
Abstract
Background
WRKY proteins play a vital role in the regulation of several imperative plant metabolic processes and pathways, especially under biotic and abiotic stresses. Although WRKY genes have been characterized in various major crop plants, their identification and characterization in pulse legumes is still in its infancy. Chickpea (Cicer arietinum L.) is the most important pulse legume grown in arid and semi-arid tropics.
Objective
In silico identification and characterization of WRKY transcription factor-encoding genes in chickpea genome.
Methods
For this purpose, a systematic genome-wide analysis was carried out to identify the non-redundant WRKY transcription factors in the chickpea genome.
Results
We have computationally identified 70 WRKY-encoding non-redundant genes which were randomly distributed on all the chickpea chromosomes except chromosome 8. The evolutionary phylogenetic analysis classified the WRKY proteins into three major groups (I, II and III) and seven sub-groups (IN, IC, IIa, IIb, IIc, IId and IIe). The gene structure analysis revealed the presence of 2–7 introns among the family members. Along with the presence of absolutely conserved signatory WRKY domain, 19 different domains were also found to be conserved in a group-specific manner. Insights of gene duplication analysis revealed the predominant role of segmental duplications for the expansion of WRKY genes in chickpea. Purifying selection seems to be operated during the evolution and expansion of paralogous WRKY genes. The transcriptome data-based in silico expression analysis revealed the differential expression of CarWRKY genes in root and shoot tissues under salt, drought, and cold stress conditions. Moreover, some of these genes showed identical expression pattern under these stresses, revealing the possibility of involvement of these genes in conserved abiotic stress–response pathways.
Conclusion
This genome-wide computational analysis will serve as a base to accelerate the functional characterization of WRKY TFs especially under biotic and abiotic stresses.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transcription factors (TFs) are proteins that play very important role during gene transcription by interacting with their corresponding cis-regulatory elements in the promoter regions. Several genes have been found in plant genomes which encode various classes of TFs (Rushton et al. 2010). Amongst them, WRKY TF family is one of the largest TF family in higher plants (Eulgem et al. 2000). WRKY TFs have been implicated in the regulation of different metabolic pathways (e.g. biosynthesis of secondary metabolites, plant senescence and signal molecule-delivery) under biotic and abiotic stresses in plants (Vom Endt et al. 2002). The first WRKY TF (SPF1) was identified from sweet potato (Ipomoea batatas) which was found to be associated with gene regulation under the abiotic stress conditions (Ishiguro and Nakamura 1994).
The WRKY proteins harbor a characteristic WRKY domain of around 60 amino acids with conserved oligopeptide sequence (WRKYGQK) at N-terminal along with Cys2His2/Cys2HisCys zinc finger motif (CX4–7CX22–23HXH/C) at C-terminal (Rushton et al. 2010; Schluttenhofer and Yuan 2015). The conservation of cysteine and histidine in the WRKY domain is responsible for the formation of unique zinc finger-like motif and sequence of WRKY amino acids can directly bind with W box (TTGACT/C) cis-regulatory element, which are found in upstream regions of target genes (Zhang et al. 2018). WRKY TFs have been classified into three major groups (I, II and III) based on the number of WRKY domains and features of zinc finger motifs (Rinerson et al. 2015). The members of group-I contain two WRKY domains along with C2H2 zinc finger motif at either C terminal or N terminal. On the other hand, members of group-II and III have only one C terminal WRKY domain (Brand et al. 2013). Group II has been further classified into five sub-groups (IIa, IIb, IIc, IId and IIe) based on evolutionary divergence and difference in conserved motifs (Rinerson et al. 2015). Group-I members are the ancient ones and have been responsible for the evolution of WRKY TFs family while group-II (IIa and IIb) has been evolved most probably from algae with a single WRKY domain and are separated from group-I derived lineage (Wu et al. 2005; Rinerson et al. 2015).
