
Abstract
Purpose of review
Necroptosis is a form of cell death regulated by specific cellular protein machinery. Although the cell death is tightly controlled like apoptosis, another type of programed cell death, the biological features of necroptosis rather resemble necrosis that is defined as an uncontrolled accidental cell death. The pathway executing necroptosis relies on a protein kinase, RIPK3, and its downstream effector molecule, MLKL. Upon necroptosis initiating signals, both RIPK3 and MLKL undergo extensive post-translation modifications to construct a death complex called necrosome, finally leading to lysis of cell membrane. Preclinical mouse models demonstrated the physiological importance of necroptosis in the progress of various inflammation-associated diseases. The objective of this brief review is to introduce a new emerging concept in cell death biology and to provide a first entry into the research field of necroptosis.
Recent findings
The uncovering of necroptosis pathway brought a fundamental change in the basic concept that necrotic cell death is passive and unregulated. Currently, multiple small molecules that can target necrotic cell death are under development and some of them are under clinical trials to evaluate their therapeutic potentials. Better understanding of the molecular mechanism leveraging necroptosis will provide an unprecedented opportunity to pathological necrosis-driven human diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cell death is a final event to cease all biological functions in unicellular organisms, but the same cellular event is a dynamic process which controls homeostasis and immunity in multicellular organisms. In humans, as many as a hundred billion cells die each day and are replaced by other cells (Nagata 2018). Therefore, the equilibrium between cell death and proliferation is crucial for the maintenance of tissue homeostasis. For example, the continuous cell death-induced shedding of intestinal epithelial cells during normal tissue homeostasis is a crucial process to maintain intestinal barrier integrity (Vereecke et al. 2011). Cell death also plays important functions in inflammation and immunity. It can restrict viral replication (Danthi 2016), but at the same time, excessive cell death can cause tissue damage and inflammation (Wallach et al. 2014). Indeed, cell death has been acknowledged as an eminent pathological feature of various inflammatory diseases (Kist and Vucic 2021). Consequently, the cell death process during immune responses is required to be tightly coordinated to ensure beneficial immunological effects without resulting in detrimental tissue damage.
The role of cell death in tissue homeostasis, immunity and inflammation have been extensively discussed in the context of two different types of cell death: apoptosis and necrosis. Apoptosis is a programed cell death whereby a cell commits a suicide by utilizing its own cellular signaling cascades in response to specific stimuli (Nagata 2018). Apoptotic cell dies by a self-destruction program coded by caspase-dependent signaling, and the cellular dead body is then cleared by the neighboring tissue-resident phagocytes. Since the process does not permit leakage of intracellular components, apoptosis is believed as a noninflammatory form of cell death.
In contrast to apoptosis, necrosis is an accidental cell death occurring in a fatally damaged cell by an external fatal insult (Galluzzi 2007). The necrotic cell death was originally regarded as a simple failure of cell survival caused by toxic interference of essential cellular functions. A necrotic cell undergoes swelling, followed by uncontrolled rupture of the cell membrane, resulting in a massive release of cellular contents to surrounding tissue environment. The unleashed cellular contents function as danger-associated molecular patterns (DAMPs) and often create inflammation in tissues (Venereau et al. 2015).
Early studies considered apoptosis as the only form of regulated cell death and necrosis was seen as an unregulated and, therefore, uncontrollable accidental cell death. The traditional concept of necrosis, which was considered as a passive cell death, has been changed since the discovery of a specific form of regulated cell death called necroptosis. As in necrosis, necroptosis executes a highly inflammatory cell death, accompanying by release of DAMPs (Newton and Manning 2016). While the phenotype and the consequence of cell death similar to necrosis, collective biochemical and genetic evidence revealed that necroptosis occur as a programed cell death regulated by Receptor Interacting Protein Kinase 3 (RIPK3) and its substrate, Mixed Lineage Kinase Like (MLKL). During necroptosis, modified MLKL triggers plasma membrane permeabilization and therefore, promotes the release of proinflammatory substances from the dead cell (Grootjans et al. 2017).
Accumulating evidence suggests that necroptosis is a physiologically relevant cell death and it is critical for tissue homeostasis, immunity, and inflammation. In this brief review, I will discuss about our current understanding of necroptosis and its implication in tissue homeostasis and inflammation.
