
Abstract
Background
Epigenetic modifications play important roles in diverse cellular processes such as X chromosome inactivation, cell differentiation, development and senescence. DNA methylation and histone modifications are major epigenetic modifications that regulate chromatin structure and gene expression without DNA sequence changes. Epigenetic alterations may induce phenotypic changes stable enough for mitotic or meiotic inheritance. Moreover, the reversibility of epigenetic marks makes the manipulation of chromatin and epigenetic signature an attractive strategy for therapeutic and breeding purposes. Targeted epigenetic manipulation, or epigenome editing, at the gene of interest commonly utilizes specific epigenetic modifiers fused with a targeting module of the conventional genome editing system.
Objective
This review aims to summarize essential epigenetic components and introduce currently available epigenetic mutants and the corresponding epialleles in plants. Furthermore, advances in epigenome editing technology are discussed while proposing its potential application to plant breeding.
Conclusions
Epimutations associated with useful traits may provide a valuable resource for crop development. It is important to explore epimutations in a variety of crop species while understanding the fundamental aspects of epigenetic regulation of agronomically important traits such as yield, quality, disease resistance and stress tolerance. In the end, plant breeding programs through epigenome editing may help not only to expand the use of limited genetic resources but also to alleviate consumers’ concerns about genetically manipulated crops.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In eukaryotic cells, DNA is not naked but interacts with histone proteins forming a compact structure called chromatin. The structural unit of chromatin is the nucleosome that consists of ~147 bp of DNA wrapped around an octamer of histone proteins H2A, H2B, H3 and H4 (Luger et al. 1997). Epigenetic modifications are covalent modifications of DNA or histones, which regulate mitotically or meiotically heritable changes in gene expression without altering the sequence of DNA itself (Heard and Martienssen 2014; Law and Jacobsen 2010). DNA methylation, a well-known epigenetic modification associated with gene silencing, refers to the addition of a methyl group onto C5 position of cytosine residues. Histone modifications include acetylation, methylation, ubiquitination and phosphorylation on specific amino acid residues of the histone N-terminal tails. Writers such as DNA methyltransferases (DNMTs), histone acetyltransferases (HATs) and histone lysine methyltransferases (HKMTs) introduce epigenetic marks, whereas erasers such as DNA demethylases, histone deacetylases (HDACs) and histone demethylases catalyze the removal of epigenetic marks. The interplay between writers and erasers is important for dynamic control of epigenetic modifications, thereby affecting chromatin conformation and transcriptional regulation (Feinberg et al. 2016; Jaenisch and Bird 2003; Torres and Fujimori 2015) Epigenetic marks are relatively stable and some of them, particularly DNA methylation, can be stably inherited over multiple generations. At the same time, most epigenetic modifications are also reversible, allowing gene expression to be switched on and off.
Epigenetic regulation plays pivotal roles in diverse cellular processes such as X chromosome inactivation, cell differentiation, development and senescence (He et al. 2011; Lamke and Baurle 2017; Ojolo et al. 2018; Zhao et al. 2019). Recent studies have revealed that aberrant epigenetic regulation is involved in many disease development in humans, including cancer, neurological disorder and autoimmune disease (Dawson and Kouzarides 2012; Kundaje et al. 2015). In some cases, the epigenetic alterations can lead to a heritable change in gene expression without change in the genome sequence, and such alterations are called an 'epimutation'. The corresponding epigenetic allele, also known as an 'epiallele', mostly arises from the alteration of DNA methylation due to its transgenerational inheritance (Hauser et al. 2011; Lloyd and Lister 2021; Quadrana and Colot 2016). In plants, several epialleles have been reported to regulate diverse developmental/physiological processes such as floral organ development (Jacobsen and Meyerowitz 1997), flowering time (Soppe et al. 2000), starch metabolism (Silveira et al. 2013), fruit ripening (Manning et al. 2006), vitamin E accumulation (Quadrana et al. 2014) and sex determination (Martin et al. 2009).
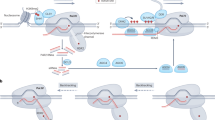
Advances in genome editing technology enable precisely targeted genome modifications using programmable DNA binding modules such as a zinc-finger (ZF) protein and transcription activator-like effector (TALE) fused with a Fok I nuclease or the bacteria-derived clustered regularly interspaced short palindromic repeat (CRISPR)-associated nuclease Cas9 (Kim 2016; Yin et al. 2017). Recently, epigenome editing technology has emerged for transcriptional regulation without altering DNA sequences. Epigenetic editing refers to directed alteration of epigenetic marks at specific genomic loci (Kungulovski and Jeltsch 2016). Programmable DNA-binding modules such as ZF, TALE, and catalytically inactive Cas9 (dCas9) can be fused with a catalytic domain of an epigenetic modifier (Fig. 1). Epigenome editing is a promising approach for selective gene activation or repression. In clinical applications, this technology was successfully used to alter specific epigenetic marks at the disease-related genes (Kungulovski and Jeltsch 2016; Nakamura et al. 2021). For example, silencing of the fragile X mental retardation-1 (FMR1) gene due to aberrant gain of DNA methylation causes a genetic disorder associated with intellectual disability (Mirabella et al. 2016; Sutcliffe et al. 1992). CRISPR/dCas9-fused ten-eleven translocation 1 (TET1) system restored normal DNA methylation levels and gene expression of FMR1 in iPSC-derived neurons (Liu et al. 2018). In addition, targeted DNA demethylation in the BRCA1 promoter decreased DNA methylation and rescued BRCA1 expression, leading to the inhibition of uncontrolled cell proliferation in breast and cervical cancer (Choudhury et al. 2016). Such studies set the stage for the potential use of epigenetic editing technology in clinical application (Goell and Hilton 2021; Nakamura et al. 2021). In plants, the manipulation of epigenetic modifications can also contribute to the creation of novel epialleles associated with useful traits for crop improvement.

