA, USA).
Terminal-deoxynucleoitidyl transferase mediated Nick end labeling (TUNEL)
The apoptosis of HT-22 cells was assessed using the TUNEL apoptosis assay kit (Beyotime Institute of Biotechnology, Shanghai, China). Simply put, 3 × 104 HT-22 cells were seeded in the 6-well plates, treated with Sev, fixed with 4% paraformaldehyde for 30 min, and incubated with 100 μL TUNEL determination reagent in dark for 1 h. After treatment with anti-fluorescence quencher, a fluorescence microscope (BX43, Olympus, Tokyo, Japan) was used to observe stained cells.
Reactive oxygen species (ROS) assay
ROS contents in cells were quantified using a DCFH2-DA fluorescence probe (S0033S, Beyotime). In brief, 3.5 × 104 HT-22 cells were seeded in the 12-well plates, exposed to Sev, and loaded with 10 μM DCFH2-DA for 45 min. Following these steps, HT-22 cells were washed and detached by trypsin, and resuspended in the buffer. Afterwards, HT-22 cells were observed using a fluorescence microscope (BX43, Olympus, Tokyo, Japan), with ImageJ software (NIH, Bethesda, MD, USA) for quantification.
Lactate dehydrogenase (LDH), superoxide dismutase (SOD), and malondialdehyde (MDA) assays
HT-22 cells (5 × 105) undergoing different treatments were harvested and lysed using cell lysis buffer (Beyotime). The supernatant was obtained after centrifugation and the contents of LDH, SOD, and MDA in the supernatant were determined using the LDH cytotoxicity assay kit (BC0680, Solarbio, Beijing, China), SOD assay kit (A001-3–2, Nanjing Jiancheng Bioengineering Institute, Nanjing, China), and MDA assay kit (A003-1–2¸ Nanjing Jiancheng Bioengineering Institute) respectively according to the manufacturer’s instructions.
Enzyme-linked immunosorbent assay (ELISA)
According to the manufacturer’s instructions (R&D System, Inc., Minneapolis, MN, USA), cells (5 × 105) were lysed at 4℃. After centrifugation, the levels of tumor necrosis factor (TNF)-α (MTA00B), interleukin (IL)-1β (MLB00C), and IL-6 (M6000B) were determined in equal amounts of supernatant.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Through treatment with the TRIzol reagent (Invitrogen), the total RNA was isolated from HT-22 cells, and 1 ng RNA was reverse-transcribed into the complementary DNA using SuperScript IV reverse transcriptase (Thermo Fisher Scientific). qRT-PCR was conducted using Taqman Universal PCR Master mix II (Thermo Fisher Scientific). The operation conditions were as follow: 5 min of pre-denaturation at 95 ℃, 15 s of denaturation at 95 ℃, and 30 s of annealing at 60 ℃. With glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the internal reference, the relative expression amount was quantified based on the 2−ΔΔCt method (Livak and Schmittgen 2001). Primers are shown in Table 1.
Western blot assay
HT-22 cells were lysed with radioimmunoprecipitation assay lysis buffer containing the mixture of proteinase inhibitor. After centrifugation, the protein concentration was tested using the bicinchoninic acid protein determination kit (P0011, Beyotime). The equal amount of protein (30 μg) was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis and transference onto polyvinylidene fluoride membranes. After a blockade with non-fat milk, the membrane incubation was performed with antibodies against HDAC3 (1:2000, ab137704, Abcam), TrkC (1:1000, ab240651, Abcam), β-actin (1:5000, ab8227, Abcam) and with the secondary horse radish peroxidase-coupled immunoglobulin G (IgG) antibody (1:1000, ab6721, Abcam). Protein signals were visualized using the enhanced chemiluminescence solution (P0018, Beyotime).
Chromatin immunoprecipitation (ChIP)
According to the protocol of the ChIP assay kit (Merck, Burlington, MA, USA), HT-22 cells (5 × 105) with different treatments were treated with 37% formaldehyde to fix the crosslink of DNA and protein, and were lysed with the lysis buffer, and were ultrasonically processed to shear DNA. The overnight incubation of DNA fragments was conducted with antibodies against HDAC3 (1:1000, ab137704, Abcam), acetylated lysine 27 on histone H3 (H3K27ac, 1:1000, ab4729, Abcam), H3K9ac (1:500, ab32129, Abcam), IgG (1:1000, ab6728, Abcam), followed by purification of DNA. The quantification analysis of purified DNA was conducted by PCR. Primers of the promoter are shown in Table 1.