Several studies have demonstrated the role of WRKY TFs in terms of increased tolerance to biotic and abiotic stresses in plants (Ali et al. 2014, 2018; Qin et al. 2015; Rushton et al. 2010). For instance, overexpression of OsWRKY45 in rice has enhanced the disease and drought tolerance (Qiu and Yu 2009), AtWRKY25 and AtWRKY33 enhanced salinity resistance in Arabidopsis (Jiang and Deyholos 2009) and VvWRKY11 increased resistance against osmotic stress in grapes (Liu et al. 2011). In kiwifruit, various AcWRKY TFs (AcWRKY29, 40, 48, 55, 95 and 96) were up-regulated under salt stress (Jing and Liu 2018). Recently, Ali et al. (2018) demonstrated the role of WRKY11 and WRKY17 in the regulation of abiotic stress responses in Arabidopsis. In Taxus chinensis, TcWRKY8 and TcWRKY47 were involved in the regulation of taxol-biosynthesis-related genes (Zhang et al. 2018). Treatment with hormones like ethylene, jasmonic acid, salicylic acid, gibberellins and abscisic acid can induce the expression of WRKY TFs, showing their involvement in phytohormone signaling cascades (Xiao et al. 2017). In cotton, WRKY transcripts have also been identified in elongating fiber ovules 3 days post-anthesis suggesting their possible role in fiber development (Wang et al. 2010). WRKY genes have also been suggested to play an important role in the development of anthers and embryos in plants (Zhang et al. 2018). WRKY TFs have also been involved in transcriptional reprogramming of plant immunity responses against various pathogens (Pandey and Somssich 2009). Many WRKY genes are regulated by miRNAs. HaWRKY6 (sunflower) is regulated by miR396 to control the damage by high temperature (Giacomelli et al. 2012).
Chickpea (Cicer arietinum L.) is the most important cool-season pulse legume primarily cultivated in arid and semi-arid regions of the world (Acharjee and Sarmah 2013; Sani et al. 2017). The availability of complete genome assembly of chickpea (Thudi et al. 2016; Varshney et al. 2013) offers an opportunity for the genome-wide computational identification and characterization of WRKY encoding genes. Here we presented genome-wide identification and classification of WRKY TF family members and described their evolution and expansion. The functions of identified proteins have also been proposed based on orthologs information from comparative phylogenetic analysis. In addition, expression profiles of identified WRKY in different tissue of chickpea plant under various abiotic stresses are also discussed. This study will help in functionally characterizing and understanding the role of chickpea WRKY family members especially in response to abiotic stresses. The candidate genes can then be employed in the stress–tolerance breeding programs.
Materials and methods
Database search and sequence retrieval
The complete genome assembly of chickpea along with complete proteome sequence file was downloaded from webpage of International Crop Research Institute for Semi-Arid Tropics (ICRISAT) http://cegsb.icrisat.org/gt-bt/ICGGC/genomedata.zip (Varshney et al. 2013). Conserved amino acids sequence (WRKYGQK) was used as a query to explore the WRKY TFs from the chickpea proteome sequence file using CLC sequence viewer (v7.6.1, Knudsen et al. 2011). Putative WRKY TFs were further confirmed by using basic local alignment search tool (blastP) at the NCBI webserver. NCBI database was further explored to find out the gene accession numbers (GI), chromosome number, genomic information and protein size of marked WRKY TFs. The genomic nucleotide sequence of all the identified WRKYs were also retrieved form NCBI. The molecular weight and isoelectric point (PI) were calculated using the ExPASy Bioinformatics Resource Portal (http://web.expasy.org/compute_pi/) (Gasteiger et al. 2005).
Chromosomal mapping, intron/exon distribution and conserved domain analysis
NCBI database was used to record the positions of these CarWRKY genes on the chromosomes, whereas Map Chart (v. 2.32) was used to construct chromosomal localization map of all the identified non-redundant chickpea WRKY genes according to scale (Voorrips 2002). In order to construct the gene structure displaying the intron–exon distribution of WRKY genes, Gene Structure Display Server (v2.0 http://gsds.cbi.pku.edu.cn/) was used (Hu et al. 2014). For this purpose, the genomic DNA as well as CDS sequences of all the WRKY genes were submitted to construct the gene structure map along with the intron phases.
In order to identify the conserved domains among all the chickpea WRKYs, their protein sequences were subjected to MEME (Multiple Em for Motif Elicitation; v5.0.3) (Bailey et al. 2009). The analyses were carried out using default parameters with following exception i.e. occurrence of motif was set at 0 or 1 per sequence; number of motifs was set as 19; optimum width of motifs 12–63 residues; minimum number of sites of motif was set as 5.
Comparative phylogenetic analysis of WRKY proteins from chickpea, Medicago and Arabidopsis
The comparative phylogenetic tree was constructed by using protein sequences of putative WRKY TFs from C. arietinum (CarWRKY), Medicago truncatula (MedtrWRKY) and Arabidosis thaliana (AtWRKY) as a reference sequence (Eulgem et al. 2000; Song and Nan 2014). For this purpose, the peptide sequences of 96 MedtrWRKYs were downloaded from plant TF database (PlantTFDB v4.0; Jin et al. 2016) webserver http://planttfdb.cbi.pku.edu.cn/family.php?sp=Mtr&fam=WRKY. On the other hand, the protein sequences of 72 AtWRKYs were obtained from Eulgem et al. (2000). The multiple sequence alignment of all the 238 WRKYs was carried out using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/; Sievers et al. 2011). The resultant alignment was used to compute the phylogenetic tree through neighbor-joining method (Tamura et al. 2013) with 1000 bootstrap replicates using Molecular Evolutionary Genetics Analysis tool (MEGA v7.0; Kumar et al. 2016). Poisson correction method was used to compute the evolutionary distances in the units of the number of amino acid substitutions per site per unit time (Saitou and Nei 1987). All positions containing < 95% site coverage were removed i.e. fewer than 5% alignment gaps, missing data and ambiguous bases were allowed at any position. There was a total of 71 amino acid positions in the final dataset. The resultant tree was then used to infer the evolutionary history and possible functional roles of WRKY TFs.
