
Abstract
Background
Drought is the major abiotic stress factor that negatively influences growth and yield in cereal grain crops such as maize (Zea mays L.). A multitude of genes and pathways tightly modulate plant growth, development and responses to environmental stresses including drought. Therefore, crop breeding efforts for enhanced drought resistance require improved knowledge of plant drought responses.
Objective
Here, we sought to elucidate the molecular and physiological mechanisms underpinning maize drought stress tolerance.
Methods
We therefore applied a 12-day water-deficit stress treatment to maize plants of two contrasting (drought tolerant ND476 and drought sensitive ZX978) hybrid cultivars at the late vegetative (V12) growth stage and performed a large-scale RNA sequencing (RNA-seq) transcriptome analysis of the leaf tissues.
Results
A comparative analysis of the two genotypes leaf transcriptomes and physiological parameters revealed the key differentially expressed genes (DEGs) and metabolic pathways that respond to drought in a genotype-specific manner. A total of 3114 DEGs were identified, with 21 DEGs being specifically expressed in tolerant genotype ND476 in response to drought stress. Of these, genes involved in secondary metabolites biosynthesis, transcription factor regulation, detoxification and stress defense were highly expressed in ND476. Physiological analysis results substantiated our RNA-seq data, with ND476 exhibiting better cell water retention, higher soluble protein content and guaiacol peroxidase activity, along with low lipid peroxidation extent than the sensitive cultivar ZX978 under drought conditions.
Conclusion
Our findings enrich the maize genetic resources and enhance our further understanding of the molecular mechanisms regulating drought stress tolerance in maize. Additionally, the DEGs screened in this study may provide a foundational basis for our future targeted cloning studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite the sharp rise in global demand for crop products, driven by population and income growth (Schils et al. 2018), agricultural productivity is challenged by numerous abiotic stress factors, often exacerbated by the changing global climate (Fahad et al. 2017). Additionally, the emerging role of crop products as raw material inputs in the transition from fossil fuels towards climate-smart low-fossil-carbon economies is putting further strain on agricultural systems (Schils et al. 2018). Among all the abiotic stress factors, drought is the most devastating (Basu et al. 2016), and the frequency of extreme drought events is estimated to increase under the changing climate scenario, further worsening damage to plants and crop yields (Zhao et al. 2016a). Therefore, to maintain sustainably higher crop yields, crop scientists need to understand how plants respond to drought stress and employ the generated knowledge in modern breeding programs.
Being an economically important cereal crop, ranked third after rice (Oryza sativa L.) and wheat (Triticum aestivum L., Thirunavukkarasu et al. 2017), maize (Zea mays L.) continue to play a critical role in evading the production gap of human food calories, animal feeds and bio-fuels in the world (Shiferaw et al. 2011; Opitz et al. 2016. However, the overall production of maize is adversely affected by drought stress (Singh et al. 2019). Drought inhibits growth by negatively impacting plant physiological processes (Prasad et al. 2008; Bhargava and Sawant, 2013; Anjum et al. 2011). It decreases plant cell turgor, inhibits CO2 exchange, decreases photosynthetic efficiency, increases post-pollination embryo abortion rate, ultimately leading to reduced yield (Farooq et al. 2009; Zhang et al. 2018).
In response to drought and other stressors, plants institute numerous adaptive responses activated at the physiological, biochemical and molecular levels. At the biochemical and physiological levels, plants activate metabolic and osmotic adjustments, antioxidant scavenging defense and phytohormones (Fang and Xiong 2015; Basu et al. 2016; Liu et al. 2019; Singh et al. 2019). The role of stress phytohormones such as abscisic acid (ABA), jasmonic acid and salicylic acid in conferring plant drought tolerance has been acknowledged (Aimar et al. 2011; Aslam et al. 2015; Fahad et al. 2017). Moreover, several researchers have identified key regulators and pathways underlying molecular mechanisms of maize drought stress response. These include calcium (Ca2+) signal sensors, calcium-dependant protein kinases (CDPKs) and mitogen-activated protein kinases (MAPKs) responsible for signal transduction, stress response proteins such as the heat shock proteins (HSPs), dehydrins, late embryogenic abundant (LEA) proteins and aquaporins (Al-Whaibi 2011; Bhargava and Sawant 2013; Harb 2016; Tai et al. 2016; Priya et al. 2019). Additionally, several TFs also regulate drought stress response including myeloblastosis (MYB), dehydration responsive element binding (DREB), abscisic acid responsive elements binding factor (ABF), [NAM, ATAF1/2, and CUC2 containing proteins] (NAC) and WRKY, among others (Singh et al. 2015; Wang et al. 2018; Priya et al. 2019). Further, epigenetic regulation mechanisms at the DNA and histone levels, as well as regulation by small non-coding RNAs in drought stress tolerance have become important (Banerjee and Roychoudhury 2017). Despite all this, however, the elucidation of drought stress response mechanisms in crop plants still remains elusive.
Previous researchers have monitored the global gene expression profiles in response to water deficit stress in different maize parts including roots, leaves, and kernels by microarray analysis (Zheng et al. 2010; Opitz et al. 2014). With the revolutionisation of the next generation sequencing (NGS) approaches came RNA sequencing (RNA-seq) method to help us decipher plants` abiotic stress responses. RNA-Seq accords for fully quantitative gene expression analysis with absolute values and capturing of very subtle expression changes. Moreover, it provides for low cost, high throughput and high sensitivity analysis of data (Kakumanu et al. 2012; Opitz et al. 2016). RNA-seq technology has been employed in drought stress response studies in maize (Kakumanu et al. 2012; Shan et al. 2013; Bhanu et al. 2016; Min et al. 2016; Zenda et al. 2019). Although these studies provided global insight into the maize drought stress responses, most of them used inbred lines as experimental materials, did not target the late vegetative (V12) crop developmental stage, and were conducted under greenhouse environments. Yet, every year, most farmers exploit the advantage of hybid vigour by planting hybrids under field conditions (Danilevskaya et al. 2019), and moisture deficit stress at the V12 stage drastically impact yield. This is so because the V12 stage is the most vigorous growth and development period in the whole maize life cycle (Wang et al. 2019a). At this phase, both number of ears per plant and number of kernel rows on an ear are being determined (Darby and Lauer 2006), so moisture deficit will have serious consequences on yield (Aslam et al. 2015). Additionally, more controlled environments are devoid of fluctuations in meteorological conditions (heat, solar radiation, wind and vapour pressure) which play vital roles in modulating plant growth and development (Danilevskaya et al. 2019). Therefore, it becomes imperative that we dissect maize hybrid cultivars` V12-stage-specific drought stress responses under conditions that closely mimic field growing environments.
In the current study, therefore, we have employed the use of the RNA-seq method, integrated with the physiological analysis approach, to understand the molecular and physiological mechanisms associated with drought stress responses in maize at the V12 stage, by way of comparative analysis of two diverse (drought-tolerant Nongdan 476 (ND476) and drought-sensitive Zhongxin 978 (ZX978) hybrid cultivars grown under rain-proof shelters. We hope that the results generated in the current paper will provide knowledge that could be useful in future crop breeding programs aimed at engineering drought tolerant crop cultivars.
Materials and methods
Plant materials and drought treatment procedure
The two contrasting (comparably drought tolerant ND476 and sensitive ZX978) maize hybrids used in this study were provided the North China Key Laboratory for Crop Germplasm Resources of Education Ministry, Hebei Agricultural University, China. Maize hybrid ND476 is a comparably drought-resistant cultivar bred by the Dryland Research Institute of Hebei Academy of Agricultural and Forestry Sciences (Hengshui, China), and ZX978,a comparably drought sensitive cultivar, was developed by the Hebei Zhongxin Seed Technology Company Limited (Handan, Hebei Province, China). The experiment was conducted between May and July 2018 in a fully automated rain-proof shelter at Qing Yuan Experimental Station, Baoding, China (115.5602790 E; 38.7950930 N; 118 m). Each experimental plot measured 25 m2 (5 m × 5 m), with 60 cm * 30 cm plant spacings. The soil water content was kept between 70 and 80% in the well-watered plots (control) and 15–20% in water-stressed plots (treatment) (Hsiao 1973). The relative soil water content of one meter underground was monitored by TZS-1 soil moisture measurement instrument (Zhejiang Tuopu Technology Co. Ltd, Zhejiang, China). To prevent the transverse infiltration of soil moisture, building waterproof membranes of one-meter depth were put between control and treatments units.
