
Abstract
Background
Intellectual disability (ID) is a neurodevelopmental condition, affecting 1–3% of the population. Genetic factors play a key role causing the limitation in intellectual functioning and adaptive behavior. The heterogeneity of ID makes it more difficult for genetic and clinical diagnosis. Mapping of variants through next generation DNA sequencing in consanguineous families would help to understand the molecular parthenogenesis of ID.
Objective
The aim of this study was to describe the genetic variants of ID in consanguineous Pakistani families.
Methods
We analyzed four unrelated consanguineous Pakistani families having an intellectual disability through whole exome sequencing (WES). Data was analyzed using different bioinformatics tools and software.
Results
We mapped four novel variants in different ID genes. Each variant is found in different family, co-segregating with a recessive pattern of inheritance. The variants found are; c.1437delG:p.Asn480Thrfs*10, mapped in FKRP, c.2041 C>A:p.Leu681Met in HIRA, c.382 C>T:p.Arg128Cys in BDH1 and c.267+1G>A:p.? identified in TRAPPC6B.
Conclusions
These variants help in demonstration of status and molecular basis of intellectual disability in Pakistani population leading to provision of genetic counseling services and a contribution in disease variant database.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Intellectual disability (ID) is a genetic disorder defined by the existence of significant limitations in intellectual capability and adaptive skills of a person. It affects children under the age of 18. The prevalence of this disorder is 1-3% of the population (Chelly et al. 2006; Basel 2007). Syndromic and nonsyndromic ID are the two main types of this disorder. In case of nonsyndromic ID, the person is suffering from mental retardation only whereas in syndromic ID, the person is suffering from some congenital anomalies such as muscular weakness, skeletal defects, physical abnormalities etc. along with mental retardation (Ropers and Hamel 2005). Today the most challenging problem faced by clinician and geneticists is to understand the molecular basis of ID. Etiologically intellectual disability is extraordinary heterogeneous including environmental as well as genetic factors. Genetic factors play a significant role causing the limitation in person intellectual functioning and adaptive behavior and consists of chromosomal and monogenic defects. Identification of monogenic variants for ID is a current challenge. Monogenic abnormalities include X linked and autosomal variants (Rasool et al. 2021). Although important advancement has been made in classification of genes involved in X-linked ID, however a little is known about genetic causes of autosomal recessive ID. To date more than sixty autosomal recessive non syndromic ID loci have been mapped with twenty-nine corresponding genes, and few are those reported earlier; PRSS12 (Molinari et al. 2002), CRBN (Higgins et al. 2004), CC2DIA (Basel et al. 2006), GRIK2 (Motazacker et al. 2007), TUSC3/N33 (Molinari et al. 2008) and CC2D2A (Nolan et al. 2008).
Limited family material and low rate of consanguinity is a major hurdle in the identification of intellectual disability genes in developed world. Due to the heterogeneity of ID, it is more difficult and challenging for clinical and genetic diagnosis but recent improvement in the area of research and technology makes it easier to study the causes of ID. An advanced combination of next generation sequencing (NGS), functional analysis and bioinformatics tools provided a high approach to sequence genome and detect a large number of pathogenic mutations. NGS has accelerated the speed of identification of novel ID causing genes and number of ID known genes and ID candidate genes has been considerably increased over the past 5 years. Genes involved in autosomal recessive ID are pleiotropic and exhibits multiple phenotypic expression. For comprehensively study of the genetics of autosomal recessive ID, investigation at the level of single genes is far better than meta-analysis (Ilyas et al. 2020). This approach will provide a new ways of treating ID. In Pakistan, rate of consanguineous marriages is very high. Approximately, 60% marriages are consanguineous in which first cousins’ marriages accounts more than 80%. This lead to high prevalence of recessive inherited disorders in children (Cheema et al. 2020; Zahoor et al. 2019, 2020). So genetic and molecular studies of such highly consanguineous population are a huge resource to identify new genes involved in ID that will eventually help to explain the molecular pathways and mechanism behind learning and cognition.
We have performed genetic analysis for causative variants of ID and identified four novel mutations in ID known and candidate genes in four unrelated consanguineous families belonging to the different regions of Pakistan using whole exome sequencing technology. In this paper, we aim to describe the molecular basis of ID and identify pathogenic variant responsible for this disease and also the pathogenic effect of all variants has been predicted by using bioinformatics tools and software.