Epigenome editing strategy for targeted manipulation of DNA methylation and histone modifications. a Fusion of DNA binding modules such as a ZF, TALE and dCas9 protein with epigenetic modifiers b Targeted deposition and removal of DNA methylation and histone modifications by epigenome editing systems. Reversible epigenome editing relies on antagonistic activities of epigenetic modifiers for transcriptional activation or repression
In this review, we briefly summarize the basic mechanism of epigenetic modifications and epigenetic modifier enzymes that can be used for epigenome editing technology. In addition, we introduce various epialleles controlling gene activity and phenotypic traits in diverse plant species. Then, we give an overview of current efforts aimed at editing the epigenome in plants and discuss the potentials of epigenetic manipulation for crop improvement.
Mechanisms of DNA methylation
In higher eukaryotes, DNA methylation is a prominent epigenetic modification that is mostly involved in transcriptional repression. DNA methylation is achieved by the addition of a methyl group to C5 position of cytosine. Unlike in mammals, DNA methylation in plants occurs in all sequence contexts: CG, CHG and CHH (H = A, T or C) (Law and Jacobsen 2010). The establishment of DNA methylation depends on the RNA-directed DNA methylation (RdDM) pathway, catalyzed by DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2), a homolog of mammalian de novo DNA methyltransferase DNMT3 (Law and Jacobsen 2010; Zhang et al. 2018). The RdDM pathway is divided into two major arms: RNA polymerase IV (RNA Pol IV)-mediated small interfering RNA (siRNA) biogenesis and RNA polymerase V (RNA Pol V)-mediated de novo DNA methylation (Zhang et al. 2018; Zhong et al. 2014). For siRNA biogenesis, RNA Pol IV is recruited to transcriptionally inactive transposable elements (TEs) which are enriched for H3 lysine 9 (H3K9) methylation. RNA Pol IV generates 30–40 nt single-stranded transcripts which are produced by RNA-DEPENDENT RNA POLYMERASE 2 (RDR2) and then processed into 24-nt siRNAs by DICER-LIKE 3 (DCL3) (Zhang et al. 2018). Next, the 24-nt siRNAs are loaded onto ARGONAUTE 4 (AGO4) or AGO6, and the complex is directed to nascent transcripts produced by RNA Pol V that are still associated with their chromatin template. RNA Pol V transcripts serve as an RNA scaffold on the chromatin that allows for siRNA-based silencing to direct de novo DNA methylation by DRM2 (Zhang et al. 2018). Once established, maintenance of DNA methylation is catalyzed by different DNA methyltransferases, depending on the sequence context (Law and Jacobsen 2010). Symmetric CG methylation, which is conserved across many organisms, requires METHYLTRANSFERASE 1 (MET1), a homolog of DNMT1 (Zhang et al. 2018). CHG and CHH methylation in heterochromatin are maintained by CHROMOMETHYLTRANSFERASE 2 (CMT2) and CMT3, respectively (Stroud et al. 2014; Zemach et al. 2013). In addition to CMT2, DRM2 is also likely involved in the maintenance of CHH methylation via the RdDM pathway (Zhang et al. 2018).
DNA methylation can be reversed by either passive or active mechanisms (Wu and Zhang 2010). Passive DNA demethylation occurs when DNA methylation maintenance mechanisms are impaired or absent during DNA replication. In contrast, active DNA demethylation takes place in a replication-independent manner and requires certain enzyme activities. Completely different enzymes are involved in mammal and plant DNA demethylation. In mammals, the TET family proteins catalyze the oxidation of 5mC to produce 5-hydroxymethylcytosine (5hmC). TET-dependent oxidation of 5hmC further produces 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), which are removed by the thymine DNA glycosylase (TDG) and then eventually replaced with unmethylated cytosine via the base excision repair pathway (Wu and Zhang 2017). On the contrary to mammals, plants have the DEMETER (DME) family of proteins that catalyze a direct removal of 5mC. As bifunctional DNA glycosylases, the DME family proteins cleave an N-glycosidic bond between the base and the ribose sugar and induce a single-strand break via β- and δ-elimination processes (Choi et al. 2002; Gehring et al. 2006; Gong et al. 2002; Penterman et al. 2007). Such DNA methylation modifiers may serve as effectors when fused with DNA-binding modules for target-specific manipulation of DNA methylation profiles.