For the drought treatment group, plants were water deprived for 12 days from eight fully-expanded-leaves (FEL) (V8) to twelve FEL (V12) stage. Leaf tissues for both RNA-seq and qRT-PCR experiments were collected from the flag leaves of ND476 and ZX978 cultivars under both control and drought treatment conditions after twelve days of drought stress exposure; each sample had three technical replicates. All the leaf samples were immediately frozen in liquid nitrogen and then stored at − 80 °C for further analysis (Jin et al. 2019).
Phenotypic and physiological characterization
Phenotypic and physiological characterizations were measured for ND476 and ZX978 cultivars at the V12 stage under well-watered (control) and water-deficit (drought) treatment conditions as previously described (Wang et al. 2019b). In brief, we estimated the leaves` relative water content (RWC) by the method of Galmés et al (2007). Leaf guaiacol peroxidase (POD) activity was determined using Han (2008) method. The leaf cell membrane stability index, estimated as the level of lipid peroxidation (MDA content) was measured by the thiobarbituric acid method (Dhindsa, et al. 1981). Further, we estimated the soluble leaf protein content (SPC) using the Coomassie Brilliant Blue G250 method (Bradford, 1976).
Total RNA extraction, cDNA library construction and RNA sequencing
Maize hybrids ND476 (drought-tolerant) and ZX978 (drought-sensitive) were grown according to the method described (Jin et al. 2019). Total RNA of the control and drought-exposed flag-leaf samples which had been stored at −80 ℃ was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer`s instructions. For the removal of genomic DNA, RNA was purified and concentrated using a RNeasy column (QIAGEN, Pudong, Shanghai, China). RNA degradation and contamination (integrity) were monitored on 1% agarose gels, whereas RNA purity and concentration were checked using the NanoDrop 2000 spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, USA). The cDNA library construction and sequencing (on an Illumina Hiseq Xten platform, San Diego, CA, USA) were conducted by Shanghai Majorbio Bio-pharm Technology Co. Ltd. (Shanghai, China).
Data quality control, genome mapping, transcript assembly and gene expression quantification
Raw data (raw reads) of FASTQ format were trimmed and quality controlled by using SeqPrep software (https://github.com/jstjohn/SeqPrep) and Sickle software (https://github.com/najoshi/sickle), which discarded reads containing adaptor sequences, ploy-N sequences and poor quality from raw data. Meanwhile, Q20, Q30, GC-content and sequence duplication level of the clean data (clean reads) were calculated. Post-processed reads (clean reads) were then mapped to the maize reference genome sequence (B73 RefGen_v4) with orientation mode using TopHat (https://tophat.cbcb.umd.edu/, version 2.1.1) software. The Tophat parameters were as follows: sequencing reads that uniquely matched to the reference genome, allowing up to 2 mismatches, without insertions or deletions. The relative abundance of transcripts in drought versus control conditions were estimated by reads per kilobase of transcript per million mapped reads (RPKM = total exon reads/mapped reads in million X exon length in kb) for each gene and log2 transformed to meet the assumptions of linear models (Mortazavi et al. 2008).
Functional annotation of unigenes
For functional annotation, the quality reads were used for BLAST (basic local alignment search) search, with the E value cut-off set to 1E-5 (Alstchul et al. 1997), against the following public databases: non-redundant protein sequence database (Nr) (ftp://ftp.ncbi.nlm.nih.gov/blast/db/), Swiss-Port (https://web.expasy.org/docs/swiss-prot_guideline.html), KOG/COG (Clusters of Orthologous Groups of proteins), Gene Ontology (GO) (https://www.geneontology.org) and the Kyoto Encyclopedia of Genes and Genomics (KEGG) (https://www.genome.jp/kegg).
Differentially expressed genes (DEGs) analysis and functional enrichment analysis
Differentially expressed genes (DEGs) sequencing libraries (for drought versus control conditions) were constructed with fragments per kilobase of exon model per million mapped reads (FPKM) as per the anticipated standards for transcriptome sequencing libraries (Mortazavi et al. 2008). DESeq2 package (version 1.12.3, Love et al. 2014) was employed for the differential expression analysis of the two groups. The analysis model uses student t test to calculate the p-values between the two groups (Wang et al. 2010); p value threshold was set at < 0.05. The raw p values were then corrected for multiple tests using the false discovery rate (FDR) according to the method of Benjamini and Hochberg (1995). Finally, the genes with an FDR < 0.05 and a fold change > 1.5 were considered to be significantly differentially expressed between the two samples. Moreover, functional-enrichment analysis including GO and KEGG were performed to identify which DEGs were significantly enriched in GO terms and metabolic pathways, respectively, at Bonferroni-corrected P-value < 0.01 compared with the whole-transcriptome background. GO functional enrichment and KEGG pathway analysis were carried out by Goatools (https://github.com/tanghaibao/Goatools) and KOBAS 2.1.1 (https://kobas.cbi.pku.edu.cn/download.php) (Xie et al. 2011).
Identification of key drought responsive genes
The expression abundances of each gene appearing in the two libraries (control versus drought treatment) were used to determine the expression changes of the genes in response to drought stress. The total DEGs identified to respond to drought stress (those that fell within the selection criteria specified above) were analysed by way of Venn diagram analysis. After filtering, the key/important drought responsive genes were specified by meeting the following criteria: the DEGs that specifically expressed in the tolerant genotype ND476 after drought treatment; DEGs shared between the drought-sensitive and drought-tolerant hybrids after drought treatment (SD_TD); DEGs of the tolerant cultivar that were also differentially expressed in SD_TD; and the common DEGs shared by the two hybrids under drought stress (TC_TD and SC_SD). This was substantiated by revealing their functional annotation and roles through GO and KEGG analyses, as well as refereeing to previously published works.
Quantitative real time-PCR (qRT-PCR) analysis
To validate the assembled sequences and the expression profiles obtained by Illumina RNA-seq, quantitative real-time PCR (qRT-PCR) was performed. Twenty (20) representative DEGs were randomly selected and gene-specific primers designed for qRT-PCR using Primer Premier 5 Designer (Premier Biosoft International, Palo Alto, CA, USA). Total RNA was isolated from V12-stage leaves as already described above in Sect. 2.3. Independent RNA from ND476 and ZX978 hybrids leaf samples, and from control and drought-stress conditions was prepared for qRT-PCR analysis. For cDNA synthesis, 1 µg of total RNA was reverse-transcribed in a total volume of 25 µL, using HiFiscript cDNA Synthesis Kit (CWBIO, Beijing, China). The qRT-PCR analysis was carried out using Bestar ® SYBR Green qPCR Mastermix (DBI ® Bioscience, Germany) in a Bio-Rad iQ5 thermo cycler (Bio-RAD, Hercules, CA, USA) (Zenda et al. 2019). A steady and constitutively expressed maize gene GAPDH (accession no. X07156) was used for the housekeeping function. The qRT-PCR program was performed with 1 µl of template cDNA, 1 µl of each prim n er (100 μmol), and 10 µl of SYBR Green mix (TOYOBO, Japan) in a total reaction volume of 20 µl. Each sample had three technical replicates. The method of Livak and Schmittgen (2001) was used to calculate the relative mRNA abundance of samples. Correlation between RNA-Seq and qRT-PCR was analyzed using SPSS 22.0 software (SPSS Institute Ltd., Armonk, NY, USA).
Statistical analysis of physiological data
All statistical analyses were performed by using SPSS statistical software (version 22.0; SPSS Institute Ltd., Armonk, NY, USA), and the data was presented as mean ± standard error of mean. A two-way analysis of variance (ANOVA) and least significant difference (LSD) tests were used to compare physiological parameters between treatments and across genotypes, whereas qRT-PCR data underwent one-way ANOVA and Duncan`s multiple range comparison tests. The significance level was set at p < 0.05.