Materials and methods
Recruitment of families and blood and data collection
We have enrolled four unrelated consanguineous families (PKID16, PKID17, PKID25 and PKID52) affected with ID from different cities of Punjab, Pakistan. Affected members as well as unaffected members of these families were participated in this study. Different parameters such as medical history, clinical presentation, general examination and family history information revealing the disease condition were collected at the time of enrollment. This research study was approved by Independent Institutional Ethical Committee (IIEC), University of Veterinary and Animal Sciences, Lahore Pakistan. We selected four unrelated consanguineous families for whole exome sequencing evaluation.
First of all, blood samples from affected as well as healthy family members are usually taken with informed written consent or guardian consent as approved by the Independent Institutional Ethics Committee. The selected four families were; PKID16 has three affected children, PKID17 currently has one affected child (three affected deceased) while PKID25 and PKID52 have one affected children. One affected sample used as an index from each family. Written informed consent was already obtained from all subjects and/or guardians to use their data in publication. Clinical information/patients phenotype of children affected with intellectual disability of all four families are described in Table 1. After the collection of blood sample of enrolled families, DNA was isolated by using commercial kit (Qiagen, Germany) following the instructions of the manufacturer.
Fragile X syndrome testing
To check all the samples for Fragile X syndrome, Fragment analysis AmplideX FMR1 kit by Asuragen, Inc. USA method were used and data was analyzed by GeneMapper software by Thermo Fisher Scientific, USA.
Whole exome sequencing and data analysis
Whole exome sequencing (WES) was performed using the IDT xGen Exome Research Panel kit (IDT) by Integrated DNA Technologies (IDT), BVBA, Leuven, Belgium. The subsequent sequencing was carried on the Illumina Novaseq-6000 by Illumina Company, Illumina Solutions Center, Berlin, Germany at the Institute of Medical Genetics, University of Zurich, Switzerland. Data were analyzed by using next generation sequencing variant analyzer tool. The variants found through NGS data analysis were then confirmed by Sanger’s sequencing along with the DNA of other family members. Functional effect of all variants has been predicted by using PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (https://sift.bii.a-star.edu.sg/) and MutationTaster (https://www.mutationtaster.org/), (Harris et al. 2021). The multiple sequence and protein domain clustal analysis were also performed for these new variants (https://asia.ensembl.org/index.html) and (https://www.ebi.ac.uk/Tools/msa/clustalo/).
Results
Four unrelated consanguineous families having ID from different cities of Punjab, Pakistan were enrolled for this study. Whole exome sequencing was performed in all four families. WES revealed four novel variants in four different ID genes present one in each unrelated consanguineous families. A variant (c.1437delG:p.(Asn480Thrfs*10) of FKRP gene identified in three affected individuals of family PKID16, c.2041 C>A:p.(Leu681Met) of HIRA gene in PKID17, c.382 C>T:p.(Arg128Cys) of BDH1 in PKID25 and variant; c.267+1G>A:p.? identified for TRAPPC6B in PKID52. All these variants were confirmed by Sanger’s sequencing and segregates through recessive pattern of inheritance. Detail of each variant and family clinical information/medical history are described as follows;
PKID16 (c.1437delG: p.Asn480Thrfs*10; FKRP)
PKID16 is a consanguineous family having affected in multiple loops but belongs to two generations. Three affected children (IV:1, IV:3, IV:5) were enrolled and all of them have severe intellectual disability, developmental delay, behavioral problems, muscles weakness, muscular dystrophy, aggression, hypotonia, spasticity, congenital anomalies include skeletal defects, and eye abnormalities (Table 1). Along with three affected, four unaffected members, (II:1, II:2, III:3 and III:4) were also enrolled.
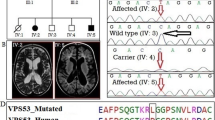
WES of (IV:1, IV:3, IV:5) revealed a novel homozygous pathogenic frameshift deletion; c.1437delG:p.(Asn480Thrfs*10) in exon 4 of FKRP gene. This variant was confirmed in other family members (II:1, II:2, III:3, III:4) by Sanger’s sequencing that showed variant co-segregating with a recessive pattern of inheritance (Fig. 1). The pathogenic variant in our family PKID16 was not found in our control samples and also not found in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) which supports the notion that this is a novel variant and not reported previously. Furthermore, the variant c.1437delG:p.(Asn480Thrfs*10) disrupt the protein production that ultimately affect the structure and function of the protein.

