
Abstract
In this study, the chloroplast genome of Hariotina reticulata was fully sequenced and compared to other Sphaeropleales chloroplast genomes. It is 210,757 bp larger than most Sphaeropleales cpDNAs. It presents a traditional chloroplast structure, and contains 103 genes, including 68 protein-coding genes, six rRNA genes and 29 tRNA genes. The coding region constitutes of 43% of the whole cpDNA. Eighteen introns are found in 11 genes and six introns are unique for Hariotina. 11 open reading frames are identified among these introns. The synteny between Hariotina and Acutodesmus cpDNAs is in general identical, while within Sphaeropleales order, high variability in cpDNA architecture is indicated by general high DCJ distances. Ankyra judayi exhibits the greatest dissimilarity in gene synteny to the others and share some unique gene clusters with Treubaria triappendiculata. The phylogenomic analyses show that A. judayi is clustered with Treubariaceae species and sister to Chlorophyceae incertae sedis and other Sphaeropleales species. The monophyly of Sphaeropleales is rejected.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Scenedesmaceae is the largest family in alga order Sphaeropleales with over three hundred species. This group exhibits high adaption to freshwater and terrestrial habitat and high level of phenotypic homoplasy which leads to taxonomic confusion and covers species diversity within this family. This family has undergone a rather complicated taxonomic history with several revisions over the years from sole genus to present 49 genera classification (Chodat 1926; Trainor et al. 1976; Hegewald 1978, 2000; Hegewald and Silva 1988; Kessler et al. 1997; An et al. 1999; Van Hannen et al. 2002; Hegewald et al. 2010; Krienitz and Bock 2012; Guiry and Guiry 2016). Krienitz and Bock (2012) listed that only 13 genera were well phylogenetically and morphologically defined. Other phylogenetically accepted genera need further test. Besides, certain relationships still remain poorly resolved (Vanormelingen et al. 2007; Eliáš et al. 2010; Hegewald et al. 2010).
Plastid genome has been proved to be a useful tool for phylogenetic studies for resolving complex evolutionary relationships. So far, Acutodesmus obliquus is the only taxon with complete chloroplast genome and mitochondrial genome within Scenedesmaceae (de Cambiaire et al. 2006). More taxon sampling is needed to gain further insight into the family evolutionary trend. Previously, we reported the mitochondrial genome of Hariotina reticulata (MMOGRB0030F) which exhibits the similar genome organization to A. obliquus except a unique CUU anticodon for tRNA-Lys (He et al. 2016). Hariotina is a typical well supported small genus including only two recognized species in subfamily Coelastroidea. In this study, we sequenced its chloroplast genome and compared to A. obliquus and other 11 cpDNAs in Sphaeropleales in the aspects of genome features, gene content and genome structure. The introduction of chloroplast genome of H. reticulata will provide material for deeper understanding of phylogeny and the genome evolution in Sphaeropleales as well as in Scenedesmaceae.
Materials and methods
Strain isolation, cultivation and microscopy
The strain was isolated from Furong Lake in Xiamen, Fujian, China and deposited in Marine Medicinal Organism Germplasm Resources Bank of Third Institute of Oceanography (MMOGRB 0030F). It is preserved under a 12:12 light:dark cycle and 40 µmol photons m− 2 s− 1 from cool-white fluorescent tubes. Cells were observed under light microscope with Zeiss Scope A1.
Genome sequencing, assembly and annotation
Approximately 1 g of fresh microalgae was collected by centrifugation to extract the total genome using DNA extraction kit (DP305, Tiangen Biotech Co., LTD). Total DNA was used to generate the 500 bp paired-end library, following the Illumina Hiseq2500 standard protocol (Illumina Inc., San Diego, IL). About 3.7 G raw data was generated with reads length 250 bp, and the chloroplast genome sequencing depth is nearly to 1259.9×.
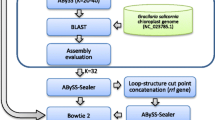
Reads were assembled with SOAPdenovo2-r240 (http://soap.genomics.org.cn). Since the assembled contigs contain a mixture of sequences from both organellar and nuclear genomes, the methods were used to isolate the chloroplast sequences based on the high correlation between contig read depth and the number of copies in the genome. Firstly, we sorted the assembled contigs by contig-read depth analysis of assemblies, that is, the raw reads sequence were mapped to the assembled contigs and the read depth of each contig was calculated through reads mapping. Taking the advantage of the difference of read depths among contigs, we could isolate the chloroplast contigs with high-coverage (more than 500×) from the nuclear contigs. Secondly, we also used all published chloroplast genome sequence to capture reads with BWA, then assembly those reads to chloroplast contigs. Finally, combined all isolated chloroplast contigs and recaptured reads again to isolate more completely chloroplast DNA reads, reassembly and contigs extended to get completely chloroplast genome sequencing.