Results
RNA-seq analysis results
RNA isolated from maize leaves that had been subjected to drought treatment for 12 days from V8 to V12 stages (as described in Sect. 2.1 above) was used for RNA-seq transcriptome analysis. Six samples for tolerant cultivar ND476 (NDC1, NDC2, NDC3, NDD1, NDD2 and NDD3) and six for sensitive cultivar ZX978 (ZXC1, ZXC2, ZXC3, ZXD1, ZXD2 and ZXD3) were categorized into four groups, viz; (NDC1, NDC2 and NDC3), (ZXC1, ZXC2 and ZXC3)—under water-abundant conditions; and (NDD1, NDD2 and NDD3), (ZXD1, ZXD2 and ZXD3)—under water-limited conditions. These four groups were named as TC, SC, TD, and SD, respectively. Then, the transcriptomes of the two hybrids in response to drought stress was analysed. The cDNA libraries were prepared from these two hybrids, before (control, C) and after drought treatment (D), and then subjected to RNA-seq profiling on the Illumina Hiseq Xten platform (San Diego, CA, USA).
After filtering, a total of 81.03 GB clean data was generated from ten samples (two unfit samples, NDC2 and NDD3, were retrieved). From the ten samples, we obtained 541.27 million total reads, with an average of 46.51 million clean reads each sample (Table 1). The clean reads were then mapped to the maize reference genome B73. The Q30 base percentage, an indicator of the overall reproducibility and quality of the assay, was above 96.0%, which met the demands for further analysis. The mapping rates ranged from 89.62 to 92.91% (Table 1). Notably, the percentage of mapped reads was lower in the ZX978 samples than in the ND476 samples (Table 1), showing that there were greater transcriptomic perturbations in ZX978 than in ND476.
We performed the principal component analysis (PCA) of all twelve samples in order to analyse the similarities and differences between the samples, using FPKM according to Mortazavi et al. (2008). The PCA results showed that a technical fault may have caused low similarity amongst the three replications of the two (one drought-stress and one control) samples (Fig. S1). Therefore, we retrieved samples NDC2 (ND476 water-sufficient) and NDD3 (ND476 water deficit) before analyzing our sequencing results. The PCA results of the remaining 10 samples showed clear separation between the drought tolerant and drought sensitive hybrids. Moreover, the replicates of each treatment clustered together (Fig. S1). These results exhibited the reproducibility and reliability of our experiment.
Analysis of transcriptomic responses and functional annotation of unigenes
The FPKM values > 1 were used to determine genes expressed. Using Cufflinks software (Trapnell et al. 2010), we identified a total of 21 566 annotated transcripts in the four treatments. The number of genes specifically expressed in each treatment, genes shared between each treatment, and genes shared among all combinations of treatments are shown in Fig. 1a. Of these 21,566 gene transcripts, 77.48% (16 709) were represented in all treatments. Before drought stress, 87.56% (18 884) and 87.18% (18 801) of the genes were expressed in the sensitive cultivar (ZX978, SC) and the tolerant cultivar (ND476, TC), respectively. After drought stress, 89.39% (19,278) and 88.94% (19 181) were expressed in ZX978 (SD) and ND476 (TD) respectively (Fig. 1a). A total of 382 genes (Group A in Fig. 1a) were specifically expressed in tolerant cultivar ND476 after drought treatment (TD). Group B, containing 558 genes, represents the genes specifically expressed in sensitive cultivar ZX978 after drought treatment (SD). Group C represents the 175 expressed genes that were shared by the tolerant and sensitive hybrids after drought treatment.

Summary analysis of gene expression profile and functional annotation of unigenes. a Venn diagram analysis of transcriptomic responses by cultivar (tolerant ND476, T; and sensitive ZX978, S) and treatment (control, C; and drought, D). The biological samples of four combinations are TC, TD, SC, and SD, respectively. The area labeled ‘A’ represents the genes exclusively expressed in TD, area labeled ‘B’ represents the genes specifically expressed in SD, and labeled ‘C’ represents the drought responsive genes shared by the tolerant and sensitive lines; b number of unigenes annotated to different sequence databases; c Venn diagram analysis of unigenes annotated to different databases; and d number of unigenes commonly annotated among databases
The unigenes were aligned against sequences in the Nr, Swiss-Prot, COG, Pfam, GO and KEGG databases to assign functional annotations. Resultantly, a total of 21,566 unigenes got annotated, 7182 (33.3%) unigenes found matches in all the six databases and 8910 (41.32%) unigenes matched sequences in at least one of the databases. However, 842 (3.90%) unigenes did not align to any of the known sequences in the public databases (Table 2; Fig. 1b). Meanwhile, 20,699 (95.84%) unigenes were found to have significant similarity with protein sequences in the Nr database at a cutoff of E ≤ 1e−05, whereas 15,241 (70.67%) unigenes were annotated by Swiss-Prot database (Tables 2; S1). Our Venn diagram analysis showed that most of the unigenes were annotated by more than one database. For instance, only 860, 32, 9 and 5 unigenes were specifically annotated in Nr, COG, Pfam and GO databases, respectively, but no unigene was specifically annotated by the KEGG and Swiss-Prot databases (Fig. 1c). At the same time, 7182 unigenes were annotated in all the six searched databases, whereas 13,284 unigenes were commonly annotated in the four (Nr, COG, Pfam and GO) databases (Fig. 1d). Overall, the results indicated that unigenes were much more easily annotated by Nr and Swiss-Prot than other databases (Fig. 1b, c). The number and proportions of unigenes assembled and annotated varied minimally between the present study and other drought stress related transcriptomic studies in maize (Li et al. 2017; Lu et al. 2017; Zenda et al. 2019), which may be as a result of the novel genes specially expressed in different organs, or probably due to technical or biological biases.
Gene differential expression analysis
The software Cuffdiff (Trapnell et al. 2012) was used to explore differentially expressed genes (DEGs) between different treatments. At a standard fold change of < 1.5 and FDR < 0.05, we identified a total of 3114 (1484 up- and 1630 down-regulated) genes to be differentially expressed between the tolerant and sensitive hybrids before drought treatment (SC_TC in Fig. 2a). Under drought conditions, 4140 (1972 up- and 2168 down-regulated) genes were identified between the tolerant and sensitive lines (SD_TD in Fig. 2a). By further comparing the differences in DEGs between the tolerant line ND476 and sensitive line ZX978, we identified 70 (29 up- and 41 down-regulated) DEGs in the tolerant line (TC_TD), and 1 199 (507 up-and 692 down-regulated) DEGs in the sensitive line (SC_SD) to be differentially expressed before and after drought treatment (Fig. 2a; Tables S2, S3).

Analysis of differentially expressed genes (DEGs). a Number of DEGs in each of the four experimental comparisons. Each compared combination is separated by an underscore (eg. TC_TD); b Venn diagram analysis of DEGs. Four critical areas, labeled I, II, III and IV, totally contain 1738 DEGs. Area I contains the tolerant treatment response DEGs, excluding others. Area II contains the line response under drought DEGs, excluding others. Area III contains both tolerance treatment response and line response under drought DEGs, excluding others. Area IV contains the treatment response DEGs within line; c clustering analysis of the tolerant line ND476 specific (Area I) DEGS. Each row indicates a gene differentially expressed (up-regulated—red, and down-regulated—blue). Columns show different samples. NDC1-2 and NDD1-2 refer to two replicates each for tolerant genotype ND476 under water sufficient (control) and drought stress conditions, respectively
Some of the Venn combinations shown in Fig. 2b are more essential than others in respect of drought tolerance. Area I represents 21 specific DEGs of TC_TD, that is, the specific drought responsive DEGs of the tolerant cultivar ND476. Of these DEGs, 9 were up-regulated and 12 were down-regulated (Table 3). Area II represents 1693 specific DEGs of SD_TD, that is, specific DEGs shared between the drought sensitive and drought tolerant hybrids after drought treatment. Of these, 811 were up-regulated whilst 882 were down-regulated (Table S4). Area III represents the 10 specifically shared DEGs between TC_TD and SD_TD, that is, drought responsive DEGs of the tolerant cultivar that were also differentially expressed between the tolerant and sensitive hybrids after drought treatment. Of these, two were up-regulated and eight were down-regulated in TC_TD comparison but showed differential expression in SD_TD comparison (Table S5). Area IV represents the 14 DEGs shared by TC_TD and SC_SD, that is, the common drought responsive DEGs of the two cultivars. These DEGs showed differential expression in the two hybrids after drought treatment, with 9 DEGs being up-regulated and 5 down-regulated in TC_TD, but showing differential expression is in SC_SD (Table S6). In total, we found 5805 DEGs to be differentially expressed among the four comparison groups, which reflect the impact of cultivars or treatment (Fig. 2a, b). Clustering analysis of the 21 DEGs specific to TC_TD comparison showed that, after drought stress exposure, DEGs were grouped into five clusters, with most of these DEGs being down-regulated than up-regulated (Fig. 2c).