Pedigrees of Pakistani families having an intellectual disability showing variants through chromatograms of enrolled individuals. Each generation in a pedigree is specified by an Arabic numeral and each individual in a generation is specified by a Roman numeral. Squares refer to males, circles to females, filled signs to affected individuals, half filled symbols to individual with other health condition and slashed lines to deceased persons
.
PKID17 (c.2041 C>A: p.Leu681Met; HIRA)
The first cousin parents of a consanguineous family having four affected monozygotic girls (IV:1, IV:2, IV:3, IV:4) but three of them are expired at an early age. One girl (IV:4) was enrolled along with her parents and maternal uncle. Parents and maternal uncle (III:5, III:6 and III:2) were healthy and participated in this study with consent. One nephew of the parents was also died at an early age with developmental delay. Clinical examination revealed that affected girl has intellectual disability, developmental delay, behavioral problems, postnatal growth retardation, dysmorphism, spasticity, and gait problem (Table 1).
Whole exome sequencing identified a homozygous missense mutation; c.2041 C>A:p.(Leu681Met) in exon 17 of HIRA gene of affected (IV:4). The parents (III:5 & III:6) are heterozygous for this variant (Fig. 1). This variant c.2041 C>A:p.(Leu681Met) was not present in our control samples and also not found in Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) which showed that this variant is not reported previously. Polyphen2 and Mutation Taster predicted variant p. (Leu681Met) as “probably damaging” (score of 0.786) and as “disease causing”. Furthermore, the wild type residue (Leucine) is non-polar aliphatic amino acid whereas methionine is polar in nature with no charge. There is a size difference in both wild type and mutant amino acids. Mutant amino acid (Methionine) is bigger in size. Due to the large size of mutant residue, this might lead to bumps and disturb the normal structure of protein. WES of affected (IV:4) also identified a novel nonframeshift deletion; c.2_4del: p.? in exon 1 of ROS1 gene. The parents (III:5 & III:6) are heterozygous for this variant. The variant in this family was not present in our control samples and was also not found in the Genome Aggregation Database (gnomAD), (https://gnomad.broadinstitute.org/), supporting the notion that this is not a population specific polymorphism. The variant c.2_4del: p.? disrupts the first codon of the ROS1 gene that ultimately affects the structure and function of the protein. Computational evaluations are not available for this variant so we say it additional candidate gene to consider for researchers of this field.
PKID25 (c.382 C>T: p.Arg128Cys; BDH1)
This consanguineous family has one affected person (IV:2). The affected have moderate intellectual disability, developmental delay, behavioral problems, muscle weakness, aggression and hypotonia (Table 1). Parents (III:1 & III:2) and healthy sister (IV:4) were also enrolled along with the affect individual. Unaffected individuals were normal. WES identified a homozygous missense mutation c.382 C>T: p.(Arg128Cys) in exon 5 of BDH1 gene in affected individual.
Variant c.382 C>T: p.(Arg128Cys) of BDH1 gene was checked in other family members by Sanger’s sequencing. Affected child is homozygous for this variant while parents (III:1 & III:2) and healthy sibling (IV:4) was heterozygous (Fig. 1). In addition, mutation (c.382 C>T: p.(Arg128Cys) depicts that the mutant residue (Cystein) is smaller than the wild type residue (Arginine). The wild type amino acid (cysteine) was positive, the mutant residue charge is neutral. Difference in charges leads can cause loss of interaction with other molecules or residues. The hydrophobicity of the wild type and mutant type residue also differs. This mutation (p.Arg128Cys) introduces a more hydrophobic residue at this position which can result in loss of hydrogen bonds and disturb correct folding, which ultimately affect the proper function of the protein. The variant (c.382 C>T: p.(Arg128Cys) was not found in our control samples and also not mentioned on Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) supporting the notion that this is a novel variant and not reported previously.
PKID52 (c.267+1G>A: p.?; TRAPPC6B)
PKID52 is a consanguineous family with one affected child (IV:4). Clinical data showed that the affected have severe intellectual disability, developmental delay, behavioral problem, muscle weakness, aggression, learning disability, hypotonia, spasticity and congenital anomalies includes skeletal defect, eye abnormalities and ear shape anomaly (Table 1). Healthy parents (III:8 & III:9) and one healthy (IV:5) sibling were also enrolled.