Mechanisms of histone modifications
Post-translational modifications at the N-terminal region of histones, often called a histone tail, include acetylation, methylation, phosphorylation and ubiquitination, which can regulate chromatin dynamics and gene expression (Li et al. 2007; Strahl and Allis 2000). Acetylation of histone lysine (K) residues neutralizes the positive charge of the histone tail, thus weakening interactions between histones and DNA while increasing the accessibility of transcription factors to DNA. Histone acetylation generally leads to transcriptional activation, which is antagonistically regulated by HATs and HDACs. In Arabidopsis, 12 HAT and 18 HDAC genes have been identified (Pandey et al. 2002). Arabidopsis HATs can be divided into four groups: GCN5-related N-acetyltransferase (GNAT), MOZ, Ybf2/Sas2 and Tip60 (MYST), CREB-binding protein (CBP)/p300 and TATA-binding protein-associated factor 1 (TAF1). HDACs can be classified into three families: the Reduced Potassium Dependency 3 (RPD3)/HDA1, Silent Information Regulator 2 (SIR2) and histone deacetylase HD2, respectively (Berr et al. 2011; Servet et al. 2010). Histone acetylation mainly occurs at K9, K14, K18, K23 and K27 residues of histone H3, and K5, K8, K12, K16 and K20 residues of histone H4. Histone acetylation regulates diverse biological functions in plants such as growth, development and responses to abiotic and biotic stresses (Earley et al. 2007; Ma et al. 2013; Zhang et al. 2007).
Lysine or arginine residues can be subjected to histone methylation. HKMTs catalyze mono-, di-, or trimethylation on K4, K9, K27 and K36 residues of H3 and K20 of H4. In general, H3K9 dimethylation and H3K27 trimethylation are associated with transcriptionally repressed genes, whereas H3K4 trimethylation and H3K36 trimethylation are associated with active genes (Berger 2007). HKMTs contain the conserved SUPPRESSOR OF VARIEGATION [SU(VAR)3–9], ENHANCER OF ZESTE and TRITHORAX (SET) domains, and the Arabidopsis genome encodes 41 SET domain proteins (Liu et al. 2010). In addition, H3K9 methylation is catalyzed by the SU(VAR)3–9 homolog (SUVH) family such as KRYPTONITE (KYP; also known as SUVH4), SUVH5 and SUVH6, and is closely associated with DNA methylation as in mammals (Du et al. 2015; Ebbs and Bender 2006). Histone methylation marks can be removed by histone demethylases. In Arabidopsis, histone demethylases are encoded by four LYSINE-SPECIFIC DEMETHYLASE 1 (LSD1) homologs and 21 JUMONJI-C-DOMAIN (JmjC) protein genes, some of which are involved in flowering time control (Berr et al. 2011; Chen et al. 2011).
Epialleles in plants
Epigenetic alterations of genes may lead to changes in their expression, and in some cases, both altered epigenetic states and expression patterns are mitotically/meiotically stable and inherited to the offspring. An individual with altered epigenetic modifications may give rise to the offspring with different phenotypes, despite the same DNA sequence at the corresponding gene. ‘Epimutants’ carry heritable phenotypic changes not caused by DNA mutation but rather by epigenetic alterations, and the corresponding epigenetic alleles are called ‘epialleles’. Epimutants may occur naturally or artificially (Table 1), and most of the epialleles are caused by changes in DNA methylation profiles.
The Arabidopsis epimutants often emerge in the mutant background defective for DNA methylation. MET1 and DECREASE IN DNA METHYLATION 1 (DDM1) encode the maintenance DNA methyltransferase and a SWI/SNF-like ATP-dependent chromatin remodeling factor, respectively (Jeddeloh et al. 1998; Vongs et al. 1993), and the met1 and ddm1 mutants have a genome-wide decrease of DNA methylation level. Several epimutations accompanied with phenotypic changes were caused by heritable changes of DNA methylation. For example, the epimutant of FLOWERING WAGENINGEN (FWA) induced in a ddm1 mutant background has loss of DNA methylation in the repeat regions upstream of the coding sequence with transcription up-regulation of FWA, leading to late flowering (Soppe et al. 2000). The bonsai (bns) allele is the ddm1-induced epiallele caused by epigenetic silencing of a subunit of the putative Anaphase-Promoting Complex 13 (Saze and Kakutani 2007). Silencing of BNS associated with DNA hypermethylation at long interspersed nuclear elements (LINEs) gives rise to a reduced plant height (Saze and Kakutani 2007). An epigenetically silenced version of SUPERMAN (SUP) is another example of an epiallele, causing increased numbers of stamens and carpels in Arabidopsis flowers (Jacobsen and Meyerowitz 1997).