DEGs annotation and functional categorization
We performed GO functional annotation and categorization of the DEGs from the four critical areas (labelled I–IV in Fig. 2b) using Blast2GO website (https://www.blast2go.com/) in order to determine their broad biological functions. The GO analysis results showed that a great number of DEGs were involved in BP functions (Fig. S3). Particularly, GO: 0009987 (cellular process); GO: 0008152 (metabolic process), GO: 0065007 (biological regulation), GO: 0050789 (regulation of biological process) and GO: 0050896 (response to stimuli) were common and top most significantly enriched level 2 GO terms in the BP category (Fig. S3a–c). Within the MF category, GO: 005488 (binding) and GO: 0003824 (catalytic activity) were most enriched among others (Fig. S3a–c). Further analysis of the Area I specific DEGs showed that GO terms related to photosynthesis (light harvesting) were the most significantly enriched under BP category whilst those related to chlorophyll binding and pigment binding were prominent under MF category (Fig. 3a). For the SD_TD specific DEGs, GO terms related to glutathione metabolic process and oxidation–reduction process were most significantly enriched under BP category (Fig. 3b). These results may suggest that the DEGs with these identified biological processes and molecular functions may be the key contributors to the drought stress responses in tolerant line ND476.

Gene ontology (GO) enrichment analysis of DEGs. Results show GO enrichment analysis of genes corresponding to a tolerant line treatment response (TC_TD) specific; and b line response under drought (SD_TD) specific. DEGs were enriched in different functional categories, viz., biological processes (BF), molecular functions (MF) and cellular component (CC)
DEGs encoding transcription factors
Our analysis of the tolerant cultivar ND476 (TC_TD) DEGs identified four TF genes that were altered in response to drought stress. Three genes; Zm00001d008869 (Homeobox-TF52), Zm00001d027900 (ZIM-TF27) and Zm00001d037769 (C3H-TF339) were up-regulated whereas one gene Zm00001d041886 (NAC-TF100) was down-regulated in response to drought stress (Table S2). Interestingly, Homeobox-TF52 and C3H-TF339 were also up-regulated in response to drought stress in sensitive line ZX978 (Table S3). Additionally, there were 98 drought responsive TF genes observed in the SD_TD specific DEGs (Table 4). These TFs were categorized into several classes, including MYB (9), NAC (2), WRKY (3), AP2-EREBP (5), bZIP (5), GARP (2), HSF (4), HAP (3) and bHLH (2) (Table 4). These TF genes are suggested to play vital roles in modulating drought stress tolerance in the tolerant maize hybrid cultivar.
‘Response to stimuli’ and ‘catalytic activity’ related DEGs
Our analysis of the DEGs that were enriched in each level 2 GO term showed that, among the tolerant cultivar ND476 specific DEGs (listed in Table 3), four were significantly enriched in GO term ‘response to stimuli’ (GO: 0050896). These genes were Zm00001d044402 (chlorophyll a-b binding protein 2), Zm00001d044401 (photosystem II light harvesting complex gene B1B2), Zm00001d000417 (photosystem Q (B) protein) and Zm00001d044399 (photosystem II light harvesting complex gene B1B2). All these four genes are involved in photosynthesis. Gene Zm00001d000417 was also significantly enriched in GO term ‘catalytic activity’ (GO: 0003824) which had 12 genes enriched in it. In common overlapping DEGs between ND476 and ZX978 (area IV in Fig. 2b), there were four genes enriched in GO term ‘response to stimuli’ (GO: 0,050,896), including Zm00001d011183 (thiamine biosynthesis1), Zm00001d037769 (C3H-transcription factor 339), Zm00001d052213 and Zm00001d024839 (glutathione S-transferase 2), and seven genes were enriched in GO term ‘catalytic activity’ (GO: 0003824), including Zm00001d020627, Zm00001d002940, Zm00001d011183 (thiamine biosynthesis1), Zm00001d033872, Zm00001d052213, Zm00001d009631 and Zm00001d024839 (glutathione S-transferase2). These genes are involved in stress signal perception and transduction, nutrient and water uptake, and cell elongation under drought stress conditions during late vegetative (V12) growth stage, and thus, are considered key actors modulating drought tolerance in maize.
KEGG metabolic pathways enrichment analysis of the DEGs
Functional involvement of the DEGs in various metabolic pathways was further investigated by mapping them to the KEGG database (available online: https://www.genome.jp/kegg/; accessed on 15 March 2019). By analysing the top twenty pathways in which most DEGs were enriched, we discovered that photosynthesis-antenna proteins (4 DEGs), ribosome (3), nitrogen metabolism (2) and photosynthesis (2) were dominant in ND476 (TC_TD) (Fig. S4a). In SD_TD comparison, ribosome (99), starch and sucrose metabolism (39), and plant hormone signal transduction (35) pathways had the greatest number of genes enriched (Fig. S4b). Comparatively, the composition of the enriched KEGG pathways in sensitive cultivar ZX978 (SC_SD) differed significantly, with carbon fixation in photosynthetic organisms (25), ribosome (51), oxidative phosphorylation (29) and plant hormone signal transduction (25) being the top most enriched (Fig. S4c). Further, we performed significant pathway enrichment analysis of these DEGs by hypergeometric test, with pathways of q value < 0.01 considered to be significantly affected by drought stress. We then observed that photosynthesis-antenna proteins and nitrogen metabolism pathways were highly enriched in TC_TD comparison (Fig. 4a). Meanwhile, ribosome, starch and sucrose metabolism, and nitrogen metabolism pathways were highly enriched in the SD_TD (Fig. 4b). In contrast, carbon fixation in photosynthetic organisms, ribosome and oxidative phosphorylation were the most significantly enriched in sensitive cultivar ZX978 (Fig. S5).

KEGG (Kyoto Encyclopedia of Genes and Genomics) pathway enrichment analysis of the DEGs. Results show the most significantly enriched pathways in a TC_TD; and b SD_TD experimental comparisons, based on the hypergeometric test. The significance of the enrichment of the KEGG pathway is based on the Student’s t test, q < 0.01. The color gradient represents the size of the q value; the color is from red to blue, and the nearer to red, the smaller the q value, the higher the significant level of enrichment. The ‘rich factor’ shows the ratio of the number of the DEGs to the total gene number in certain pathways
Validation of RNA-seq by quantitative real-time PCR (qRT-PCR)
In order to validate the accuracy of the RNA-Seq results, we randomly chose a sample of 20 DEGs for qRT-PCR analysis. We designed gene specific primers (Table S7) for qRT-PCR analysis using Primer Premier 5.0 software (Premier Biosoft International, Palo Alto, CA, USA). Our qRT-PCR analysis results were congruent to our RNA-seq based findings. Notably, RNA-seq expression patterns of all the sampled DEGs were closely replicated by the qRT-PCR approach, with an R2 (correlation coefficient of qRT-PCR log2FC versus RNA-seq log2FC) of 92.98% (Fig. 5; Table S8; Fig. S6). This confirmed that our RNA-seq data was reliable.

qRT-PCR validation of the RNA-seq data of the 20 randomly selected maize V12-stage leaf DEGs. a TC_TD specific DEGs; b SD_TD specific DEGs; c DEGs shared between TC_TD and SD_TD; and (d) DEGs specific to SC_SD. The y axis represents the gene relative expression levels (Log2 fold changes) in the real-time PCR analysis and Log2 fold changes in the RNA-Seq data. All the genes with negative values of expression level means that they were down-regulated in response to drought stress. Maize gene GAPDH (Accession No. X07156) was used as the internal reference. Error bars represent the SE (n = 3)
Phenotypic and physiological responses of two maize hybrids to drought stress
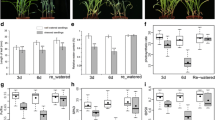
Expectedly, no apparent phenotypic differences were observed between ND476 and ZX978 maize plants under water-sufficient conditions. They both exhibited vigorous and intact plant structures (Fig. S7a). However, post drought treatment exposure, considerable variations were noted between the two hybrids. Whilst the tolerant cultivar ND476 maintained erect green leaves and considerably intact plant stature, the sensitive line ZX978 exhibited pendulous leaves and a drooping plant stature (Fig. S7b). Meanwhile, in both maize lines, the RWC significantly (p < 0.05) decreased with increasing number of days post water deprivation exposure. Evidently, the rate of decline of RWC was greater in ZX978 than in ND476 under drought stress conditions (Fig. 6a). The SPC showed gradual increase in both cultivars, from day 1 until day 6 under drought conditions. Thereafter, SPC started to drop steadily. Notably, the tolerant genotype ND476 maintained significantly higher SPC values than the sensitive genotype ZX978 at any particular time point (Fig. 6b). The POD activity showed an almost similar trend to SPC in both maize lines, increasing from day 3 to 7 in drought-stressed ND476 and day 3–6 in drought-stressed ZX978, and gradually dropping thereafter (Fig. 6c).Under drought treatment, MDA content gradually increased in both maize genotypes, starting from day 3 and becoming more significantly different between genotypes from day 6 onwards. Of note, the MDA content in ZX978 was significantly greater than in ND476 (Fig. 6d).