Statistical analysis
Data analysis was performed with help of SPSS 21.0 software (SPSS, Inc, Chicago, IL, USA). Data were normally distributed as tested by Kolmogorov–Smirnov and presented as mean ± standard deviation. The t test was adopted for pairwise comparisons and one-way/two-way analysis of variance (ANOVA) was employed for multi-group comparisons, with Tukey’s multiple comparison test used for pairwise comparisons after ANOVA. P values were obtained from the two-sided test, and P < 0.05 denoted statistical significance.
Result
HDAC3 is upregulated in Sev-induced HT-22 cells
Under exposure to 1%, 2%, and 4% Sev, the viability of HT-22 cells was gradually decreased (P < 0.05, Fig. 1A). HDAC3 expression levels in HT-22 cells were determined and were found to be increased with the increases in Sev concentration (P < 0.05, Fig. 1B, C). Then, 4% Sev was selected for the subsequent assays. The apoptosis rate of HT-22 cells was significantly increased under exposure to 4% Sev (P < 0.05, Fig. 1D), and cytotoxic marker LDH levels were notably elevated (P < 0.05, Fig. 1E).

HDAC3 is upregulated in Sev-induced HT-22 cells. HT-22 cells were exposed to 0%, 1%, 2%, and 4% Sev. A: Cell viability was tested by the CCK-8 assay; B, C: HDAC3 expression levels were determined by qRT-PCR and Western blot assay; 4% Sev was selected for the subsequent assays; D: Apoptosis was tested by TUNEL staining; E: LDH levels in cells were determined by the assay kit. Cell experiments were performed 3 times independently. * P < 0.05. Data in panels A, B, and C were analyzed by one-way ANOVA, with Tukey’s multiple comparison test used for pairwise comparisons after ANOVA. Data in panels D and E were analyzed by the t test
Inhibition of HDAC3 alleviates Sev-induced neurotoxicity
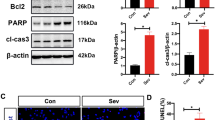
To explore the role of HDAC3 in Sev-induced neurotoxicity, two strands of siRNAs targeting HDAC3 were synthesized and transfected into HT-22 cells. According to the transfection efficiency, si-HDAC3-1 was selected for the subsequent assays (P < 0.05, Fig. 2A, B). Then, transfected cells were exposed to 4% Sev. Compared with the si-NC group, the viability of HT-22 cells was elevated (P < 0.05, Fig. 2C), apoptosis rate was decreased (P < 0.05, Fig. 2D), and cytotoxic marker LDH levels were reduced (P < 0.05, Fig. 2E) after HDAC3 downregulation. The above results illustrated that inhibition of HDAC3 alleviated Sev-induced neurotoxicity.

Inhibition of HDAC3 alleviates Sev-induced neurotoxicity. HT-22 cells were transfected with si-HDAC3-1 and si-HDAC3-2, with si-NC as the control. A: Transfection efficiency was tested by qRT-PCR; si-HDAC3-1 with better transfection effectiveness was selected for the subsequent assays, and transfected cells were exposed to 4% Sev; B: HDAC3 expression levels were determined by Western blot assay; C: Cell viability was assessed by the CCK-8 assay; D: Apoptosis was tested by TUNEL staining; E: LDH levels in cells were determined by the assay kit. Cell experiments were performed 3 times independently. * P < 0.05. Data were analyzed by one-way ANOVA, with Tukey’s multiple comparison test used for pairwise comparisons after ANOVA
Inhibition of HDAC3 alleviates Sev-induced OS and inflammation in neurons
Meanwhile, OS in HT-22 cells was determined. Based on our results, Sev exposure triggered OS in HT-22 cells by increasing ROS and MDA levels and decreasing SOD levels (P < 0.05, Fig. 3A, B), and promoted the secretion of inflammatory cytokines (TNF-α, IL-1β, and IL-6) (P < 0.05, Fig. 3C). After inhibition of HDAC3, OS was moderated (P < 0.05, Fig. 3A, B), and the secretion of inflammatory cytokines (TNF-α, IL-1β, and IL-6) was reduced (P < 0.05, Fig. 3C).