WES revealed a pathogenic splicing mutation c.267+1G>A:p.? in intron 3 of TRAPPC6B gene of family PKID52. Affected (IV:4) is homozygous while parents (III:8 & III:9) and healthy sibling (IV:5) are heterozygous for this variant when checked by Sanger’s sequencing (Fig. 1). Furthermore, the variant in our family was not present in our control samples and also not found on Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) supporting the notion that this variant is not reported previously in any population. The variant (c.267+1G>A:p.?) in our family may result in a defective RNA splicing and ultimately affect the resulting protein. It may also affect regulatory sequences, resulting in decreased/premature translation and low gene product. Mutation damage the protein structure chemically and abolish its proper function.
Homology analysis
Protein homology alignment for these variants of FKRP, HIRA, and BDH1 was performed with nine different species (Homo sapiens (Hs), Pan troglodytes (Pt), Canis lupus familiaris (Cl), Mus musculus (Mm), Gallus gallus (Gg), Danio rerio (De), Drosophila melanogaster (Dm), Ciona intestinalis (Ci), Caenorhabditis elegans (Ce) and Saccharomyces cerevisiae (Sc) performed with Clustal omega showing a high degree of conservation shown in Fig. 2.

Variants are annotated by using Clustal Omega alignment tool with order of multi to unicellular model organism i.e. Homo sapiens (Hs), Pan troglodytes (Pt), Canis lupus familiaris (Cl), Mus musculus (Mm), Gallus gallus (Gg), Danio rerio (De), Drosophila melanogaster (Dm), Ciona intestinalis (Ci), Caenorhabditis elegans (Ce) and Saccharomyces cerevisiae (Sc) respectively
Multiple sequence alignment analysis by Clustal omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) showed that wildtype amino acid Asparagine (N) at 480 position in FKRP gene is conserved in following species; Homo sapiens (Hs), Pan troglodytes (Pt), Canis lupus familiaris (Cl), Mus musculus (Mm) and Danio rerio (De) and not conserved in Drosophila melanogaster (Dm). Orthologues of FKRP gene are not present in Gallus gallus (Gg), Ciona intestinalis (Ci), Caenorhabditis elegans (Ce) and Saccharomyces cerevisiae respectively (Fig. 2).
In HIRA gene, the wild type amino acid Leucine (L) at 681 position is conserved in primates and also in Danio rerio (De) only. On the other hand, this variant is not conserved in other unicellular model organisms as Drosophila melanogaster, Caenorhabditis elegans, Ciona intestinalis and Saccharomyces cerevisiae respectively. Furthermore, the amino acid Arginine (R) at position 128 in BDH1gene is conserved in all primates except Canis lupus familiaris (Cl), Gallus gallus (Gg), and Danio rerio (De). Variant, p.(Arg128Cys) is located in KR domain of D-beta-hydroxybutyrate dehydrogenase enzyme encoded by the BDH1 gene (Fig. 2). Orthologues of BDH1 gene are not present in Drosophila melanogaster, Ciona intestinalis, Caenorhabditis elegans, and Saccharomyces cerevisiae respectively.
Discussion
Four unrelated consanguineous Pakistani families exhibiting low IQ level, developmental delay, behavioral problems and other congenital abnormalities such as skeletal defects, were investigated (Table 1). These families were diagnosed by psychologist on the basis of their clinical presentation, medical history, general examination and family history. We identified four different novel variants in different ID genes present in four different families. A deleterious variant (c.1437delG:p.(Asn480Thrfs*10) of FKRP gene identified in PKID16, c.2041 C>A:p.(Leu681Met) of HIRA gene in PKID17, c.382 C>T:p.(Arg128Cys) of BDH1 in PKID25 and c.267+1G>A:p.? identified for TRAPPC6B in PKID52 (Table 2).