In addition to artificially induced epimutations, several studies reported epimutants and epialleles that spontaneously occur in nature. The naturally occurring epiallele was first reported in Linaria vulgaris, in which an asymmetric flower was transformed into a radially symmetric (peloric) flower (Cubas et al. 1999). The abnormal flower structure is caused by DNA hypermethylation and silencing of the Lcyc gene, which encodes a CYCLOIDEA gene that determines the dorsoventral asymmetry of flowers in Antirrhinum. The epimutant of Lcyc was originally described more than 250 years ago by Linnaeus, suggesting that an epiallele can be stable enough for transgenerational inheritance over multiple generations. In tomato (Solanum lycopersicum), fruit ripening is inhibited in the Colorless non-ripening (Cnr) mutant (Manning et al. 2006). The Cnr gene encodes the SBP-box transcription factor, and its promoter in the Cnr mutant was hypermethylated, leading to gene silencing (Manning et al. 2006). The follow-up study revealed that DNA methylation at the Cnr promoter regions gradually decreases during fruit ripening in wild type, indicating that DNA methylation is also developmentally regulated (Zhong et al. 2013). In natural populations of Arabidopsis, expression of Qua-Quin Starch (QQS), the gene involved in starch metabolism, is negatively correlated with the DNA methylation level in its promoter region. Natural epigenetic variations can be stably inherited for several generations (Silveira et al. 2013). In rice, Epi-d1 is the first identified epigenetic mutant and has a metastable dwarf phenotype caused by epigenetic silencing of the DWARF1 gene (Miura et al. 2009). Epi-df is a gain-of-function epiallele in rice caused by DNA hypermethylation in the promoter of FERTILIZATION INDEPENDENT ENDOSPREM 1 (FIE1), inducing floral defects (Zhang et al. 2012). In the epimutant of Epi-RAV6, larger lamina inclination and smaller grain size phenotypes result from hypermethylation in the 5’ region of RELATED TO ABSCISIC ACID INSENSITIVE 3 VIVIPAROUS1 6 (RAV6) (Zhang et al. 2015).
In many cases, epimutations are associated with TE insertions. In Arabidopsis, the wild type FWA gene and bna epiallele are silenced due to DNA hypermethylation of short interspersed nuclear elements (SINEs) and LINEs, respectively (Saze and Kakutani 2007; Soppe et al. 2000). Recent study showed that DNA methylation of retrotransposon NMR19 near the PHEOPHYTIN PHEOPHORBIDE HYDROLASE (PPH) gene leads to gene silencing and is negatively associated with leaf senescence (He et al. 2018). The methylation patterns of NMR19 are correlated with local climates, suggesting the roles of epialleles for environmental adaptation. In melon (Cucumis melo), an insertion of hAT transposon near the CmWIP1 gene induces the spreading of DNA methylation to the promoter leading to gene silencing (Martin et al. 2009). Expression of CmWIP1 promotes carpel abortion and development of male flowers, whereas its silencing suppresses anther development, mainly producing female flowers (Martin et al. 2009). Therefore, CmWIP1 acts as a sex determination switch for melon flower development. In tomato, expression of VITAMIN E PATHWAY GENE 3 (VTE3), encoding 2-methyl-6-phytylquinol methyltransferase that catalyzes the final steps of vitamin E biosynthesis, is modulated by differential DNA methylation of a SINE element located in the promoter region. DNA methylation of the VTE3 promoter is spontaneously reverted, generating different epialleles with varying expression levels. This indicates that naturally occurring epialleles are also involved in the regulation of metabolite contents. In African oil palm (Elaeis guineensis), the Karma TE was inserted in the intron of the DEFICIENS (DEF1) gene, which encodes the B-class MADS-box transcription factor. DNA hypomethylation of Karma TE is associated with alternative splicing of DEF1, resulting in the mantled fruit phenotype (Ong-Abdullah et al. 2015).