Inhibition of HDAC3 alleviates Sev-induced OS and inflammation in neurons. A: ROS levels were determined using a DCFH2-DA probe; B: SOD and MDA levels were determined by the assay kits; C: Levels of TNF-α, IL-1β, and IL-6 were determined by ELISA. Cell experiments were performed 3 times independently. *P < 0.05. Data were analyzed by one-way ANOVA, with Tukey’s multiple comparison test used for pairwise comparisons after ANOVA
HDAC3 inhibits H3 acetylation on the TrkC promoter to repress TrkC expression
To provide insights into the downstream mechanism of HDAC3, we firstly analyzed the enrichment of HDAC3 on the TrkC promoter and found that HDAC3 was abundantly enriched on the TrkC promoter (P < 0.05, Fig. 4A, B), and the enrichment was increased with the increases in Sev concentration (P < 0.05, Fig. 4A) and was decreased upon inhibition of HDAC3 (P < 0.05, Fig. 4B). Furthermore, Sev treatment was found to notably reduce the enrichment of H3K27ac and H3K9ac on the TrkC promoter (P < 0.05, Fig. 4A, B). Consistent with the results caused by HDAC3 inhibitor (RGFP966; with the addition of DMSO as the control), the enrichment of H3K27ac and H3K9ac were markedly augmented in si-HDAC3-tarnsfected cells (P < 0.05, Fig. 4B). In addition, the expression levels of TrkC were found to be inhibited by Sev treatment and increased after inhibition of HDAC3 (P < 0.05, Fig. 4C, D). The above results elicited that HDAC3 inhibited the enrichment of H3K27ac and H3K9ac on the TrkC promoter to repress TrkC expression.

HDAC3 inhibits the enrichment of H3K27ac and H3K9ac on the TrkC promoter to repress TrkC expression. A, B: Enrichments of HDAC3, H3K27ac, and H3K9ac on the TrkC promoter in HT-22 cells with different treatments were determined by the ChIP assay; cells were incorporated with HDAC3 inhibitor RGFP966, with DMSO as the control; C, D: TrkC expression levels in HT-22 cells with different treatments were determined by qRT-PCR and Western blot assay. Cell experiments were performed 3 times independently. *P < 0.05. Data in panels A and B were analyzed by two-way ANOVA and data in panels C and D were analyzed by one-way ANOVA, with Tukey’s multiple comparison test used for pairwise comparisons after ANOVA
TrkC downregulation reverses the alleviative role of HDAC3 inhibition in Sev-induced neurotoxicity
Eventually, to validate the above mechanism, HT-22 cells were transfected with two strands of si-TrkC (P < 0.05, Fig. 5A, B). si-TrkC-1 was found to have the better silencing effectiveness and as a result was selected for combined treatment with si-HDAC3-1. The result showed that after TrkC downregulation, cell viability was decreased (P < 0.05, Fig. 5C), apoptosis rate was elevated (P < 0.05, Fig. 5D), levels of LDH, ROS, and MDA were elevated, SOD levels were reduced (P < 0.05, Fig. 5E, F), and the secretion of inflammatory cytokines was intensified (P < 0.05, Fig. 5G). The above findings suggested that TrkC downregulation reversed the alleviative role of HDAC3 inhibition in Sev-induced neurotoxicity.

TrkC downregulation reverses the alleviative role of HDAC3 inhibition in Sev-induced neurotoxicity. HT-22 cells were transfected with si-TrkC-1 and si-TrkC-2, with si-NC as the control. A: Transfection efficiency was tested by qRT-PCR; si-TrkC-1 with better silencing effectiveness was selected for combined treatment with si-HDAC3-1, and transfected cells were exposed to 4% Sev; B: TrkC expression levels were determined by Western blot assay; C: Cell viability was assessed by the CCK-8 assay; D: Apoptosis was tested by TUNEL staining; E: ROS levels were determined using a DCFH2-DA probe; F: LDH, SOD and MDA levels were determined by the assay kits; G: Levels of TNF-α, IL-1β, and IL-6 were determined by ELISA. Cell experiments were performed 3 times independently. *P < 0.05. Data were analyzed by one-way ANOVA, with Tukey’s multiple comparison test used for pairwise comparisons after ANOVA
Discussion
Sevoflurane (Sev) is a popular general anesthetic commonly used in pediatric surgery but its excessive exposure can induce neurotoxicity that would damage neuronal growth and cognitive performance (Sun 2022). Histone acetylation is a vital epigenetic mechanism responding to environmental stresses. Sev is known to repress histone acetylation (acetylated H3 and H4) in the hippocampus, reducing the expression of synaptic proteins (Chai et al. 2022), while HDAC inhibitor sodium butyrate can reverse the inhibition of neurogenesis caused by Sev (Jia et al. 2020). However, the mechanism of HDAC3-mediated histone deacetylation in Sev-induced neurotoxicity remains unknown. Our study is the first to explore the role of HDAC3/TrkC in Sev-induced neurotoxicity, providing a theoretical reference to overcome Sev-induced neurotoxicity (Fig. 6).