Overview of necroptosis
Apoptosis, which is a form of caspase-dependent programed cell death, is characterized by several distinct biological features such as shrinkage of the cell, forming membrane blebs, and nuclear fragmentation (Nagata 2018). This cell death can be triggered by a wide variety of stimuli and conditions such as genotoxic stress, deprivation of growth factors, and occupation of death receptors by their specific ligands. Apoptosis is believed to be a non-inflammatory cell death since apoptotic cells generate "eat me" signals on their cell surface assisting rapid phagocytosis by surrounding tissue-resident immune cells. Because of this silent nature of cell death, apoptosis is utilized during normal development to balance cell populations in tissues (Voss and Strasser 2020).
The term, necroptosis was first introduced when a group of researchers recognized a cell death accompanied by necrotic morphological features was caspase-independent and inhibited by a small molecule called, necrostatin-1(Nec-1) (Degterev 2005). In contrast to apoptotic cell death, which is governed by caspase-mediated series of molecular events, necroptosis deploys apical protein kinases for its execution and does not require caspases. Although it was initially thought that RIPK1 is a mandatory driver of necroptosis, currently necroptosis is defined as RIPK3-and MLKL-dependent cell death (Galluzzi 2018). The main reason why RIPK1 was thought to be required for necroptosis is that Nec‐1, a RIPK1 inhibitor, potently inhibits TNF-dependent necroptosis. In fact, many studies still utilize Nec‐1 to define a cell death as necroptosis. However, it is now very clear that RIPK1 can function both as an inhibitor and a promoter of necroptosis depending on the cellular contexts (Voss and Strasser 2020) and therefore the absence of RIPK1 often results in necroptosis-driven inflammation in vivo due to lack of the kinase-independent survival function of RIPK1 (Ito 2016; Lin 2016; Newton 2016a).
The physiological role of RIPK1 in cell survival and death has been well-demonstrated by mouse genetic studies. RIPK1 deficient mice display postnatal lethality (Kelliher 1998). It was initially thought that the lethal phenotype is delivered by excessive apoptosis in the animals. But later, it was found that the deficiency of Fas-associated protein with death domain (FADD), which would inactivate apoptosis, did not rescue the lethal phenotype of RIPK1 KO mice (Zhang 2011). Furthermore, loss of RIPK3 also did not prevent the lethality resulted from RIPK1 deficiency in the animals. The postnatal death of RIPK1 KO mice was eventually rescued by the combined ablation of Caspase-8 and RIPK3, indicating that RIPK1 can repress both caspase‐8‐dependent apoptosis and RIPK3‐dependent necroptosis (Dillon 2014; Rickard 2014). Interestingly, animals with a kinase-inactive form of RIPK1 are viable, unlike RIPK1 KO mice (Berger 2014; Polykratis 2014). The result demonstrates that a kinase-independent scaffolding function of RIPK1 is responsible for the pro-survival effect of RIPK1. Taken together, RIPK1 can either promotes apoptosis and necroptosis via its kinase activity or suppress cell death by its kinase-independent function. In this regard, it is important to keep it in mind that inhibition of a cell death by RIPK1 inhibitors does not necessarily indicate the mode of cell death is necroptosis.
Activation of necroptosis
Necroptosis can be triggered by different initiating signals such as death receptors, some toll-like receptors, and unknown ligands sensed by protein, Z-DNA binding protein 1(ZBP1) (Grootjans et al. 2017). All these necroptotic signals employ proteins contain RIP Homotypic Interaction Motif (RHIM) and the RHIM provides a critical protein–protein interaction interface that regulates the formation of death-inducing protein complexes (Rebsamen 2009). There are only four proteins that contain RHIM in the mammalian system: RIPK1, RIPK3, ZBP1 and TIR-domain-containing adapter-inducing interferon-β (TRIF).
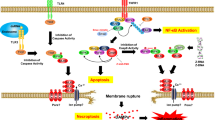
The molecular mechanism of necroptosis is most well-established in TNFR1 signaling pathway(Fig. 1) (Wajant and Siegmund 2019). TNFR1 is a member of the death receptor family. Upon the ligation of TNF, the death domain (DD) in TNFR1 enables to recruit DD-containing proteins including TNFR1-associated death domain protein (TRADD) and RIPK1 (Haas 2009). The ubiquitin ligases, cIAP1/2, which preexist as cytoplasmic complexes with TNF receptor-associated factor 2 (TRAF2) (Zheng et al. 2010), are then recruited to the TFNR1 membrane complex, followed by LUBAC, an E3 ubiquitin ligase complex composed of proteins, SHARPIN, HOIP, and HOIL-1 (Spit et al. 2019). The ubiquitin chains assembled by the coordination of cIAP1/2 and LUBAC allow the recruitment of ubiquitin binding proteins such as TAK1-binding protein 2/3 (TAB2/3) and NF-κB essential modulator (NEMO), resulting in the activation of NF-kB signaling pathway. Once TNF-mediated proinflammatory and cell survival signals are established, later the TNFR1 membrane complex transforms into a cell death-inducting cytoplasmic complex associated with FADD, caspase-8 and RIPK3 (Grootjans et al. 2017). The cytoplasmic death complex can induce caspase-8-dependent apoptosis and normally, RIPK3 is inactivated in the complex by its cleavage by caspase-8 (Feng 2007). Nevertheless, under caspase-inactive conditions, RIPK3-dependent necroptotic pathway begins by the autophosphorylation of RIPK3, which enables interaction with its effector, MLKL. The RIPK3-dependent phosphorylation of MLKL allows oligomerization of MLKL, leading to lysis cell membranes (Sun 2012; Petrie et al. 2019).