Targeted DNA methylation in plants
Several studies on targeted DNA methylation have been reported in plants (Table 2). The first targeted epigenetic manipulation in Arabidopsis utilized a ZF protein and an RdDM component SUVH9 (Johnson et al. 2014). This study demonstrated that the fusion of ZF and SUVH9 successfully induced gain of DNA methylation at an unmethylated epiallele of fwa-4 via the RdDM pathway, leading to FWA silencing and an early flowering phenotype (Johnson et al. 2014). This study also showed that the altered DNA methylation pattern was stably transmitted to the next generation even without a transgene module (Johnson et al. 2014). The following study utilized the ZF-based targeting approach to elucidate how other RdDM components act in de novo DNA methylation (Gallego-Bartolome et al. 2019). The fusion of ZF and RdDM components showed that co-targeting of RNA Pol IV and RNA Pol V complexes enhanced the efficiency of targeted DNA methylation (Gallego-Bartolome et al. 2019). In addition to the plant-specific RdDM components, Spiroplasma sp. strain MQ1 CG methyltransferase M.SssI (SssI) fused with the ZF protein can establish heritable CG methylation at the FWA promoter and repress its expression (Liu et al. 2021). However, transgenic lines expressing the ZF fusions with SssI and RdDM components exhibited genome-wide ectopic DNA methylation. In order to avoid target non-specific hypermethylation, the CRISPR/dCas9-based system employed a variant of MQ1 (MQ1v) having reduced affinity to DNA (Ghoshal et al. 2021). Compared to the fusion of ZF and SssI, the CRISPR/dCas9-mediated MQ1v caused heritable DNA methylation at FWA promoters with minimal off-target effects. Moreover, a combination of the SunTag system that recruits multiple copies of the same modifier with CRISPR/dCas9 enhanced the efficiency of FWA-targeted DNA methylation (Ghoshal et al. 2021). Similarly, the SunTag-CRISPR/dCas9 system fused with the catalytic domain of Nicotiana tabacum DRM methyltransferase (NtDRMcd) efficiently targeted DNA methylation to the FWA promoter (Papikian et al. 2019). These studies demonstrate that a variety of DNA methyltransferases with DNA-binding modules can efficiently induce heritable DNA methylation at target sites. Occasionally, a few examples showed incomplete inheritance of modified DNA methylation and off-target effects, suggesting that an efficient and precise DNA-binding module is needed for successful manipulation of target DNA methylation.
Targeted DNA demethylation in plants
For targeted DNA demethylation, DNA demethylases can serve as effectors when fused to DNA-binding modules (Table 2). In plants, the first example of targeted DNA demethylation utilized the human TET1 catalytic domain (TET1cd) along with the ZF protein or the SunTag-CRISPR/dCas9 system (Gallego-Bartolome et al. 2018). When TET1cd was fused to the ZF protein targeting the FWA promoter (hereafter called ZFFWA-TET1cd), the fusion protein decreased DNA methylation levels at the FWA promoter and induced the corresponding late-flowering phenotype (Gallego-Bartolome et al. 2018). Such loss of DNA methylation was heritable even when the DNA demethylation transgene module was segregated out (Gallego-Bartolome et al. 2018). Additionally, the ZF-TET1cd targeting the CACTA1 transposon within a heterochromatic region (hereafter called ZFCACTA1-TET1cd) exhibited loss of DNA methylation and TE reactivation. However, on the contrary to the ZFFWA-TET1, DNA methylation at CACTA1 was re-established and its silencing was restored in the absence of ZFCACTA1-TET1cd (Gallego-Bartolome et al. 2018). It indicates that the incomplete removal of DNA methylation at the heterochromatic regions is likely to attract the RdDM machinery, leading to the re-establishment of DNA methylation. This study also reported that the SunTag-CRISPR/dCas9-TET1cd system induced DNA demethylation at the FWA and CACTA1 loci with marginal off-target effects (Gallego-Bartolome et al. 2018). Compared to the ZFCACTA1-TET1cd showing genome-wide hypomethylation, the SunTag-CRISPR/dCas9-TET1cd improved DNA demethylation at targets (Gallego-Bartolome et al. 2018). In these selective DNA methylation/demethylation systems, the SunTag can efficiently recruit MQ1, NtDRMcd or TET1cd to the target site, resulting in heritable alteration of DNA methylation. Alternatively, DME family proteins that catalyze a direct removal of 5mC base may be considered a powerful tool to edit DNA methylation.
Manipulation of histone modifications in plants
Besides manipulation of DNA methylation, epigenome editing with histone-modifying enzymes has been demonstrated in several organisms. In mouse primary neurons, the TALE-KYP fusion was found to induce H3K9 monomethylation at the promoter and repress the expression of the target gene (Konermann et al. 2013). Recently, several cases of CRISPR/dCas9-mediated manipulation of histone modifications have been reported in plants (Table 2). In an effort to increase the editing efficiency, the sgRNA scaffold in CRISPR/dCas9 system was engineered to recruit multiple MS2 bacteriophage coat proteins to concentrate effectors with a single guiding complex (Konermann et al. 2015). The catalytic domains of histone modifying enzymes such as H3K9 methyltransferases G9a and KYP for histone methylation and human HAT p300 for histone acetylation were fused with MS2 to target the flowering time gene FLOWERING LOCUS T (FT) (Lee et al. 2019). While MS2-G9a failed to cause late flowering, the MS2-KYP transgenic plants exhibited the late flowering phenotype inherited to the next generation (Lee et al. 2019). The MS2-KYP transgenic plants did not exhibit any significant enrichment for H3K9 dimethylation at the FT promoter, but FT expression was significantly decreased compared to the wild-type (Lee et al. 2019). The MS2-p300 was able to increase H3K27 acetylation at the FT promoter despite the minor effects on FT expression and flowering time (Lee et al. 2019). These results indicate that transcriptional regulation is not always correlated with the alteration of histone modifications at target loci. Furthermore, the dCas9-based epigenome editing tool was used to improve drought stress tolerance (Paixao et al. 2019). Arabidopsis HAT1 in fusion with dCas9 was employed for histone acetylation at the promoter region of ABSCISIC ACID-RESPONSIVE ELEMENT BINDING PROTEIN1 (AREB1; also known as ABF2), a key determinant in drought stress response. With an increase in AREB1 expression, the dCas9-HAT1 transgenic plants displayed enhanced drought stress tolerance, although histone acetylation enrichment was elusive (de Melo et al. 2020; Paixao et al. 2019). Nevertheless, editing of histone code will be a fascinating approach to manipulate the chromatin state in conjunction with a dramatic change in the corresponding phenotype.