Physiological responses of two contrasting maize hybrid cultivars to drought stress after 12 days of exposure to water-sufficient (control) or water-limited (drought) conditions. a RWC; b SPC; c POD activity; and d MDA content. Data are presented as mean ± SE of mean (n = 3). Different letters above line graphs show significant difference (p < 0.01) among treatments at a particular treatment time point
Discussion
In face of global climate change, the major goal for global agriculture is to develop drought-tolerant crops (Edmeades 2013; Min et al. 2016). To this end, a full understanding of physiological, biochemical, and gene regulatory networks relating to drought tolerance in plants becomes imperative. Here, using an integration of RNA-seq based technology and physiological parameters in comparative analysis of transcriptomes of two contrasting maize (drought-tolerant ND476 and drought-sensitive ZX978) hybrid cultivars, we report key differentially expressed genes and regulatory mechanisms involved in maize drought stress tolerance. In addition, we have conducted some physiological analyses to support the RNA-seq data. Our results enhance our further understanding of the mechanisms modulating drought tolerance in maize, in addition to aiding as foundational base to our future targeted cloning studies.
Maize hybrid cultivars differential drought stress responses at the physiological level
Cultivars within crop species are known to differ in their responses to drought and other stresses. These responses can occur at different levels, including physiological and molecular, as well as at different plant growth phases (Prasad et al. 2008). To avoid these deleterious effects of oxidative stress emanating from ROS, plants maintain cellular structures, including membrane and protein stabilities by several strategies such as cell turgor maintenance and osmotic adjustment (Oliver et al. 2007; Jogaiah et al. 2013). Here, our results revealed that tolerant genotype ND476 maintained comparably higher leaf RWC and SPC than sensitive genotype ZX978 under drought stress conditions (Fig. 6a, b). This resulted in limited loss of cell turgor and integrity, and less stress at the cellular level. Contrastingly, sensitive genotype ZX978 experienced significant decline in RWC, SPC and POD from day 6 onwards (Fig. 6a–c), causing a rapid loss of cell turgor and cell integrity. This possibly explains why, at the transcriptome level, fewer (70) genes were differentially expressed in ND476 than 1199 genes in ZX978 in response to drought stress (Fig. 2a). Thus, there was limited transcriptome perturbation in the tolerant cultivar as compared to wider transcriptome perturbation in the sensitive line at the same drought stress intensity. The role of POD in ROS scavenging and detoxification is well documented (Sharma et al. 2012). Here, we speculate that the sustained increase of POD in ND476 hybrid cultivar enhanced ROS quenching than in ZX978 under drought stress. Consequently, ND476 had better cell membrane stability and drought stress tolerance than ZX978, in conformity to previous studies (Moussa and Abdel-Aziz 2008; Zenda et al. 2018). Additionally, MDA content, which reflects the extent of lipid peroxidation and cell membrane stability (Sharma et al. 2012), was more significantly increased in ZX978 than in ND476 as the drought stress exposure period progresses, particularly from day 6 onwards (Fig. 6d). This may suggest that ND476 cells had better ROS quenching capacity than ZX978 cells, providing for enhanced cell membrane stability in ND476 than in ZX978 (Min et al. 2016). Taken together, our results reveal some differential drought stress responses between the two hybrid cultivars at the physiological level.
Secondary metabolites biosynthesis and carbohydrates metabolism related enzymes under drought
Among the top most up-regulated drought responsive genes specific to the tolerant line ND476 (TC_TD) were secondary metabolite biosynthesis (SMB) related enzymes, including probable O-methlytransfarase 2 (OMT2; Zm00001d019613), tyrosine decarboxylase 1(Zm00001d033024) and benzoxazinless1 (Zm00001d048709), as well as one carbohydrate (CHO) metabolism related enzyme stachyose synthetase (SSG; Zm00001d019163) (Table 3). OMT2 is involved in SMB in Sorghum bicolor L (https://www.uniprot.org/uniprot/A8QW51). Tyrosine decarboxylase 1 constitutes isoquinoline alkaloids, one of the quite divergent natural chemicals produced by higher plants, including alkaloids, terpenoids and phenylpropanoids. These compounds are biogenetically derived from phenylalanine and tyrosine, and are essential in conferring combined pathogenic and abiotic stress resistance in plants (Edreva et al. 2007). Benzoxazinless1, together with other enzymes such as cytochrome P450 (Zm00001d040764 in Table S2) and OMT, is functionally recruited in the benzoxazinoid biosynthesis pathway (Glawischnig et al. 1999). These genes participate in phenylalanine metabolism, a stage in the phenylpropanoid metabolism, which is the first step of the SMB (Shinde et al. 2018); coordinated activation of these genes is considered vital to improved stress tolerance in plants (Ma et al. 2014). CHO metabolism is essential in bio-molecular metabolism, via CHO breakdown providing important saccharides and energy that are critical for cell growth and survival under stress conditions (Min et al. 2016; Yang et al. 2019). SSG enzyme participates in galactose metabolism processes. Stachyose, as a tetrasaccharide, is recognized as an important transport CHO in a large number of woody plants, cucurbits and legumes (Peterbauer et al. 1999). Therefore, SSG is a key player in CHO metabolism and energy provision in response to drought stress. Taken collectively, the SMB and CHO related genes are suggested to balance cell growth, defense and drought stress response in maize.
Transcription factor related genes are critical in regulating drought stress response
The role of TFs in various stress tolerances in different crop plants including maize is well-documented (Wang et al. 2016). In maize, different TF families have been identified as responsible for modulating the gene regulation in response to drought stress (Joshi et al. 2016; Kimotho et al. 2019). TFs such as MYB, NAC, WRKY, bZIP, bHLH, HD-zip via ABA-dependent or ABA-independent pathways play a significant role in drought tolerance (Mittal et al. 2018). They achieve this by regulating numerous physiological and molecular processes, including stomatal opening and closure, hormone signalling and osmoregulation (Mittal et al. 2017; Jin et al. 2019). In the current report, more than 33 drought-responsive TF families were differentially expressed under drought conditions (Tables 4, S9). Among these were nine MYB (4 up- and 5 down-regulated), two NAC (1 up- and 1- down), three WRKY (1 up- and 2 down-), five AP2-EREBP (all down-), five bZIP (3 up- and 2 down-), two GARP (both down-), and three bHLH (2 up- and 1 down-) that were regulated in response to drought stress (Tables S4 and S9). Previously, Shinde et al. (2018) identified several TF genes to be up-regulated in response to drought stress in foxtail millet (Setaria italic L.), including five NACs and three MYBs. On the other hand, Bianchi et al. (2015) revealed that WRKY TF genes were down-regulated under drought stress. Zhang et al. (2014) identified numerous drought responsive TF genes in Medicago truncatula L., including eight NACs, eight MYBs, six AP2/EREBPs, six bZIPs, five HDs, and four bHLHs, among others. Taken together, this discussion fortifies the important role TFs play in regulating drought stress tolerance in maize, with various TF families exhibiting differential responses, and interacting with each other in complex networks.