Molecular route of sevoflurane-induced neurotoxicity. Sevoflurane promotes HDAC3 to deacetylate H3K27ac and H3K9ac on the TrkC promoter, thereby repressing TrkC expression and exacerbating neurotoxicity
Deficiency of HDAC3 in neurons and glia is detrimental for proper brain development, promoting astrogliogenesis while reducing oligodendrocytes (Norwood et al. 2014). On the other hand, the aberrant expression of HDAC3 results in brain injuries and cognitive dysfunction. For instance, HDAC3 is a contributor to ischemia/reperfusion-induced neuroinflammation and brain injury (Liao et al. 2020); HDAC3 in dorsal hippocampus is likely to induce postoperative cognitive dysfunction (Yang et al. 2022); HDAC3 overexpression in the hippocampus can increase amyloid burden and impair spatial memory in mice with Alzheimer’s disease (Zhu et al. 2017). Most importantly, Sev has been previously shown to upregulate HDAC3 levels in the hippocampus and HDAC3 can promote Sev-induced neurotoxicity by degrading the BDNF signaling (Yu, 2020). Likewise, HDAC3 downregulation is associated with alleviation of isoflurane-induced neuronal apoptosis and cognitive dysfunction caused by paeonol (Jin et al. 2020). When exposed to toxic stimulation, the plasma membrane ruptures and LDH is rapidly released outside cells (Parhamifar et al. 2019). Therefore, LDH serves as the most widely used marker of cytotoxicity. According to our results, Sev exposure augmented HDAC3 expression levels in a concentration-dependent manner and increased LDH levels, while HDAC3 downregulation inhibited Sev-induced neurotoxicity as manifested by increased cell viability, decreased apoptosis, and reduced LDH levels.
OS and inflammation are two key events that activate neurotoxicity (Oztas et al. 2019). OS occurs as a result of an imbalance between ROS production and the activity of the cellular antioxidant system, leading to lipid peroxidation (e. g. MDA) and inhibited activity of enzymatic antioxidants (e. g. SOD) (Islam 2017). SOD and MDA are commonly used as markers of OS. TNF-α, IL-1β, and IL-6 are pro-inflammatory cytokines detectable in hippocampal neurons (Liu et al. 2018). The effects of Sev on OS and inflammation impact its safety in surgery (Lee et al. 2015). HDAC3 knockdown can decrease OS in cortical neurons by inactivating the c-Abl/MST1/YAP pathway (Yu et al. 2018). Besides, HDAC3 can coordinate with nuclear factor-kappa B to induce brain inflammation in subacute Alzheimer’s disease rat model (Yu et al. 2021). In accordance, we found that Sev exposure increased ROS and MDA levels while decreased SOD levels and elevated the secretion of TNF-α, IL-1β, and IL-6, whereas HDAC3 inhibition reduced OS and inflammation. Therefore, HDAC3 inhibitor may be a potential adjuvant of Sev in surgery to reduce neurotoxicity, OS, and inflammation and increase anesthetic safety.