FKRP (OMIM # 606,596) encodes fukutin related protein which is a member of a class of molecules that mediate O-linked glycosylation. The main target of these glycosyltransferase is posttranslational modification of dystroglycan. Mutation in this gene leads to wide spectrum of disease including muscular dystrophy, intellectual disability and cerebellar cysts (MacLeod et al. 2007). The severity of disease is depending on the level of abnormal glycosylation of dystroglycan (Mercuri et al. 2006). This gene also reported as a ID known gene (https://sysid.cmbi.umcn.nl/) in multiple families having muscular dystrophy as well as intellectual disability and congenital anomalies. A novel variant; c.1387 A>G (p.N463D) has been reported in FKRP gene in two unrelated mexican children with intellectual disability and muscular dystrophy (MacLeod et al. 2007). A homozygous FKRP gene mutations; (c.663 C>A;p.Ser221Arg) & (c.946 C>A;p.Pro316Thr) have been identified in two patients having muscular dystrophy and intellectual disability (Topaloglu et al. 2003). Moreover, two homozygous mutations; (c.1213G>T;p.Val405Leu) & (c.1364 C>A;p.Ala455Asp) have been identified in an Algerian boy, child of consanguineous parents and six unrelated Tunisian families (Louhichi et al. 2004). The variants of this gene have reported in other populations too (Mercuri et al. 2003).
HIRA (OMIM # 600,237) gene encodes a histone cell cycle regulator protein that play an important role in transcriptional regulation or histone and chromatin metabolism (Magnaghi et al. 1998). This gene is expressed in a range of fetal tissues (Halford et al. 1993) and also in the neural tube, developing neural plate, the neural crest and the mesenchyme of the head and branchial arch structures (Roberts et al. 1997). Besides act as a histone chaperone, this gene also reported as ID candidate gene on SysID database; a publicly accessible database of published neurodevelopmental disorders associated genes (https://sysid.cmbi.umcn.nl/). A homozygous missense variant; (c.41 A>G;p.Lys14Arg) in a patient having ID, severe postnatal growth retardation, global developmental delay, strabismus, dysmorphism and congenital anomalies (Anazi et al. 2016).
BDH1 (OMIM # 603,063) encodes a 3-hydroxybutyrate dehydrogenase which is a mitochondrial enzyme and required phosphatidylcholine (PC) for maximum enzyme activity (allosteric activator). The optimum activity of the enzyme is obtained only with membranes containing PC. The encoded protein plays a significant role in fatty acid catabolism which includes interconversion of acetoacetate and (R)-3-hydroxybutyrate (Marks et al. 1992). In addition, this gene also reported as a ID candidate gene on SysID database (https://sysid.cmbi.umcn.nl/). A homozygous missense variant; (c.668G>A;p.Arg223His) has been mapped in two affected from one family. They have severe ID, short stature, microcephaly, hypotonia and spasticity. BDH1 reported as a novel recessive candidate gene involved in neurodevelopmental disorders (Reuter et al. 2017).
TRAPPC6B (OMIM # 610,397) (Trafficking protein particle complex subunit 6B) gene is a component of TRAPP complexes which regulates membrane trafficking through Golgi apparatus. These complexes are tethering complexes which play important role in vesicle transport (Kummel et al. 2005). There are three TRAPP complexes; TRAPP I, TRAPP II, TRAPP III, with additional subunits required for proper functioning. Any mutation in this gene leads to neurodevelopmental disorder. A homozygous splice mutation (c.82-2 A>G;p.E28Vfs*11) has been identified in six individuals from three unrelated consanguineous Egyptian families. All six children manifested a neurodevelopmental disorder which is characterized by severe ID, microcephaly, hypotonia, no speech and autistic features (Marin-Valencia et al. 2018). They also designed the zebra fish model to check the expression of TRAPPC6B. In human, TRAPPC6B gene is expressed in many tissues including the fetal brain and different parts of adult brain and spinal cord. Another loss of function mutation (c.124 C>T;p.Arg42*) has been mapped in two patients from Iranian consanguineous family (Harripaul et al. 2018).
As the conclusion, we have mapped four novel variants in four different ID genes of four unrelated consanguineous Pakistani families. These variants will help in provision of an opportunity for genetic counselling, prenatal/preimplantation diagnosis, carrier testing in the extended family members and within-family marriage planning. This will be an addition to existing genomic data of our population. The implementation of newborn screening program will also help in early diagnosis and management of intellectual disability thus improving the overall status of this disorder in consanguineous populations and as well as in sporadic cases.
Data availability statement
All the data related with this project is available with the corresponding author and will be provided upon request.