Aquaporins and glutathione S-transferases play crucial roles in drought tolerance
Detoxification and stress responsive proteins such as aquaporins, HSPs, glutathione S-transferases (GSTs), and LEA proteins play crucial roles in drought tolerance (Al-whaibi, 2011; Bhargava and Sawant, 2013; Harb, 2016; Jan et al. 2017). Here, our analysis of the TC_TD and the SD_TD specific DEGs showed that genes encoding aquaporins, GSTs and HSPs were differentially regulated in response to drought stress (Tables 3, S4). Aquaporins are membrane channels that facilitate the transport of water across biological membranes in most living organisms (Maurel et al. 2015). In plants, aquaporins are required for the facilitation of transport of water across cell membranes, and hence play an important role in water homeostasis by turgor regulation (Aslam et al. 2015; Min et al. 2016). Previously, aquaporins were also identified to be involved in drought tolerance in sheep grass [Leymus chinensis (Trin.) Tzvel] (Zhao et al. 2016a). Here, we suggest that aquaporin PIP2-4 (Zm00001d017288) functions to increase water absorption in tolerant genotype ND476 under drought stress, thus maintaining better cellular redox homeostasis under such conditions. GSTs play a crucial role in the plants` response to various types of abiotic stresses. GSTs quench reactive molecules with the addition of glutathione and protect the cell from ROS-induced oxidative damage (Kumar and Trivedi 2018). Their role in oxidation–reduction processes in plant cells in response to drought stress has been reported (Ahmad et al. 2016; Min et al. 2016). Taken together, this discussion reveals that detoxification and stress defense genes, including those involved in facilitating water transport across membranes and cellular oxidation–reduction processes constitute a vital drought stress response strategy in maize leaves.
Metabolic pathways significantly enriched under drought stress conditions
Photosynthesis antenna proteins pathway (PAPP) and nitrogen metabolism pathway (NMP) were the most significantly enriched in the tolerant genotype ND476 under drought stress (Fig. 4a). The PAPP is highly sensitive to drought stress (Ghannoum 2009; Zhao et al. 2016b). Photosynthesis antenna proteins, a part of the light harvesting complexes and the electron transport components of the photosystem II (PSII), act as peripheral antenna systems enabling more efficient absorption of light energy (Zhang et al. 2018). They are also involved in the intracellular non-photochemical quenching processes of the plant photosynthesis machinery (Murata et al. 2012). Previously, PAPP has been recognized as a critical pathway in drought stress response (Zhao et al. 2016b; Dudhate et al. 2018; Zenda et al. 2018). Here, the down-regulation of several photosynthesis-related genes in both genotypes implies the high sensitivity of the PSII to drought stress; with drought stress impeding photosynthesis process. The role of NMP in drought stress response has been acknowledged (Zenda et al. 2019). By directly influencing the formation of cellular components and regulation of cellular activities, NMP becomes the most basic and important physiological metabolic process during plant growth. Additionally, NMP is critical in the transformation of photosynthetic products, mineral nutrient absorption, and protein biosynthesis (Ahmadi et al. 2010). Previously, NMP has been implicated in salt stress response (Luo et al. 2018).
In the SD_TD experimental comparison, pathways related to ‘ribosome’, starch and sucrose’ and ‘cysteine and methionine metabolism’ were the most significantly enriched in response to drought stress (Fig. 4b). The importance of starch and sucrose metabolism pathway in drought stress response in maize has been reported (Jin et al. 2019; Wang et al. 2019b). Starch and sucrose degradation are vital for the provision of cellular energy to help plants tolerate stressful conditions. Ribosomes are the site for protein synthesis, one of the fundamental biological processes that are influenced by drought stress (Shinde et al. 2018). Amino acids cysteine and methionine are essential in protein synthesis; hence the enrichment of these pathways is directly linked with protein biogenesis under drought stress conditions.
Conclusion
We have herein presented the results of our comprehensive comparative analysis of the transcriptomic and physiological responses of two contrasting maize hybrids to drought stress at the V12 growth stage. Phenotypically, the tolerant genotype ND476 had better cell water retention, higher metabolites, and low lipid peroxidation extent than the sensitive genotype ZX978 under drought conditions. Our RNA-seq results identified total 3114 DEGs were expressed in response to drought, with 21 DEGs being uniquely found in ND476. We found out that genes associated with previously reported pathways involved in drought stress response were altered upon drought-stress exposure, including those associated with secondary metabolites biosynthesis, TF regulation, detoxification and stress defense. Our results enhance our further understanding of the mechanisms regulating drought tolerance in maize, in addition to aiding as foundational base to our future targeted cloning studies.
References
Ahmad N, Malagoli M, Wirtz M, Hell R (2016) Drought stress in maize causes differential acclimation responses of glutathione and sulfur metabolism in leaves and roots. BMC Plant Biol 16:247. https://doi.org/10.1186/s12870-016-0940-z
Ahmadi A, Emam Y, Pessarakli M (2010) Biochemical changes in maize seedlings exposed to drought stress conditions at different nitrogen levels. J Plant Nutr 33:541–556. https://doi.org/10.1080/01904160903506274
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res 25(17):3389–3402. https://doi.org/10.1093/nar/25.17.3389
Al-Whaibi MH (2011) Plant heat-shock proteins: A mini review. J King Saud Univ Sci 23:139–150. https://doi.org/10.1016/j.jksus.2010.06.022
Anjum SA, Xie XY, Wang LC, Saleem MF, Man C, Lei W (2011) Morphological, physiological and biochemical responses of plants to drought stress. Afr J Agric Res 6:2026–2032
Aslam M, Maqbool MA, Cengiz R (2015) Drought stress in maize (Zea mays L.): Effects, resistance mechanisms, global achievements and biological strategies for improvement. Springer: Cham, ISBN 978-3-319-25440-1.
Banerjee A, Roychoudhury A (2017) Abscisic-acid-dependent basic leucine zipper (bZIP) transcription factors in plant abiotic stress. Protoplasma 254(1):1–14. https://doi.org/10.1007/s00709-015-0920-4
Basu S, Ramegowda V, Kumar A, Pereira A (2016) Plant adaptation to drought stress. F1000 Res 5:1554. https://doi.org/10.12688/f1000research.7678.1
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B 57:289–300
Bhanu BB, Ulaganathan K, Shanker AK, Desai S (2016) RNA-seq analysis of irrigated vs water stressed transcriptomes of Zea mays cultivar Z59. Front Plant Sci 7:239. https://doi.org/10.3389/fpls.2016.00239
Bhargava S, Sawant K (2013) Drought stress adaptation: Metabolic adjustment and regulation of gene expression. Plant Breed 132:21–32. https://doi.org/10.1111/pbr.12004
Bianchi VJ, Rubio M, Trainotti L, Verde I, Bonghi C, Martínez-Gómez P (2015) Prunus transcription factors: breeding perspectives. Front Plant Sci 6:443. https://doi.org/10.3389/fpls.2015.00443
Bradford MM (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. https://doi.org/10.1016/0003-2697(76)90527-3
Danilevskaya ON, Yu G, Meng X, Xu J, Stephenson E, Estrada S, Chilakamarri S, Hayes ZG, Thatcher S (2019) Developmental and transcriptional responses of maize to drought stress under field conditions(Article). Plant Direct 3(5):1–20. https://doi.org/10.1002/pld3.129
Darby H, Lauer J (2006) Plant physiology: critical stages in the life of a corn plant. Department of Agronomy, Winsconsin University