Gene duplication and evolutionary divergence analysis
The putative paralogous WRKY gene pairs were identified from the phylogenetic tree. Their coding sequences were used to construct pairwise alignment using inbuilt ClustalW and PAM protein weight matrix of MEGA7. The resultant alignments were subjected to DNA sequence polymorphism software (DnaSP v5.10.01) (Librado and Rozas 2009) to compute the synonymous substitution rates (Ks) and non-synonymous substitution rates (Ka). The Ks/Ka was also calculated to determine which type of codon selection operated during evolution. The values of Ks were further used to calculate the approximate period of duplication event by using a formula T = Ks/2λ assuming a clock rate (λ) of 6.05 × 10−9 substitutions/synonymous site/year for chickpea (Jain et al. 2013).
In silico gene expression analysis under abiotic stresses
The transcriptome data of chickpea were obtained from chickpea transcriptome database (CTDB, Verma et al. 2015) and from Garg et al. (2015), where the plants were treated with various abiotic stresses. Briefly, salt stress was imposed by treating 10 days old seedlings with 150 mM NaCl (Garg et al. 2015). Drought stress was imposed by transferring 10 days old seedlings on folded dry tissue paper, while cold stress was induced by keeping the seedlings at 4 °C and the samples were collected for total RNA extraction (Garg et al. 2015). We retrieved the expression data of WRKY genes from root and shoot tissues under salinity, drought and cold stresses from Garg et al. (2015). The hierarchical clustering and the heatmap-based expression profiles of CarWRKY genes were performed using Genesis (v1.7.6) (Sturn et al. 2002).
Results
Identification and distribution of WRKY TFs encoding genes in chickpea genome
Taking the advantage of availability of complete genome assembly of chickpea, we initially identified 72 non-redundant WRKY genes encoding putative WRKY TFs from chickpea genome using WRKYGQK as a query. The identified genes were further scanned for the presence of WRKY-specific principal domains in the encoding proteins. For this purpose, the presence of complete WRKY domain was opted as a basic criterion for the inclusion of gene in WRKY family. Thus, two WRKY genes were removed due to incomplete WRKY domains in their amino acid sequence. These could be considered as pseudogenes which might have lost the part of functional domain during the evolution process (Panchy et al. 2016). The remaining 70 non-redundant CarWRKY genes were renamed in ascending order according to their positions on the chromosomes (CarWRKY1 to CarWRKY70) as described in various other species (Eulgem et al. 2000; Li et al. 2015; Jing and Liu 2018).
The peptide length of CarWRKY proteins varied from 162 to 763 amino acids within an average of 371 amino acids (detailed in Table 1). The isoelectric point value had a range from 5.01 to 9.91, while the molecular weight ranged from 18.6 to 84.6 kDa, with an average of 41.48 kDa in chickpea WRKYs (Table 1).
The distribution of identified CarWRKY genes on the chromosomes was carried out using MapChart. The chromosome map revealed the uneven distribution of 65 WRKY genes on 7 out of 8 chromosomes (Fig. 1). The position of remaining five WRKY genes i.e. WRKY 66–70 could not be mapped to any chromosome, as they were present on scaffold regions. All the 7 chromosomes share almost the same number of WRKY genes among them. For examples chromosomes 1, 3, 6 and 7 contained an equal number of WRKY genes i.e. ten genes each, while chromosome 8 has no WRKY gene at all. Interestingly, it was observed that most of these WRKY genes are present on the chromosome in the form of clusters, which suggest that the genes within a cluster may be a part of single QTL.