TNFR1 signaling pathway. Upon ligation of TNF, TNFR1 forms a large membrane-associated complex that is responsible for inflammation and cell survival-promoting cellular signaling pathways. During the process, if the survival-promoting signal is attenuated, death-inducing cytoplasmic complex can potentiate caspase-8 dependent apoptosis. RIPK3-dependent necroptosis is believed as an alternative backup cell death program, which occurs when apoptosis is compromised. The RIPK3-dependent modification of MLKL induces the translocation of MLKL to cell membrane resulting in lysis of a cell.
Pathological implications of necroptosis
Necrotic cell death is found in various pathological conditions (Nieminen 2003). Despite its presence in a broad range of clinical states, necrosis in human disease was not perceived as a potential therapeutic target until very recently since it was thought that necrotic cell death is an uncontrollable process. However, the discovery of necroptosis, which is regulated by cellular machinery, provided a new opportunity to target pathological necrosis.
Recent studies performed in preclinical animal models have revealed the potential benefit of targeting necroptosis in various human diseases, including sepsis, tissue injuries, chronic inflammatory disease, and neurodegenerations (Spit et al. 2019; Khoury et al. 2020; Molnar 2019). Notably, some studies utilized only specific RIPK1 inhibitors or RIPK1-deficient mice in testing necroptosis as a potential therapeutic target. Importantly, as discussed before, RIPK1 can activate both apoptosis and necroptosis, and can potentiate NF-kB-dependent inflammation. Therefore, RIPK1-dependent phenotypes do not necessarily correlate with necroptosis-driven pathological outcomes. In this section, I will briefly discuss only studies that confirmed the pathological contribution of necroptosis in the absence of activity of RIPK3 or MLKL.
The inflammatory nature of necroptosis proposes its potential involvement in the pathologies of acute inflammation. A mouse model of TNF-induced systemic inflammatory response syndrome (SIRS) showed that loss of RIPK3 in animals were protected against lethal SIRS (Duprez 2011). In the same study, ablation of RIPK3 also prevented cecal ligation and puncture-induced sepsis.
In addition to acute inflammation models, necroptosis has been implicated in chronic inflammation-associated disease models. A model of nonalcoholic fatty liver disease (NAFLD) demonstrated that when RIPK3-deficient mice were fed with methionine- and choline-deficient (MCD), MCD diet-induced liver injury, steatosis and fibrosis were attenuated (Afonso 2015). Moreover, the high levels of RIPK3 expression in patient livers were correlated with poor prognosis of alcoholic cirrhosis (Zhang 2018). In another chronic inflammation-associated animal model, RIPK3-deficiency reduced atherosclerotic lesions, which were promoted by loss of LDL receptor (Lin 2013).
Necroptosis also appears to contribute to multiple acute tissue injury models. A study using a model of kidney I/R reported protection of RIPK3-deficient mice but interestingly, the protection was minimal in MLKL-deficient mice (Newton 2016b). Another acute kidney model induced by toxic folic acid demonstrated that RIPK3 or MLKL did not contribute to the early stage of renal injury, but later when the TWEAK signaling-dependent necroptosis occurred, ablation of RIPK3 or MLKL reduced the late injury of kidney in the model (Martin-Sanchez 2018). In addition to kidney models, RIPK3-deficient mice were protected from long-term adverse post-infarct remodeling following I/R injury-induced myocardial infarction (Luedde 2014). Furthermore, the contribution of RIPK3 to lung injury was reported in a murine model of acute lung injury (ALI). The study demonstrated that both necroptosis-dependent and -independent functions of RIPK3 can contribute to the phenotype of an LPS-induced ALI model. Importantly, RIPK3 inhibitor ameliorated lung injury and reduced inflammation in this model (Chen 2018).