Conclusions
In this review, we summarized currently available epialleles and epigenome editing tools in plants. Some epigenetic mutations without a change in DNA sequence can be transgenerationally inherited to the offspring. Thus, epigenetic variations may serve as a novel source for plant breeding with genetic/phenotypic stability and heritability. Although few studies on targeted epigenetic modifications have been reported in plants, the epigenome editing technology can be applied to the artificial creation of epialleles, producing breeding materials in the future.
Despite epigenome engineering is considered a powerful technology for arbitrary control of gene expression, further investigations are needed for the precise manipulation of epigenetic marks. In plants, targeted DNA methylation was mainly performed at the FWA locus as a reference but many other loci remain to be investigated. Moreover, fewer cases of targeted histone manipulation were reported in plants. Some histone marks seem unlikely to be affected by epigenetic modifiers, and the transgenerational epigenetic inheritance of modified histone marks is still unveiled. This suggests that an epigenetic crosstalk and the reversibility of histone modifications may compromise the reliability of epigenome manipulation in plants. To increase the efficiency of epigenome editing, a multifaceted strategy targeting various epigenetic modifications is required, and advances in epigenome editing technology will provide new opportunities for crop improvement.
References
Bender J, Fink GR (1995) Epigenetic control of an endogenous gene family is revealed by a novel blue fluorescent mutant of Arabidopsis. Cell 83:725–734
Berger SL (2007) The complex language of chromatin regulation during transcription. Nature 447:407–412
Berr A, Shafiq S, Shen WH (2011) Histone modifications in transcriptional activation during plant development. BBA-Gene Regul Mech 1809:567–576
Blevins T, Wang J, Pflieger D, Pontvianne F, Pikaard CS (2017) Hybrid incompatibility caused by an epiallele. Proc Natl Acad Sci U S A 114:3702–3707
Chen XS, Hu YF, Zhou DX (2011) Epigenetic gene regulation by plant Jumonji group of histone demethylase. BBA-Gene Regul Mech 1809:421–426
Choi YH, Gehring M, Johnson L, Hannon M, Harada JJ, Goldberg RB, Jacobsen SE, Fischer RL (2002) DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 110:33–42
Choudhury SR, Cui Y, Lubecka K, Stefanska B, Irudayaraj J (2016) CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 7:46545–46556
Cubas P, Vincent C, Coen E (1999) An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401:157–161
Dawson MA, Kouzarides T (2012) Cancer epigenetics: from mechanism to therapy. Cell 150:12–27
de Melo BP, Lourenco-Tessutti IT, Paixao JFR, Noriega DD, Silva MCM, de Almeida-Engler J, Fontes EPB, Grossi-De-Sa MF (2020) Transcriptional modulation of AREB-1 by CRISPRa improves plant physiological performance under severe water deficit. Sci Rep 10:16231
Du JM, Johnson LM, Jacobsen SE, Patel DJ (2015) DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol 16:519–532
Durand S, Bouche N, Strand EP, Loudet O, Camilleri C (2012) Rapid establishment of genetic incompatibility through natural epigenetic variation. Curr Biol 22:326–331
Earley KW, Shook MS, Brower-Toland B, Hicks L, Pikaard CS (2007) In vitro specificities of Arabidopsis co-activator histone acetyltransferases: implications for histone hyperacetylation in gene activation. Plant J 52:615–626
Ebbs ML, Bender J (2006) Locus-specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell 18:1166–1176
Feinberg AP, Koldobskiy MA, Gondor A (2016) Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 17:284–299
Gallego-Bartolome J, Gardiner J, Liu WL, Papikian A, Ghoshal B, Kuo HY, Zhao JMC, Segal DJ, Jacobsen SE (2018) Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain. Proc Natl Acad Sci U S A 115:E2125–E2134
Gallego-Bartolome J, Liu W, Kuo PH, Feng S, Ghoshal B, Gardiner J, Zhao JM, Park SY, Chory J, Jacobsen SE (2019) Co-targeting RNA polymerases IV and V promotes efficient de novo DNA methylation in Arabidopsis. Cell 176:1068–1082e1019
Gehring M, Huh JH, Hsieh TF, Penterman J, Choi Y, Harada JJ, Goldberg RB, Fischer RL (2006) DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell 124:495–506
Ghoshal B, Picard CL, Vong B, Feng S, Jacobsen SE (2021) CRISPR-based targeting of DNA methylation in Arabidopsis thaliana by a bacterial CG-specific DNA methyltransferase. Proc Natl Acad Sci U S A 118:e2125016118