Gene annotation was carried out with Organellar GenoMe Annotator (Wyman et al. 2004) and CPGAVAS (Cheng et al. 2013) with plastid/bacterial genetic code and default conditions. The cpDNA of A. obliquus chloroplast genome was applied as the referee. To verify the exact gene and exon boundaries, putative gene sequences and protein sequences were BLAST searched in Nt and Nr database. All tRNA genes were further confirmed through online tRNAscan-SE and tRNADB-CE search sever (Griffiths-Jones et al. 2003; Schattner et al. 2005; Abe et al. 2011). The graphical map of the circular plastome was drawn with Organellar Genome DRAW v1.2 (Lohse et al. 2007). UniMoG (Hilker et al. 2012) was applied to run the minimal histories of rearrangements among the pairwise aligned genomes under a double cut and join (DCJ) model.
Phylogenomic analyses
Phylogenomic analyses were based on amino acid sequences of 48 shared protein-coding genes of chloroplast genomes from 33 members in Chlorophyceae. Alignments were performed by MUSCLE v3.8 (Edgar 2004) with gaps or missing data eliminated. Amino acids substitution was estimated using ProtTest v3.4 (Darriba et al. 2011). Maximum likelihood (ML) analyses were conducted by RAxML v.8 (Stamatakis 2014) with LG + G + I + F model. Branch support was evaluated by 1000 replications of bootstrap. Bayesian analyses were applied with MrBayes v3.2.2 (Ronquist and Huelsenbeck 2003; Ronquist et al. 2012) running one million generations with four independent runs under CpREV+I+G+F model. The first 25% of samples were discarded as burn-in.
Results and discussion
Genome features
The complete Hariotina cpDNA (KX131180) assembles as a double-stranded circular with 210,757 bp larger than A. obliquus and other 12 cpDNAs in Sphaeropleales except Bracteacoccus giganteus and Pseudomuriella schumacherensis. It displays a typical quardripartite structure as most Chlorophyta species including a large single copy (LSC, 104,555 bp), a small single copy (SSC, 74,102 bp) and two 16,050 bp inverted repeats (IRA and IRB). 103 genes were identified, including 68 protein coding genes (PCGs), 29 tRNA genes and six rRNA genes (Table 1; Fig. 1). Of the 103 genes, we identified that one PCG, six rRNA genes and six tRNA genes are located within IRs. The LSC region contains 34 PCGs and eight tRNA genes while the SSC region has 33 PCGs and 15 tRNA genes. The psbC is the unique gene located in the boundary region between IRB and LSC. The total coding region accounts for 43.6% of the whole cp genome and the overall GC content was about 29.2% which is in the range of Sphaeropleales cpDNAs (Table 1).

Gene map of the chloroplast genome of Hariotina reticulata. Genes shown on the outside of the circle are transcribed counterclockwise. Annotated genes are colored according to the functional categories shown in the legend bottom left. (Color figure online)
The cpDNA of Hariotina strain exhibits great similarity with that of A. obliquus in the gene content with few genes absent e.g. rps2a and a copy of trnL. The differentiation of psaA gene structure is also observed between two cpDNAs. In A. obliquus, this gene is separated to two parts by five genes. The separation of psaA gene into two to four parts is common among Sphaeropleales cpDNAs. However, in Hariotina, it keeps intact with one intron and an ORF inserted which encodes putative maturase and HNH homing endonuclease.
Introns
Among the known Sphaeropleales cpDNAs, Kirchneriella aperta has the highest proportion of introns up to 20.94% of the whole plastid genome while Mychonastes homosphaera the least with only 0.14% (Table 1). In Hariotina chloroplast genome, eighteen introns are identified in 11 genes (Table 2), comprising 7.69% (including intronic ORFs) of the total cpDNA. 11 introns were identified as group I type, and seven as group II. Except introns found in psaA, psbA and rrn23S, other introns are rarely found or have not yet been observed in previously reported Sphaeropleales plastids (Supplementary Table S1). 11 open reading frames (ORFs) are distinguished within these introns (Table 2, Supplementary Table S2). Only one ORF encoding HNH is present in group II introns and ten ORFs in group I introns.