Beside acute tissue injury models, necroptosis has been reported in neurodegenerative diseases. RIPK3 deficiency markedly improves neurological and systemic disease in a mouse model of Gaucher’s disease (GD). When GD was induced by daily injection of a GlcCerase inhibitor, RIPK3-deficient mice showed considerably improved survival and motor coordination. In a mouse amyotrophic lateral sclerosis (ALS) model, the RIPK1, RIPK3 and MLKL–dependent necroptosis was observed in the central nerve system. In this optineurin-deficient ALS mice, the axonal pathology was rescued by RIPK3-deficiecy (Ito 2016).
Collectively, the studies discussed above synchronously propose crucial roles of necroptosis in the development of pathophysiology. It is worth to note that some studies suggested there are MLKL-independent, therefore necroptosis-independent roles of RIPK3 (Chen 2018; Alvarez-Diaz 2016). In this regard, when studying the role of necroptosis in vivo, it is important to confirm if the identified RIPK3-dependent phenotypes are also MLKL-dependent, therefore, ensuring bona fide necroptosis-driven pathology.
Conclusion
Investigating the details of the mechanism regulating necroptosis is critical to understand physiological roles of necroptosis in the pathogenesis of human diseases. The key signaling molecules in necroptotic pathway are largely distinct from those involved in other pathways including apoptosis and inflammation. So far, therapeutic potentials of targeting necroptosis have been reported mainly in mouse models. It remains to be demonstrated if blocking necroptosis is a viable strategy to treat human diseases.
The development of specific inhibitors against RIPK3 was initially perused to achieve specific therapeutic benefits in the necroptosis-associated pathophysiology of the disease. But it turned out that the inhibition of RIPK3 either by specific inhibitors or mutations leading to inactivation of its kinase activity not only block necroptosis but promotes spontaneous apoptosis (Mandal 2014). Hence, it seems a specific inhibitor against MLKL will be a better strategy to target necroptosis in the clinic.
References
Afonso MB et al (2015) Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin Sci (Lond) 129:721–739. https://doi.org/10.1042/CS20140732
Alvarez-Diaz S et al (2016) The pseudokinase MLKL and the kinase RIPK3 have distinct roles in autoimmune disease caused by loss of death-receptor-induced apoptosis. Immunity 45:513–526. https://doi.org/10.1016/j.immuni.2016.07.016
Berger SB et al (2014) Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol 192:5476–5480. https://doi.org/10.4049/jimmunol.1400499
Chen J et al (2018) RIP3 dependent NLRP3 inflammasome activation is implicated in acute lung injury in mice. J Transl Med 16:233. https://doi.org/10.1186/s12967-018-1606-4
Choi ME, Price DR, Ryter SW, Choi AMK (2019) Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight. https://doi.org/10.1172/jci.insight.128834
Danthi P (2016) Viruses and the diversity of cell death. Annu Rev Virol 3:533–553. https://doi.org/10.1146/annurev-virology-110615-042435
Degterev A et al (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1:112–119. https://doi.org/10.1038/nchembio711
Dillon CP et al (2014) RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157:1189–1202. https://doi.org/10.1016/j.cell.2014.04.018
Duprez L et al (2011) RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35:908–918. https://doi.org/10.1016/j.immuni.2011.09.020
Feng S et al (2007) Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal 19:2056–2067. https://doi.org/10.1016/j.cellsig.2007.05.016
Galluzzi L et al (2007) Cell death modalities: classification and pathophysiological implications. Cell Death Differ 14:1237–1243. https://doi.org/10.1038/sj.cdd.4402148
Galluzzi L et al (2018) Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25:486–541. https://doi.org/10.1038/s41418-017-0012-4
Grootjans S, Vanden Berghe T, Vandenabeele P (2017) Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ 24:1184–1195. https://doi.org/10.1038/cdd.2017.65
Haas TL et al (2009) Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell 36:831–844. https://doi.org/10.1016/j.molcel.2009.10.013
Ito Y et al (2016) RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353:603–608. https://doi.org/10.1126/science.aaf6803
Kelliher MA et al (1998) The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 8:297–303. https://doi.org/10.1016/s1074-7613(00)80535-x
Khoury MK, Gupta K, Franco SR, Liu B (2020) Necroptosis in the pathophysiology of disease. Am J Pathol 190:272–285. https://doi.org/10.1016/j.ajpath.2019.10.012