As a HDAC, HDAC3 can induce histone deacetylation to repress gene transcription. Trk receptors-mediated neurotrophic trafficking plays a role in neurogenesis (Scott-Solomon and Kuruvilla 2018). The BDNF/Trk/Akt pathway participates in general anesthesia-induced neuronal apoptosis (Lu et al. 2006). A previous study has elucidated that the TrkC activity is more dynamic in Sev-induced pluripotent stem cell-derived neurons compared to TrkA and TrkB and protects against neurotoxicity (Zhang, 2020). Moreover, TrkC upregulation is associated with the use of HDAC inhibitor and H3 acetylation (Dedoni, 2021). Our experimentation revealed that the TrkC promoter presented the enrichment of HDAC3, indicating an affinity between them. Then, si-HDAC3 and HDAC3 inhibitor (RGFP966) were shown to increase the enrichment of H3K27ac and H3K9ac on the TrkC promoter. The expression levels of TrkC were uncovered to be diminished by Sev treatment and elevated as a result of silencing HDAC3. These results suggested that HDAC3 can inhibit TrkC expression by exerting HDAC activity to reduce the enrichment of H3K27ac and H3K9ac. Eventually, TrkC was silenced by si-TrkC-1, upon which cell viability was reduced while apoptosis, LDH levels, OS, and inflammation were enhanced. Overall, these evidences suggested that TrkC may be the predominant target of HDAC3 to enhance Sev-induced neurotoxicity.
To conclude, our findings unraveled that 1) under the condition of 6 h treatment with 4% Sev, HDAC3 mRNA and protein levels were increased by about one fold in HT-22 cells, and H3K27ac and H3K9ac on the TrkC promoter were reduced by about half and TrkC expression was reduced to 1/3; 2) after transfection with si-HDAC3, HDAC3 expression was decreased by three folds, and in response to si-HDAC3 and 4% Sev treatment, the H3K27ac and H3K9ac levels on the TrkC promoter were increased by three folds, and TrkC expression was increased by about one fold; 3) transfection with si-HDAC3 reduced Sev-induced neurotoxicity. However, our study was limited to the cellular level, with experiments only conducted on HT-22 cell line. Meanwhile, we only analyzed TrkC and other downstream targets of HDAC3 in HT-22 cells were not clear. In the future, we will employ more animal species and other neuron types and establish the animal models to validate our mechanism.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Apai C, Shah R, Tran K, Pandya Shah S (2021) Anesthesia and the developing brain: a review of sevoflurane-induced neurotoxicity in pediatric populations. Clin Ther 43:762–778
Chai G et al (2022) Sevoflurane inhibits histone acetylation and contributes to cognitive dysfunction by enhancing the expression of ANP32A in aging mice. Behav Brain Res 431:113949
Chelladurai P et al (2021) Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br J Pharmacol 178:54–71
Chen YT et al (2012) Expression patterns of histone deacetylases in experimental stroke and potential targets for neuroprotection. Clin Exp Pharmacol Physiol 39:751–758
D’Mello SR (2020) Histone deacetylase-3: Friend and foe of the brain. Exp Biol Med (maywood) 245:1130–1141
De Hert S, Moerman A (2015) Sevoflurane. F1000Res 4: 626
Dedoni S, Marras L, Olianas MC, Ingianni A, Onali P (2021) The neurotrophin receptor trkc as a novel molecular target of the antineuroblastoma action of valproic acid. Int J Mol Sci 22:7790
Gonzalez S et al (2022) Small molecule modulation of TrkB and TrkC neurotrophin receptors prevents cholinergic neuron atrophy in an Alzheimer’s disease mouse model at an advanced pathological stage. Neurobiol Dis 162:105563
Guo J et al (2017) Antidepressant imipramine protects bupivacaine-induced neurotoxicity in dorsal root ganglion neurons through coactivation of TrkA and TrkB. J Cell Biochem 118:3960–3967
Gupta R, Ambasta RK, Kumar P (2021) Histone deacetylase in neuropathology. Adv Clin Chem 104:151–231
Islam MT (2017) Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res 39:73–82
Jia J et al (2020) The Role of Histone acetylation in the sevoflurane-induced Inhibition of neurogenesis in the hippocampi of young mice. Neuroscience 432:73–83
Jin H et al (2020) Paeonol attenuates isoflurane anesthesia-induced hippocampal neurotoxicity via modulation of JNK/ERK/P38MAPK pathway and regulates histone acetylation in neonatal rat. J Matern Fetal Neonatal Med 33:81–91