In Sphaeropleales, the introns in six genes including rps3, chlB, cemA, clpP, rps18 and ccsA are only present in Hariotina cpDNA. All are identified as group II type and no ORF is recognized. The other group II intron in psaA is consistent in Sphaeropleales with the exception of M. jurisii.
The psbA gene in Sphaeropleales chloroplast genomes comprises a large number of introns. For examples, nine introns are present in Neochloris aquatica cpDNA, eight in K. aperta and seven in B. giganteus. In six reported cpDNAs, e.g. Chlromochloris zofingiensis, B. minor, B. aerius, the psbA introns are missing. Four group IA introns are identified in Hariotina psbA gene containing four ORFs: three encoding HNH homing endonuclease and one encoding GIY-YIG endonuclease. The majority of psbA introns in Sphaeropleales are group I introns with the exception of four group II introns in K. aperta.
The introns in rrn23S gene are only found in P. schumacherensis and C. zofingiensis clade (Fig. 3) with both IA and IB types identified. Only IA type is present in Hariotina and two introns exhibits 74% similarity to that of A. obliquus. The intron in psbC is rarely observed in Sphareopleales plastid genome. Besides Hariotina, it is only observed in N. aquatica. All are identified as group IA type.
The group-I intron in tRNA (Leu) gene was reported to be conservative through Rhodophyta to high plants. This intron was thought to have been inherited by vertical transmission from the common ancestor of all chloroplasts (Besendahl et al. 2000). In Sphaeropleales order, it is present in most species as IB type except the IC3 type in A. obliquus cpDNA, while in Hariotina, this intron is lost as well as in A. judayi and two Mychonastes species.
Synteny and gene clusters
High variability in cpDNA architecture is widespread in Chlorophyceae (de Cambiaire et al. 2006; Turmel et al. 2015) as well as in Sphaeropleals as indicated by high DCJ distances (Table 3). Except A. judayi, the average DCJ distance among the Sphaeropleales cpDNAs is 28. When exluding all the tRNA genes from the analysis, the average DCJ distance reduces to 18. The cpDNA of A. judayi exhibits the highest variability in gene synteny to the others with the DCJ distance of 46 on average (31 excluding all tRNA genes).
The gene order of Hariotina cpDNA is identical to that of Acutodesmus with the DCJ distance of 5 (3 excluding all tRNA genes), indicating that the genome organization of cpDNAs is conservative at the level of the family Scenedesmaceae. The DCJ distance among the three Bracteacoccus taxon is higher than that between Hariotina and Acutodesmus. The average distance value of the three Bracteacoccus species reduces from 13 to 6 after all tRNA genes reduced. It might indicate a higher level of gene rearrangement in the family Bracteacoccaceae than in Scenedesmaceae.
Six gene clusters conserved throughout Chlorophyta cpDNAs (de Cambiaire et al. 2006) are also conserved in Sphaeropleales, e.g. psbH–psbN–psbT–psbB cluster, rrs–trnI–trnA–rrl–rrf cluster. Besides the above ones, ten unique gene clusters are found to be conservative throughout Sphaeropleales order, e.g. psbM–psbZ–ccsA–psaB, psbE–rps9–ycf4–ycf3, and rps12–psaJ–atpI–psbJ–psaA (Fig. 2). The plastid genome of A. judayi generates the most dissimilarity to other Sphaeropleales cpDNAs in gene clusters and exhibits similarity to Treubaria triappendiculata instead. Some unique gene clusters are observed between the two plastid genomes, e.g. psaA–psbJ–atpI–psaJ–rpl36–rps12–chlL–rps2 cluster.

Comparison of conserved genes clusters in Sphaeropleales. Black connected circles represent gene clusters. Grey circles indicate that genes are not adjacent, and white circles indicate genes that are absent from the cpDNA
Phylogenomic analyses
The Bayesian topology is almost identical to that of maximum likelihood analyses and is presented in Fig. 3. The topology of the trees obtained in this study was similar to that from the previous studies based on few markers or phylogenomic analyses (Wolf et al. 2002; Keller et al. 2008; Fučíková et al. 2016). The Ulvophyceae is sister to Chlorophyceae which includes fiver orders, e.g. OCC clade, Chlamydomonadales and Sphaeropleales. Hariotina reticulata was tightly linked with A. obliquus, forming the deepest branch with Neochloridaceae subclade (Fig. 3) and Neochloridaceae species (Fučíková et al. 2016) within Sphaeropleales clade.