References
Anazi S, Maddirevula S, Faqeih E, Alsedairy H, Alzahrani F, Shamseldin HE, Patel N, Hashem M, Ibrahim N, Abdulwahab F et al (2016) Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol Psychiatry 22(4):615–624
Basel VL (2007) Genetics of autosomal recessive non-syndromic mental retardation: recent advances. Clin Genet 72:167–174
Basel VL, Attia R, Yahav M, Ferland RJ, Anteki L, Walsh CA, Olender T, Straussberg R, Magal N, Taub E et al (2006) The CC2D1A, a member of a new gene family with C2 domains, is involved in autosomal recessive nonsyndromic mental retardation. J Med Genet 43:203–210
Cheema HA, Rasool IG, Anjum MN, Zahoor MY (2020) Mutational spectrum of SMPD1 gene in Pakistani Niemann-Pick disease patients. Pak J Med Sci 36(3):479–484
Chelly J, Khelfaoui M, Francis F, Chérif B, Bienvenu T (2006) Genetics and pathophysiology of mental retardation. Eur J Hum Genet 14:701–713
Halford S, Wilson DI, Daw SCM, Roberts C, Wadey R, Kamath S, Wickremasinghe A, Burn J, Goodship J, Mattei MG et al (1993) Isolation of a gene expressed during early embryogenesis from the region of 22q11 commonly deleted in DiGeorge syndrome. Hum Mol Genet 2:1577–1582
Harripaul R, Vasli N, Mikhailov A, Rafiq MA, Mittal K, Windpassinger C (2018) Mapping autosomal recessive intellectual disability: combined microarray and exome sequencing identifies 26 novel candidate genes in 192 consanguineous families. Mol Psychiatry 23:973–984
Harris HK, Nakayama T, Lai J, Zhao B, Argyrou N, Gubbels CS, Soucy A, Genetti CA, Suslovitch V, Rodan LH (2021) Disruption of RFX family transcription factors causes autism, attention-deficit/hyperactivity disorder, intellectual disability, and dysregulated behavior. Genet Med 23:1028–1040
Higgins JJ, Pucilowska J, Lombardi RQ, Rooney JP (2004) A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology 63(10):1927–1931
Ilyas M, Mir A, Efthymiou S, Houlden H (2020) The genetics of intellectual disability: advancing technolog and gene editing. F1000 Res 5:5. https://doi.org/10.12688/f1000research.163151.2
Kummel D, Muller JJ, Roske Y, Misselwitz R, Bussow K, Heinemann U (2005) The structure of the TRAPP subunit TPC6 suggests a model for a TRAPP subcomplex. EMBO Rep 6:787–793
Louhichi N, Triki C, Quijano-Roy S, Richard P, Makri S, Meziou M, Estournet B, Mrad S, Romero NB, Ayadi H et al (2004) New FKRP mutations causing congenital muscular dystrophy associated with mental retardation and central nervous system abnormalities: identification of a founder mutation in Tunisian families. Neurogenetics 5:27–34
MacLeod H, Pytel P, Wollmann R, Chelmicka-Schorr E, Silver K, Anderson RB, Waggoner D, McNally EM (2007) A novel FKRP mutation in congenital muscular dystrophy disrupts the dystrophin glycoprotein complex. Neuromusul Disor 17:285–289
Magnaghi P, Roberts C, Lorain S, Lipinski M, Scambler PJ (1998) HIRA, a mammalian homologue of Saccharomyces cerevisiae transcriptional co-repressors interacts with Pax3. Nat Genet 20:74–77
Marin-Valencia I, Novarino G, Johansen A, Rosti B, Issa MY, Musaev D, Bhat G, Scott E, Silhavy JL, Stanley V et al (2018) A homozygous founder mutation in TRAPPC6B associates with a neurodevelopmental disorder characterized by microcephaly, epilepsy and autistic features. J Med Genet 55:48–54
Marks AR, McIntyre JO, Duncan TM, Erdjument-Bromage H, Tempst P, Fleischer S (1992) Molecular cloning and characterization of (R)-3-hydroxybutyrate dehydrogenase from human heart. J Biol Chem 267:15459–15463
Mercuri E, Brockington M, Straub V, Quijano-Roy S, Yuva Y, Herrmann R, Brown SC, Torelli S, Dubowitz V, Blake DJ (2003) Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann Neurol 53:537–542