Goell JH, Hilton IB (2021) CRISPR/Cas-based epigenome editing: advances, applications, and clinical utility. Trends Biotechnol 39:678–691
Gong ZH, Morales-Ruiz T, Ariza RR, Roldan-Arjona T, David L, Zhu JK (2002) ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA glycosylase/lyase. Cell 111:803–814
Hauser MT, Aufsatz W, Jonak C, Luschnig C (2011) Transgenerational epigenetic inheritance in plants. BBA-Gene Regul Mech 1809:459–468
He L, Wu WW, Zinta G, Yang L, Wang D, Liu RY, Zhang HM, Zheng ZM, Huang H, Zhang QZ et al (2018) A naturally occurring epiallele associates with leaf senescence and local climate adaptation in Arabidopsis accessions. Nat Commun 9:460
Heard E, Martienssen RA (2014) Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157:95–109
Jacobsen SE, Meyerowitz EM (1997) Hypermethylated SUPERMAN epigenetic alleles in Arabidopsis. Science 277:1100–1103
Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33:245–254
Jeddeloh JA, Bender J, Richards EJ (1998) The DNA methylation locus DDM1 is required for maintenance of gene silencing in Arabidopsis. Genes Dev 12:1714–1725
Johnson LM, Du J, Hale CJ, Bischof S, Feng S, Chodavarapu RK, Zhong X, Marson G, Pellegrini M, Segal DJ et al (2014) SRA- and SET-domain-containing proteins link RNA polymerase V occupancy to DNA methylation. Nature 507:124–128
Kim JS (2016) Genome editing comes of age. Nat Protoc 11:1573–1578
Konermann S, Brigham MD, Trevino A, Hsu PD, Heidenreich M, Cong L, Platt RJ, Scott DA, Church GM, Zhang F (2013) Optical control of mammalian endogenous transcription and epigenetic states. Nature 500:472–476
Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H et al (2015) Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517:583–U332
Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ et al (2015) Integrative analysis of 111 reference human epigenomes. Nature 518:317–330
Kungulovski G, Jeltsch A (2016) Epigenome editing: state of the art, concepts, and perspectives. Trends Genet 32:101–113
Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11:204–220
Lee JE, Neumann M, Duro DI, Schmid M (2019) CRISPR-based tools for targeted transcriptional and epigenetic regulation in plants. PLoS ONE 14:e022778
Li B, Carey M, Workman JL (2007) The role of chromatin during transcription. Cell 128:707–719
Liu CY, Lu FL, Cui X, Cao XF (2010) Histone methylation in higher plants. Annu Rev Plant Biol 61:395–420
Liu XS, Wu H, Krzisch M, Wu XB, Graef J, Muffat J, Hnisz D, Li CH, Yuan BB, Xu CY et al (2018) Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell 172:979–
Liu W, Gallego-Bartolome J, Zhou Y, Zhong Z, Wang M, Wongpalee SP, Gardiner J, Feng S, Kuo PH, Jacobsen SE (2021) Ectopic targeting of CG DNA methylation in Arabidopsis with the bacterial SssI methyltransferase. Nat Commun 12:3130
Lloyd JPB, Lister R (2021) Epigenome plasticity in plants. Nat Rev Genet 2:1–14
Luan X, Liu SC, Ke SW, Dai H, Xie XM, Hsieh TF, Zhang XQ (2019) Epigenetic modification of ESP, encoding a putative long noncoding RNA, affects panicle architecture in rice. Rice 12:20
Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251–260
Ma XJ, Lv SB, Zhang C, Yang CP (2013) Histone deacetylases and their functions in plants. Plant Cell Rep 32:465–478
Manning K, Tor M, Poole M, Hong Y, Thompson AJ, King GJ, Giovannoni JJ, Seymour GB (2006) A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat Genet 38:948–952
Martin A, Troadec C, Boualem A, Rajab M, Fernandez R, Morin H, Pitrat M, Dogimont C, Bendahmane A (2009) A transposon-induced epigenetic change leads to sex determination in melon. Nature 461:1135–U1237
Mirabella AC, Foster BM, Bartke T (2016) Chromatin deregulation in disease. Chromosoma 125:75–93
Miura K, Agetsuma M, Kitano H, Yoshimura A, Matsuoka M, Jacobsen SE, Ashikari M (2009) A metastable DWARF1 epigenetic mutant affecting plant stature in rice. Proc Nati Acad Sci U S A 106:11218–11223
Nakamura M, Gao YC, Dominguez AA, Qi LS (2021) CRISPR technologies for precise epigenome editing. Nat Cell Biol 23:11–22
Ong-Abdullah M, Ordway JM, Jiang N, Ooi SE, Kok SY, Sarpan N, Azimi N, Hashim AT, Ishak Z, Rosli SK et al (2015) Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature 525:533–537
Paixao JFR, Gillet FX, Ribeiro TP, Bournaud C, Lourenco-Tessutti IT, Noriega DD, de Melo BP, de Almeida-Engler J, Grossi-de-Sa MF (2019) Improved drought stress tolerance in Arabidopsis by CRISPR/dCas9 fusion with a histone acetyltransferase. Sci Rep 9:8080