Dhindsa RS, Plumb-Dhindsa P, Thorpe TA (1981) Leaf senescence: correlated with increased leaves of membrane permeability and lipid peroxidation, and decreased levels of superoxide dismutase and catalase. J Exp Bot 32:93–101. https://doi.org/10.1093/jxb/32.1.93
Dudhate A, Shinde H, Tsugama D, Liu S, Takano T (2018) Transcriptomic analysis reveals the differentially expressed genes and pathways involved in drought tolerance in pearl millet [Pennisetum glaucum (L.) R. Br]. PLoS One 13:e0195908. https://doi.org/10.1371/journal.pone.0195908
Edmeades GO (2013) Progress in achieving and delivering drought tolerance in maize—an update. ISAA, Ithaca, pp 1–39
Edreva AM, Velikova VB, Tsonev TD (2007) Phenylamides in plants. Russ J Plant Physl 54(3):287–301. https://doi.org/10.1134/S1021443707030016
Fahad S, Bajwa AA, Nazir U, Anjum SA, Farooq A, Zohaib A, Sadia S, Nasim W, Adkins S, Saud S (2017) Crop production under drought and heat stress: plant responses and management options. Front Plant Sci 8:1147. https://doi.org/10.3389/fpls.2017.01147
Fang Y, Xiong L (2015) General mechanisms of drought response and their application in drought resistance improvement in plants. Cell Mol Life Sci 72:673–689. https://doi.org/10.1007/s00018-014-1767-0
Farooq M, Wahid A, Kobayashi N, Fujita D, Basra SMA (2009) Plant drought stress: effects, mechanisms and management. Agron Sustain Dev 29(1):185–212. (Springer Verlag/EDP Sciences/INRA)
Galmés J, Flexas J, Savé R, Medrano H (2007) Water relations and stomatal characteristics of Mediterranean plants with different growth forms and leaf habits: responses to water stress and recovery. Plant Soil 290:139–155. https://doi.org/10.1007/s11104-006-9148-6
Ghannoum O (2009) C4 photosynthesis and water stress. Ann Bot 103:635–644. https://doi.org/10.1093/aob/mcn093
Glawischnig E, Grun S, Frey M, Gierl A (1999) Cytochrome P450 monooxygenases of DIBOA biosynthesis: specificity and conservation among grasses. Phytochemistry 50:925–930. https://doi.org/10.1016/s0031-9422(98)00318-5
Han LB, Song GL, Zhang X (2008) Preliminary observation of physiological responses of three turfgrass species to traffic stress. Hort Technol 18(1):139–143. https://doi.org/10.21273/HORTTECH.18.1.139
Harb A (2016) Identification of candidate genes for drought stress tolerance. In: Hossain MA (ed) Drought stress tolerance in plants 2. Springer International Publishing, Berlin
Hsiao T (1973) Plant responses to water stress. Annu Rev Plant Physiol 24:519–570
Jan AU, Hadi F, Midrarullah AA, Rahman K (2017) Role of CBF/DREB gene expression in abiotic stress tolerance. Int J Hort Agric 2(1):1–12
Jin HY, Liu ST, Zenda T, Wang X, Liu G, Duan HJ (2019) Maize leaves drought-responsive genes revealed by comparative transcriptome of two cultivars during the filling stage. PLoS One 14(10):e0223786. https://doi.org/10.1371/journal.pone.0223786
Jogaiah S, Govind SR, Tran LS (2013) Systems biology-based approaches toward understanding drought tolerance in food crops. Crit Rev Biotechnol 33:23–39. https://doi.org/10.3109/07388551.2012.659174
Joshi R, Wani SH, Singh B, Bohra A, Dar ZA, Lone AA, Pareek A, Pareek SLS (2016) Transcription factors and plants response to drought stress: current understanding and future directions. Front Plant Sci 7:1029. https://doi.org/10.3389/fpls.2016.01029
Kakumanu A, Ambavaram MMR, Klumas C, Krishnan A, Batlang U, Myers E, Grene R, Pereira A (2012) Effects of drought on gene expression in maize reproductive and leaf meristem tissue revealed by RNA-seq. Plant Physiol 160:846–867. https://doi.org/10.1104/pp.112.200444
Kimotho RN, Baillo EH, Zhang ZB (2019) Transcription factors involved in abiotic stress responses in maize (Zea mays L.) and their roles in enhanced productivity in the post genomics era. Peer J 7:e7211. https://doi.org/10.7717/peerj.7211
Kumar S, Trivedi PK (2018) Glutathione S-transferases: role in combating abiotic stresses including arsenic detoxification in plants. Front Plant Sci 9:751. https://doi.org/10.3389/fpls.2018.00751
Li P, Cao W, Fang H, Xu S, Yin S, Zhang Y, Lin D, Wang J, Chen Y, Xu C, Yang Z (2017) Transcriptomic profiling of the maize (Zea mays L.) leaf response to abiotic stresses at the seedling stage. Front Plant Sci 8:290. https://doi.org/10.3389/fpls.2017.00290
Liu ST, Zenda T, Dong AY, Yang YT, Liu XY, Wang YF, Li J, Tao YS, Duan HJ (2019) Comparative proteomic and morpho-physiological analyses of maize wild-type Vp16 and mutant vp16 germinating seed responses to PEG-induced drought stress. Int J Mol Sci. https://doi.org/10.3390/ijms20225586
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
Lu X, Zhou X, Cao Y, Zhou M, McNeil D, Liang S, Yang C (2017) RNA-seq analysis of cold and drought responsive transcriptomes of Zea mays ssp. mexicana L. Front Plant Sci 8:136. https://doi.org/10.3389/fpls.2017.00136
Luo M, Zhao Y, Wang Y, Shi Z, Zhang P, Zhang Y, Song W, Zhao J (2018) Comparative proteomics of contrasting maize genotypes provides insights into salt-stress tolerance mechanisms. J Proteome Res 17:141–153. https://doi.org/10.1021/acs.jproteome.7b00455
Ma D, Sun D, Wang C, Li Y, Guo T (2014) Expression of flavonoid biosynthesis genes and accumulation of flavonoid in wheat leaves in response to drought stress. Plant Physiol Biochem 80:60–66. https://doi.org/10.1016/j.plaphy.2014.03.024
Maurel C, Boursiac Y, Luu DT, Santoni V, Shahzad Z, Verdoucq L (2015) Aquaporins in plants. Physiol Rev 95(4):1321–1358. https://doi.org/10.1152/physrev.00008.2015
Min H, Chen C, Wei S, Shang X, Sun M, Xia R, Liu XG, Hao DY, Chen HB, Xie Q (2016) Identification of drought tolerant mechanisms in maize seedlings based on transcriptome analysis of recombination inbred lines. Front Plant Sci 7:1080. https://doi.org/10.3389/fpls.2016.01080
Mittal S, Arora K, Ramakrishna RA, Mallikarjuna MG, Gupta HS, Nepolean T (2017) Genomic selection for drought tolerance using genome-wide SNPs in maize. Front Plant Sci 8:550. https://doi.org/10.3389/fpls.2017.00550
Mittal S, Banduni P, Mallikarjuna MG, Rao AR, Jain PA, Dash PK, Thirunavukkarasu N (2018) Structural, functional, and evolutionary characterization of major drought transcription factors families in maize. Front Chem 6:177. https://doi.org/10.3389/fchem.2018.00177
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Meth 5:621–628. https://doi.org/10.1038/nmeth.1226
Moussa HR, Aziz SMA (2008) Comparative response of drought tolerant and drought sensitive maize genotypes to water stress. Aust J Crop Sci 1(1):31–36
Murata N, Allakhverdiev SI, Nishiyama Y (2012) The mechanism of photoinhibition in vivo: re-evaluation of the roles of catalase, α-tocopherol, non-photochemical quenching, and electron transport. Biochim Biophys 1817:1127–1133. https://doi.org/10.1016/j.bbabio.2012.02.020
Oliver SN, Dennis ES, Dolferus R (2007) ABA regulates apoplastic sugar transport and is a potential signal for cold-induced pollen sterility in rice. Plant Cell Physiol 48:1319–1330. https://doi.org/10.1093/pcp/pcm100
Opitz N, Paschohold A, Marcon C, Malik WA, Lanz C, Piepho HP, Hochholdinger F (2014) Transcriptomic complexity in young maize primary roots in response to low water potentials. BMC Genom 15:1471–2164. https://doi.org/10.1186/1471-2164-15-741
Opitz N, Marcon C, Paschold A, Malik WA, Lithio A, Brandt R, Piepho H, Nettleton D, Hochholdinger F (2016) Extensive tissue-specific transcriptomic plasticity in maize primary roots upon water deficit. J Ex Bot 67(4):1095–1107. https://doi.org/10.1093/jxb/erv453
Peterbauer T, Mucha J, Mayer U, Popp M, Glossl J, Richter A (1999) Stachyose synthesis in seeds of adzuki bean (Vigna angularis): molecular cloning and functional expression of stachyose synthase. Plant J Cell Mol Biol 20:509–518. https://doi.org/10.1046/j.1365-313X.1999.00618.x