Bayesian phylogenetic tree of Hariotina reticulata and other Chlorophyceae species based on amino acids of 48 chloroplast protein-coding genes. Values above branches indicate Bayesian posterior probabilities and maximum likelihood bootstrap values. Asterisks indicate full support in each analysis. Values below 70 and 0.60, respectively, are not shown
To our surprise, the monophyly of Sphaeropleales is rejected. A. judayi, which had been strongly assigned to Sphaeropleales with other ‘Sphaeroplea’ species in several studies (Wolf et al. 2002; Keller et al. 2008; Buchheim et al. 2012; Caisová et al. 2013; Fučíková et al. 2016), is not clustered with Sphaeropleales members. Instead, it is shown to be closely related to Treubariaceae member, T. triappendiculata and sister to Chlorophyceae incertae sedis and Sphaeropleales. Besides, the high DCJ distance between A. judayi and other Sphaeropleales members (Table 3) also suggests A. judayi exhibits the greatest dissimilarity in cpDNA architecture. It also shares a few unique gene clusters with T. triappendiculata, e.g. psaA–psbJ–atpI–psaJ–rpl36–rps12–chlL–rps2 cluster. These evidences bring about questions to reconsider the Sphaeropleales definition.
Conclusion
H. reticulata is the second Scenedesmaceae species to have its complete chloroplast and mitochondrial genomes sequenced. It provides important information to explore plastid evolution and phylogeny in Sphaeropleales as well as in Scenedesmaceae. Our most surprising advance is the sister relationship between Treubaria and Ankyra. We suggest a broader sampling of the Golenkinia, Chlorophyceae incertae sedis and Sphaeropleales and a probable reconsideration of the definition of Sphaeropleales. Additional chloroplast genome sequencing of Scenedesmaceae species is also necessary in order to explore a robust intrafamilial phylogenetic topology.
References
Abe T, Ikemura T, Sugahara J, Kanai A, Ohara Y, Uehara H et al (2011) tRNADB-CE 2011: tRNA gene database curated manually by experts. Nucleic Acids Res 39:210–213. https://doi.org/10.1093/nar/gkq1007
An SS, Friedl T, Hegewald E (1999) Phylogenetic relationships of Scenedesmus and Scenedesmus-like coccoid green algae as inferred from ITS-2 rDNA sequence comparisons. Plant Biol 1:418–429
Besendahl A, Qiu YL, Lee J, Palmer JD, Bhattacharya D (2000) The cyanobacterial origin and vertical transmission of the plastid tRNALeu group-I intron. Curr Genet 37:12–23
Buchheim MA, Sutherland DM, Schleicher T, Forster F, Wolf M (2012) Phylogeny of Oedogoniales, Chaetophorales and Chaetopeltidales (Chlorophyceae): inferences from sequence-structure analysis of ITS2. Ann Bot 109:109–116
Caisová L, Marin B, Melkonian M (2013) A consensus secondary structure of ITS2 in the Chlorophyta identified by phylogenetic reconstruction. Protist 164:482–496
Cheng J, Zeng X, Ran G, Liu Z (2013) CGAP: a new comprehensive platform for the comparative analysis of chloroplast genomes. BMC Bioinform 14:95. https://doi.org/10.1186/1471-2105-14-95
Chodat R (1926) Scenedesmus. Étude de génétique, de systématique expérimentale et d’hydrobiologie. Rev Hydrobiol 3:71–258
Darriba D, Taboada GL, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165
de Cambiaire JC, Otis C, Lemieux C, Turmel M (2006) The complete chloroplast genome sequence of the chlorophycean green alga Scenedesmus obliquus reveals a compact gene organization and a biased distribution of genes on the two DNA strands. BMC Evol Biol 6:37. https://doi.org/10.1186/1471-2148-6-37
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Eliáš M, Němcová Y, Škaloud P, Neustupa J, Kaufnerová V, Šejnohová L (2010) Hylodesmus singaporensis gen. et sp. nov., a new autosporic subaerial green alga (Scenedesmaceae, Chlorophyta) from Singapore. Int J Syst Evol Microbiol 60:1224–1235
Fučíková K, Lewis PO, Lewis LA (2016) Chloroplast phylogenomic data from the green algal order Sphaeropleales (Chlorophyceae, Chlorophyta) reveal complex patterns of sequence evolution. Mol Phylogenet Evol 98:176–183
Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR (2003) Rfam: an RNA family database. Nucleic Acids Res 31:439–441
Guiry MD, Guiry GM (2016) AlgaeBase. World-wide electronic publication. National University of Ireland, Galway