Pandey R, Muller A, Napoli CA, Selinger DA, Pikaard CS, Richards EJ, Bender J, Mount DW, Jorgensen RA (2002) Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic Acids Res 30:5036–5055
Papikian A, Liu WL, Gallego-Bartolome J, Jacobsen SE (2019) Site-specific manipulation of Arabidopsis loci using CRISPR-Cas9 SunTag systems. Nat Commun 10:729
Penterman J, Zilberman D, Huh JH, Ballinger T, Henikoff S, Fischer RL (2007) DNA demethylation in the Arabidopsis genome. Proc Natl Acad Sci U S A 104:6752–6757
Quadrana L, Almeida J, Asis R, Duffy T, Dominguez PG, Bermudez L, Conti G, da Silva JVC, Peralta IE, Colot V et al (2014) Natural occurring epialleles determine vitamin E accumulation in tomato fruits. Nat Commun 5:4027
Quadrana L, Colot V (2016) Plant transgenerational epigenetics. Annu Rev Genet 50:467–491
Saze H, Kakutani T (2007) Heritable epigenetic mutation of a transposon-flanked Arabidopsis gene due to lack of the chromatin-remodeling factor DDM1. EMBO J 26:3641–3652
Servet C, Silva NCE, Zhou DX (2010) Histone acetyltransferase AtGCN5/HAG1 is a versatile regulator of developmental and inducible gene expression in Arabidopsis. Mol Plant 3:670–677
Silveira AB, Trontin C, Cortijo S, Barau J, Del Bem LEV, Loudet O, Colot V, Vincentz M (2013) Extensive natural epigenetic variation at a de novo originated gene. Plos Genet 9:e1003437
Soppe WJJ, Jacobsen SE, Alonso-Blanco C, Jackson JP, Kakutani T, Koornneef M, Peeters AJM (2000) The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol Cell 6:791–802
Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403:41–45
Stroud H, Do T, Du J, Zhong X, Feng S, Johnson L, Patel DJ, Jacobsen SE (2014) Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nature Structural & Molecular Biology 21(1) 64–72. https://doi.org/10.1038/nsmb.2735
Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, Warren ST (1992) DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet 1:397–400
Torres IO, Fujimori DG (2015) Functional coupling between writers, erasers and readers of histone and DNA methylation. Curr Opin Struct Biol 35:68–75
Vongs A, Kakutani T, Martienssen RA, Richards EJ (1993) Arabidopsis thaliana DNA methylation mutants. Science 260:1926–1928
Wu SC, Zhang Y (2010) Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol 11:607–620
Wu X, Zhang Y (2017) TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 18:517–534
Yin KQ, Gao CX, Qiu JL (2017) Progress and prospects in plant genome editing. Nat Plants 3:17107
Zemach A, Kim MY, Hsieh PH, Coleman-Derr D, Eshed-Williams L, Thao K, Harmer SL, Zilberman D (2013) The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153:193–205
Zhang K, Sridhar VV, Zhu J, Kapoor A, Zhu JK (2007) Distinctive core histone post-translational modification patterns in Arabidopsis thaliana. PLoS ONE 2:e1210
Zhang LG, Cheng ZJ, Qin RZ, Qiu Y, Wang JL, Cui XK, Gu LF, Zhang X, Guo XP, Wang D et al (2012) Identification and characterization of an Epi-Allele of FIE1 reveals a regulatory linkage between two epigenetic marks in rice. Plant Cell 24:4407–4421
Zhang XQ, Sun J, Cao XF, Song XW (2015) Epigenetic mutation of RAV6 affects leaf angle and seed size in rice. Plant Physiol 169:2118–2128
Zhang H, Lang Z, Zhu JK (2018) Dynamics and function of DNA methylation in plants. Nat Rev Mol Cell Biol 19:489–506
Zhong S, Fei Z, Chen YR, Zheng Y, Huang M, Vrebalov J, McQuinn R, Gapper N, Liu B, Xiang J et al (2013) Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat Biotechnol 31:154–159
Zhong X, Du J, Hale CJ, Gallego-Bartolome J, Feng S, Vashisht AA, Chory J, Wohlschlegel JA, Patel DJ, Jacobsen SE (2014) Molecular mechanism of action of plant DRM de novo DNA methyltransferases. Cell 157:1050–1060
Acknowledgements
This work was supported by the National Agricultural Genome Program (PJ013440) and the New breeding technologies development Program (PJ01480103) by Rural Development Administration (RDA), Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that thy have no conflicts of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shin, H., Choi, W.L., Lim, J.Y. et al. Epigenome editing: targeted manipulation of epigenetic modifications in plants. Genes Genom 44, 307–315 (2022). https://doi.org/10.1007/s13258-021-01199-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-021-01199-5