Mercuri E, Topaloglu H, Brockington M, Berardinelli A, Pichiecchio A, Santorelli F, Rutherford M, Talim B, Ricci E, Voit T et al (2006) Spectrum of brain changes in patients with congenital muscular dystrophy and FKRP gene mutations. Arch Neurol 63:251–257
Molinari F, Rio M, Meskenatic V, Encha RF, Auge V, Bocq D, Briault S, Vekemans M, Munnich A, Attie BT et al (2002) Truncating neurotrypsin mutation in autosomal recessive non- syndromic mental retardation. Science 298:1779–1781
Molinari F, Foulquier F, Tarpey PS, Morelle W, Boissel S, Teague J, Edkins S, Futreal PA, Stratton MR, Turner G et al (2008) Oligosaccharyltransferase-subunit mutations in nonsyndromic mental retardation. Am J Hum Genet 82:1150–1157
Motazacker MM, Rost BR, Hucho T, Garshasbi M, Kahrizi K, Ullmann B, Abedini SS, Nieh SE, Amini SH, Goswami C et al (2007) A defect in the ionotropic glutamate receptor 6 gene (GRIK2) is associated with autosomal recessive mental retardation. Am J Hum Genet 81:792–798
Nolan DK, Chen P, Das S, Ober C, Waggoner D (2008) Fine mapping of a locus for nonsyndromic mental retardation on chromosome 19p13. Am J Med Genet 146A:1414–1422
Rasool IG, Zahoor MY, Iqbal M, Anjum AA, Ashraf F, Abbas HQ, Baig HMA, Mahmood T, Shehzad W (2021) Whole exome sequencing revealed novel variants in consanguineous Pakistani families with intellectual disability. Gene Genom 43(5):503–512
Reuter MS, Tawamie H, Buchert R, Gebril OH, Froukh T, Thiel C, Uebe S, Ekici AB, Krumbiegel M, Zweier C et al (2017) Diagnostic yield and novel candidate genes by exome sequencing in 152 consanguineous families with neurodevelopmental disorders. JAMA Psychiatry 74(3):293–299
Roberts C, Daw SCM, Halford S, Scambler PJ (1997) Cloning and developmental expression analysis of chick Hira (Chira), a candidate gene for DiGeorge syndrome. Hum Mol Genet 6:237–245
Ropers HH, Hamel BC (2005) X-linked mental retardation. Nat Rev Genet 6:46–57
Topaloglu H, Brockington M, Yuva Y, Talim B, Haliloglu G, Blake D, Torelli S, Brown SC, Muntoni F (2003) FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology 60:988–992
Zahoor MY, Cheema HA, Ijaz S, Anjum MN, Ramzan K, Bhinder MA (2019) Mapping of IDUA gene variants in Pakistani patients with mucopolysaccharidosis type 1. J Pediatr Endocrinol Metab 32(11):1221–1227
Zahoor MY, Cheema HA, Ijaz S, Fayyaz Z (2020) Genetic analysis of tyrosinemia type 1 and fructose-1, 6 bisphosphatase deficiency affected in Pakistani cohorts. Fetal Pediatr Pathol 39(5):430–440
Acknowledgements
We are thankful to the families who participated in the study.
Funding
This work was supported by departmental support as PhD scholar of Institute of Biochemistry and Biotechnology, University of Veterinary and Animal Sciences, Lahore and Higher Education Commission, Pakistan.
Author information
Authors and Affiliations
Contributions
IGR performed major lab work as her PhD thesis work. MYZ, IGR and WS planned and executed project. MI and SS did data collection and families enrollment. IA did families enrollment and performed some lab work. AAA performed statistical data analysis.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest of any sort with anyone.
Statement of Ethics
This study was approved by Independent Institutional Ethics Committee (IIEC), University of Veterinary and Animal Sciences, Lahore Pakistan (Ethical approval letter. DAS/1259). Informed written consent or guardian consent also approved by committee. This is the original article which did not published in any other journal/publication.
.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rasool, I.G., Zahoor, M.Y., Ahmed, I. et al. Description of novel variants in consanguineous Pakistani families affected with intellectual disability. Genes Genom 45, 457–465 (2023). https://doi.org/10.1007/s13258-022-01219-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-022-01219-y