He LJ, Lou SL, Zhang F, Yang SJ, Zhang C, Lin XZ, Yang LH (2016) The complete mitochondrial DNA sequence of the green algae Hariotina sp. F30 (Scenedesmaceae, Sphaeropleales, Chlorophyceae). Mitochondrial DNA Part B: Resour 1:132–133
Hegewald E (1978) Eine neue Unterteilung der Gattung Scenedesmus Meyen. Nova Hedwig 30:343–376
Hegewald E (2000) New combinations in the genus Desmodesmus (Chlorophyceae, Scenedesmaceae). Algol Stud 96:1–18
Hegewald E, Silva PC (1988) Annotated catalogue of Scenedesmus and nomenclaturally related genera including original descriptions and figures. Bibl Phycol 80:1–587
Hegewald E, Wolf M, Keller A, Friedl T, Krienitz L (2010) ITS2 sequence-structure phylogeny in the Scenedesmaceae with special reference to Coelastrum (Chlorophyta, Chlorophyceae), including the new genera Comasiella and Pectinodesmus. Phycologia 49:325–335
Hilker R, Sickinger C, Pedersen CNS, Stoye J (2012) UniMoG—a unifying framework for genomic distance calculation and sorting based on DCJ. Bioinformatics 28:2509–2511
Keller A, Schleicher T, Förster F, Ruderisch B, Dandekar T, Müller T, Wolf M (2008) ITS2 data corroborate a monophyletic chlorophycean DO-group (Spaheropleales). BMC Evol Biol 8:218. https://doi.org/10.1186/1471-2148-8-218
Kessler E, Schäfer M, Hümmer C, Kloboucek A, Huss VAR (1997) Physiological, biochemical, and molecular characters for the taxonomy of the subgenera of Scenedesmus (Chlorococcales, Chlorophyta). Bot Acta 110:244–250
Krienitz L, Bock C (2012) Present state of the systematic of planktonic coccoid green algae of inland waters. Hydrobiologia 698:295–326
Lohse M, Drechsel O, Bock R (2007) Organellar Genome DRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet 52:267–274
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, HÓ§hna S et al (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Schattner P, Brooks AN, Lowe TM (2005) The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33:W686–W689
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Trainor FR, Cain JR, Shubert EL (1976) Morphology and nutrition of the colonial green alga Scenedesmus: 80 years later. Bot Rev 42:5–25
Turmel M, Otis C, Lemieux C (2015) Dynamic evolution of the chloroplast genome in the green algal classes Pedinophyceae and Trebouxiophyceae. Genome Biol Evol 7:2062–2080
Van Hannen EJ, Fink P, Lürling M (2002) A revised secondary structure model for the internal transcribed spacer 2 of the green algae Scenedesmus and Desmodesmus and its implication for the phylogeny of these algae. Eur J Phycol 37:203–208
Vanormelingen P, Hegewald E, Braband A, Kitschke M, Friedl T, Sabbe K, Vyverman W (2007) The systematics of small spineless Desmodesmus species, D. costato-granulatus (Sphaeropleales, Chlorophyceae), based on ITS2 rDNA sequence analyses and cell wall morphology. J Phycol 43:378–396
Wolf M, Buchheim M, Hegewald E, Krienitz L, Hepperle D (2002) Phylogenetic position of the Sphaeropleaceae (Chlorophyta). Plant Syst Evol 230:161–171
Wyman SK, Jansen RK, Boore JL (2004) Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20:3252–3255
Acknowledgements
This study was supported by Demonstration Project of Marine Economy Innovative Development Project (Nos. 12PYY001SF08, 16PYY005SF09), Natural Science Foundation of Fujian Province (No. 2016J05100), Xiamen Southern Oceanographic Center (No. 14CZP028HJ02) and the Xiamen Science and Technology Project (No. 3502Z20172017).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Lijuan He declares that she does not have conflict of interest. Zhaokai Wang declares that he does not have conflict of interest. Sulin Lou declares that she does not have conflict of interest. Xiangzhi Lin declares that he does not have conflict of interest. Fan Hu declares that he does not have conflict of interest.
Research involving with human and animal participants
This article does not contain any animal or human subjects. The research material is not in the endangered species list.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
He, L., Wang, Z., Lou, S. et al. The complete chloroplast genome of the green algae Hariotina reticulata (Scenedesmaceae, Sphaeropleales, Chlorophyta). Genes Genom 40, 543–552 (2018). https://doi.org/10.1007/s13258-018-0652-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-018-0652-x