Prasad PVV, Staggenborg SA, Ristic Z (2008) Impacts of drought and/or heat stress on physiological, developmental, growth, and yield processes of crop plants. In: Ahuja LR, Reddy VR, Saseendran SA, Qiang Yu (eds) Response of crops to limited water: understanding and modeling water stress effects on plant growth processes. Advances in Agricultural Systems Modeling Series 1 (2008), Agronomy Society of America (ASA), Crop Science Society of America (CSSA), Soil Science Society of America (SSSA), Madison, USA. https://doi.org/10.2134/advagricsystmodel1.c11
Priya M, Dhanker OP, Siddique KHM, Rao BH, Nair RM, Pandey S, Singh S, Varshney RK, Prasad PVV, Nayyar H (2019) Drought and heat stress-related proteins: an update about their functional relevance in imparting stress tolerance in agricultural crops. Theor Appl Genet Springer Verlag GmbH Germany. https://doi.org/10.1007/s00122-019-03331-2
Schils R, Olesen JE, Kersebaum K, Rijk B, Oberforster M, Kalyada V, Khitrykau M, Gobin A, Kirchev H, Manolova V, Manolov I, Trnka M, Hlavinka P, Palosuo T, Peltonen-Sainio P, Jauhiainen L, Lorgeou J, Marrou H, Danalatos N, Archontoulis S, Fodor N, Spink J, Roggero PP, Bassu S, Pulina A, Seehusen T, Uhlen AK, Żyłowska K, Nieróbca A, Si VJ (2018) Cereal yield gaps across Europe. Eur J Agron 101:109–120. https://doi.org/10.1016/j.eja.2018.09.003
Shan X, Li Y, Jiang Y, Jiang Z, Hao W, Yuan Y (2013) Transcriptome profile analysis of maize seedlings in response to high-salinity, drought and cold stresses by deep sequencing. Plant Mol Biol Rep 31:1485–1491. https://doi.org/10.1007/s11105-013-0622-z
Sharma P, Jha AB, Dubey RS, Pessarakli M (2012) Reactive oxygen species, oxidative damage, and antioxidative defense mechanism in plants under stressful conditions. J Bot 10:26. https://doi.org/10.1155/2012/217037
Shiferaw B, Prasanna BM, Hellin J, Bänziger M (2011) Crops that feed the world 6. Past successes and future challenges to the role played by maize in global food security. Food Sec 3(3):307–327. https://doi.org/10.1007/s12571-011-0140-5
Shinde H, Tanaka K, Dudhate A, Tsugama D, Mine Y, Kamiya T, Gupta SK, Liu S, Takano T (2018) Comparative de novo transcriptomic profiling of the salinity stress responsiveness in contrasting pearl millet lines. Environ Exp Bot 155:619–627. https://doi.org/10.1016/j.envexpbot.2018.07.008
Singh D, Laxmi A (2015) Transcriptional regulation of drought response: a tortuous network of transcriptional factors. Front Plant Sci 6:895. https://doi.org/10.3389/fpls.2015.00895
Singh S, Kumar V, Kapoor D, Kumar S, Singh J (2019) Revealing on hydrogen sulfide and nitric oxide signals cordination for plant growth under stress conditions. Physiol Plant. https://doi.org/10.1111/ppl.13002
Tai F, Yuan Z, Li S, Wang Q, Liu F (2016) ZmCIPK8, a CBL-interacting protein kinase, regulates maize response to drought stress. Plant Cell Tiss Org 124:59–469. https://doi.org/10.1007/s11240-015-0906-0
Thirunavukkarasu N, Sharma R, Singh N, Shiriga K, Mohan S, Mittal S, Mittal S, Mallikarjuna MG, Rao AR, Dash PK, Hossain F, Gupta HS (2017) Genomewide expression and functional interactions of genes under drought stress in maize. Int J Genet. https://doi.org/10.1155/2017/2568706. (Article ID 2568706)
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515. https://doi.org/10.1038/nbt.1621
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. https://doi.org/10.1038/nprot.2012.016
Wang L, Feng Z, Wang X, Wang X, Zhang X (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26:136–138. https://doi.org/10.1093/bioinformatics/btp612
Wang H, Wang H, Shao H, Tang X (2016) Recent advances in utilizing transcription factors to improve plant abiotic stress tolerance by transgenic technology. Front Plant Sci 7:67. https://doi.org/10.3389/fpls.2016.00067
Wang CT, Ru JN, Liu YW, Yang JF, Li M, Xu ZS, Fu JD (2018) The maize WRKY transcription factor ZmWRKY40 confers drought resistance in transgenic Arabidopsis. Int J Mol Sci 19:2580. https://doi.org/10.3390/ijms19092580
Wang BM, Liu C, Zhang DF, He CM, Zhang JR, Li ZX (2019a) Effects of maize organ-specific drought stress response on yields from transcriptome analysis. BMC Plant Biol 19:335. https://doi.org/10.1186/s12870-019-1941-5
Wang X, Zenda T, Liu ST, Liu G, Jin HY, Dai L, Dong AY, Yang YT, Duan HJ (2019b) Comparative proteomics and physiological analyses reveal important maize filling-kernel drought-responsive genes and metabolic pathways. Int J Mol Sci 20:3743. https://doi.org/10.3390/ijms20153743
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L (2011) KOBAS2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39:W316–W322. https://doi.org/10.1093/nar/gkr483
Yang M, Geng MY, Shen PF, Chen XH, Li YJ, Wen XX (2019) Effect of post-silking drought stress on the expression profiles of genes involved in carbon and nitrogen metabolism during leaf senescence in maize (Zea mays L.). Plant Physiol Biochem 135:304–309
Zenda T, Liu ST, Wang X, Jin HY, Liu G, Duan HJ (2018) Comparative proteomic and physiological analyses of two divergent maize inbred lines provide more insights into drought-stress tolerance mechanisms. Int J Mol Sci 19:3225. https://doi.org/10.3390/ijms19103225
Zenda T, Liu ST, Wang X, Liu G, Jin HY, Dong AY, Yang YT, Duan HJ (2019) key maize drought-responsive genes and pathways revealed by comparative transcriptome and physiological analyses of contrasting inbred lines. Int J Mol Sci 20(6):1268. https://doi.org/10.3390/ijms20061268
Zhang JY, Cruz de Carvalho MH, Torres-Jerez I, Kang Y, Allen SN, Huhman DV, Tang Y, Murray J, Sumner LW, Udvardi MK (2014) Global reprogramming of transcription and metabolism in Medicago truncatula during progressive drought and after rewatering. Plant Cell Environ 37:2553–2576. https://doi.org/10.1111/pce.12328
Zhang X, Lei L, Lai J, Zhao H, Song W (2018) Effects of drought stress and water recovery on physiological responses and gene expression in maize seedlings. BMC Plant Biol 18:68. https://doi.org/10.1186/s12870-018-1281-x
Zhao F, Zhang D, Zhao Y, Wang W, Yang H, Tai F, Li C, Hu X (2016a) The difference of physiological and proteomic changes in maize leaves adaptation to drought, heat, and combined both stresses. Front Plant Sci 7:1–19. https://doi.org/10.3389/fpls.2016.01471
Zhao P, Liu P, Yuan GX, Jia JT, Li XX, Qi D, Chen SY, Ma T, Liu GS, Cheng LQ (2016b) New insights on drought stress response by global investigation of gene expression changes in sheepgrass (Leymus chinensis). Front Plant Sci 7:954. https://doi.org/10.3389/fpls.2016.00954
Zheng J, Fu JJ, Gou MY, Huai JL, Liu Y, Jian M, Huang QS, Guo XY, Dong ZG, Wang HZ, Wang GY (2010) Genome-wide transcriptome analysis of two maize inbred lines under drought stress. Plant Mol Biol 72:407–421. https://doi.org/10.1007/s11103-009-9579-6
Acknowledgements
This research was supported by the National Key Research and Development Project of China (Selection and Efficient Combination Model of Wheat and Maize Water Saving, High Yield and High Quality Varieties) (Grant No. 2017YFD0300901).
Author information
Authors and Affiliations
Contributions
GL, TZ, and HD conceived and designed the experiment; GL, TZ, SL, XW, HJ, AD and YY performed the investigations and collected data; GL, TZ, SL and XW analyzed the data; GL and TZ wrote the original manuscript; All authors made the revision of the manuscript and approved this submission.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest. Furthermore, the founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, G., Zenda, T., Liu, S. et al. Comparative transcriptomic and physiological analyses of contrasting hybrid cultivars ND476 and ZX978 identify important differentially expressed genes and pathways regulating drought stress tolerance in maize. Genes Genom 42, 937–955 (2020). https://doi.org/10.1007/s13258-020-00962-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-020-00962-4