Kist M, Vucic D (2021) Vucic D (2021)) Cell death pathways: intricate connections and disease implications. EMBO J. https://doi.org/10.15252/embj.2020106700
Lin J et al (2013) A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep 3:200–210. https://doi.org/10.1016/j.celrep.2012.12.012
Lin J et al (2016) RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 540:124–128. https://doi.org/10.1038/nature20558
Luedde M et al (2014) RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res 103:206–216. https://doi.org/10.1093/cvr/cvu146
Mandal P et al (2014) RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell 56:481–495. https://doi.org/10.1016/j.molcel.2014.10.021
Martin-Sanchez D et al (2018) TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc Natl Acad Sci U S A 115:4182–4187. https://doi.org/10.1073/pnas.1716578115
Molnar T et al (2019) Current translational potential and underlying molecular mechanisms of necroptosis. Cell Death Dis 10:860. https://doi.org/10.1038/s41419-019-2094-z
Nagata S (2018) Apoptosis and clearance of apoptotic cells. Annu Rev Immunol 36:489–517. https://doi.org/10.1146/annurev-immunol-042617-053010
Newton K (2020) Multitasking kinase RIPK1 regulates cell death and inflammation. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a036368
Newton K, Manning G (2016) Necroptosis and inflammation. Annu Rev Biochem 85:743–763. https://doi.org/10.1146/annurev-biochem-060815-014830
Newton K et al (2016a) RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 540:129–133. https://doi.org/10.1038/nature20559
Newton K et al (2016b) RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ 23:1565–1576. https://doi.org/10.1038/cdd.2016.46
Nieminen AL (2003) Apoptosis and necrosis in health and disease: role of mitochondria. Int Rev Cytol 224:29–55. https://doi.org/10.1016/s0074-7696(05)24002-0
Petrie EJ, Czabotar PE, Murphy JM (2019) The structural basis of necroptotic cell death signaling. Trends Biochem Sci 44:53–63. https://doi.org/10.1016/j.tibs.2018.11.002
Polykratis A et al (2014) Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol 193:1539–1543. https://doi.org/10.4049/jimmunol.1400590
Rebsamen M et al (2009) DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep 10:916–922. https://doi.org/10.1038/embor.2009.109
Rickard JA et al (2014) RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157:1175–1188. https://doi.org/10.1016/j.cell.2014.04.019
Spit M, Rieser E, Walczak H (2019) Linear ubiquitination at a glance. J Cell Sci. https://doi.org/10.1242/jcs.208512
Sun L et al (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148:213–227. https://doi.org/10.1016/j.cell.2011.11.031
Venereau E, Ceriotti C, Bianchi ME (2015) DAMPs from cell death to new life. Front Immunol 6:422. https://doi.org/10.3389/fimmu.2015.00422
Vereecke L, Beyaert R, van Loo G (2011) Enterocyte death and intestinal barrier maintenance in homeostasis and disease. Trends Mol Med 17:584–593. https://doi.org/10.1016/j.molmed.2011.05.011
Voss AK, Strasser A (2020) The essentials of developmental apoptosis. F1000Res. https://doi.org/10.12688/f1000research.21571.1
Wajant H, Siegmund D (2019) TNFR1 and TNFR2 in the control of the life and death balance of macrophages. Front Cell Dev Biol 7:91. https://doi.org/10.3389/fcell.2019.00091
Wallach D, Kang TB, Kovalenko A (2014) Concepts of tissue injury and cell death in inflammation: a historical perspective. Nat Rev Immunol 14:51–59. https://doi.org/10.1038/nri3561
Zhang H et al (2011) Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 471:373–376. https://doi.org/10.1038/nature09878
Zhang Z et al (2018) RIPK3-mediated necroptosis and neutrophil infiltration are associated with poor prognosis in patients with alcoholic cirrhosis. J Immunol Res 2018:1509851. https://doi.org/10.1155/2018/1509851
Zheng C, Kabaleeswaran V, Wang Y, Cheng G, Wu H (2010) Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: affinity, specificity, and regulation. Mol Cell 38:101–113. https://doi.org/10.1016/j.molcel.2010.03.009
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2020R1F1A1074601).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The author declares that there is no conflict of interest.
Human and animal rights
This review article contains published animal studies, which were performed under institutional and national guidelines.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kim, C. The modulation of necroptosis and its therapeutic potentials. Mol. Cell. Toxicol. 17, 93–97 (2021). https://doi.org/10.1007/s13273-021-00129-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13273-021-00129-6