Lee YM, Song BC, Yeum KJ (2015) Impact of volatile anesthetics on oxidative stress and inflammation. Biomed Res Int 2015:242709
Liao Y et al (2020) HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics 10:9644–9662
Liu M, Li M, Zhou Y, Zhou Q, Jiang Y (2020) HSP90 inhibitor 17AAG attenuates sevoflurane-induced neurotoxicity in rats and human neuroglioma cells via induction of HSP70. J Transl Med 18:166
Liu Y et al (2018) Galantamine improves cognition, hippocampal inflammation, and synaptic plasticity impairments induced by lipopolysaccharide in mice. J Neuroinflammation 15:112
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408
Louis Sam Titus ASC, Sharma D, Kim MS, D’Mello SR (2019) The Bdnf and Npas4 genes are targets of HDAC3-mediated transcriptional repression. BMC Neurosci 20:65
Lu LX, Yon JH, Carter LB, Jevtovic-Todorovic V (2006) General anesthesia activates BDNF-dependent neuroapoptosis in the developing rat brain. Apoptosis 11:1603–1615
Norwood J, Franklin JM, Sharma D, D’Mello SR (2014) Histone deacetylase 3 is necessary for proper brain development. J Biol Chem 289:34569–34582
Oztas E, Abudayyak M, Celiksoz M, Ozhan G (2019) Inflammation and oxidative stress are key mediators in AKB48-induced neurotoxicity in vitro. Toxicol in Vitro 55:101–107
Parhamifar L, Andersen H, Moghimi SM (2019) Lactate dehydrogenase assay for assessment of polycation cytotoxicity. Methods Mol Biol 1943:291–299
Reichardt LF (2006) Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci 361:1545–1564
Sartor GC et al (2019) Enhancement of BDNF expression and memory by HDAC inhibition requires BET bromodomain reader proteins. J Neurosci 39:612–626
Scott-Solomon E, Kuruvilla R (2018) Mechanisms of neurotrophin trafficking via Trk receptors. Mol Cell Neurosci 91:25–33
Sun M, Xie Z, Zhang J, Leng Y (2022) Mechanistic insight into sevoflurane-associated developmental neurotoxicity. Cell Biol Toxicol 38:927–943
Xiao H, Liu B, Chen Y, Zhang J (2016) Learning, memory and synaptic plasticity in hippocampus in rats exposed to sevoflurane. Int J Dev Neurosci 48:38–49
Yang L et al (2022) HDAC3 of dorsal hippocampus induces postoperative cognitive dysfunction in aged mice. Behav Brain Res 433:114002
Yu L et al (2018) Lentivirus-mediated HDAC3 inhibition attenuates oxidative stress in APPswe/PS1dE9 mice. J Alzheimers Dis 61:1411–1424
Yu X, Yu W, Wu L, Yang W, Lu Y (2021) Chitotriosidase attenuates brain inflammation via HDAC3/NF-kappaB pathway in D-galactose and aluminum-induced rat model with cognitive impairments. Neurosci Res 172:73–79
Yu Z et al (2020) Enriched environment improves sevoflurane-induced cognitive impairment during late-pregnancy via hippocampal histone acetylation. Braz J Med Biol Res 53:e9861
Zhang J, Chen Z, Luo X, Yang Z (2020a) TrkC overexpression protects sevoflurane-induced neurotoxicity in human induced pluripotent stem cell-derived neurons. Dev Neurosci 42:105–113
Zhang N, Wang D, Yang X, Hou D (2020b) Long noncoding RNA small nucleolar RNA host gene 1 contributes to sevoflurane-induced neurotoxicity through negatively modulating microRNA-181b. NeuroReport 31:416–424
Zhou X et al (2016) Dose-dependent effects of sevoflurane exposure during early lifetime on apoptosis in hippocampus and neurocognitive outcomes in Sprague-Dawley rats. Int J Physiol Pathophysiol Pharmacol 8:111–119
Zhu X et al (2017) HDAC3 negatively regulates spatial memory in a mouse model of Alzheimer’s disease. Aging Cell 16:1073–1082
Acknowledgement
Not applicable.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
Conceptualization: JZ, XF, Dan Wang; Methodology: JZ, XF; Data curation: JZ, XF, DW; Validation: JZ; Supervision: XF; Writing – original draft: JZ; Writing – review and editing: JZ, XF, DW.
Corresponding author
Ethics declarations
Conflict of interest
Jiegang Zhou, Xinwei Feng and Dan Wang declare that there is no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhou, J., Feng, X. & Wang, D. HDAC3 deacetylates H3K27ac and H3K9ac on the TrkC promoter to exacerbate sevoflurane-induced neurotoxicity. Mol. Cell. Toxicol. (2023). https://doi.org/10.1007/s13273-023-00394-7
Accepted:
Published:
DOI: https://doi.org/10.1007/s13273-023-00394-7