Distribution of 65 CarWRKY genes on chickpea chromosomes. The Chr (1–8) at the top of each bar represents the chromosome number of the chickpea. The scale is shown in Mb at the left of the figure. The location of each CarWRKYs is indicated on the right side of the respective chromosome
Classification, gene structure and conserved domains analyses of chickpea WRKYs
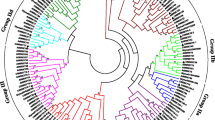
The chickpea WRKY TFs were classified into three major groups based on an unrooted phylogenetic tree. This classification followed the same pattern as observed in various other crop species (Eulgem et al. 2000; Zhang and Wang 2005) i.e. group I, II and III. Group-I contains 14 members, and further divided into two distinct subgroups (IN and IC with 10 and 4 members respectively). Group-II contains 48 members and was further classified into five sub-groups i.e. IIa, IIb, IIc, IId, and IIe with 5, 11, 15, 7 and 10 members respectively. Group III represents a monophyletic group with eight members (Fig. 2a).

a Phylogenetic tree-based classification of CarWRKYs. An unrooted phylogenetic tree was created based on the full-length peptide sequences of CarWRKYs with 1000 bootstraps. Classification is shown on the base of phylogenetic tree using different colors into groups (I, IIa–e and III). b Exon–intron structure analyses of CarWRKY genes. Grey line represents introns, while yellow boxes represent exons. The blue boxes represent the untranslated region (UTR). The asterisks (“***”) represent the exclusion of the CarWRKY66 from the graphical presentation due to its large genomic size. c Conserved domains of CarWRKY proteins. The conserved domains of WRKY proteins identified by MEME, are shown with colored boxes according to the scale. Gray lines represent the non-conserved sequences, and each domain is indicated by a colored box numbered at the bottom. d Gene structure of CarWRKY66 along with scale. (Color figure online)
The gene structure i.e. the intron/exon distribution pattern of all the WRKY genes was also determined to gain further insights into the evolution of WRKY family in chickpea. The ordinal position and intron–exon distribution pattern in the genomic region of a gene is used as a supporting evidence of expansion pattern of a gene family and its evolutionary relationship with its ancestors.
Chickpea WRKY genes exhibited a diversity with respect to the number of introns e.g. ranging from 1 to 7. Despite the difference in the size of their genomic regions, a somewhat conserved gene structure within the groups of phylogenetic tree has been observed (Fig. 2b) except CarWRKY66, a member of group IIb. The gene structure of CarWRKY66 was drawn separately due to extra-large size of its genomic DNA sequence i.e. 18.14 Kb (Fig. 2d). It contains a total of 6 introns, of which four introns have comparable length with other WRKY genes (i.e. intron 1, 3, 4 and 5), while the remaining two introns are unusually long i.e. intron 2 has 5.5 kb length, and intron 6 has 9.5 kb length.
All the members of group IIC have 2 introns except CarWRKY23, CarWRKY68 and CarWRKY69. Both CarWRKY23 and CarWRKY68 (which seems to be paralogous genes) might have lost one intron during the speciation process. On the other hand, CarWRKY69 have three introns, which probably had resulted by a recent intron gain event. Similarly, group IId, IIe and group III members also have 2 introns except CarWRKY47 in group IIe which might have lost one intron during evolution from the ancestral sequence. Overall, 39 out of 69 CarWRKYs contained two introns.
In addition to intron/exon distribution pattern, the intron phases i.e. the position of introns within codons were also determined. Three types of intron phases (0, 1 and 2) were found but overall type 2 was most common and frequent in CarWRKYs (Fig. 2b). In group I, mostly intron phase 0 was observed in first two introns, while all the remaining introns were in phase 2. Sub-group IIa and IIb carried intron phase 0 predominantly. On the other hand, intron phase 2 was abundant among members of sub-group IIc, IId, IIe and group III. Overall, a total of 180 introns were present among all the 70 WRKYs of chickpea. Among them, 42% (i.e. 75 introns) were phase 0 introns, 12% (i.e. 22 introns) were phase 1 and remaining 46% (i.e. 83) were phase 2 introns.
The identification of conserved domains in a gene family also serves as a supporting evidence to verify the gene duplication events during evolution; and to dissect their functional conservation. For this purpose, the peptide sequences of all the CarWRKY TFs were subjected to MEME, for the identification of conserved domains. A total of 19 conserved domains (ranging from 12 to 42 residues in length) were identified among 70 WRKY TFs (Table 2). Among them, domain 1 and 2 represent the characteristic WRKY DNA-binding domain which is absolutely conserved among all the 70 WRKYs (Fig. 2c). The conserved domain analysis further revealed the presence of conserved domain in a phylogenetic group-specific manner. For example, the members of group I have 7 conserved domains (domain 1, 2, 3, 4, 5, 11 and 16); the members of group IIa have 4 conserved domains i.e. 1, 2, 6 and 7; member of group IIb have 7 conserved domains (domain 1, 2, 5, 6, 7, 8 and 10); 11 members of group IIc have 3 conserved domains (domain 1, 2 and 5); 5 members of group IId showed 7 conserved domains (domain 1, 2, 9, 12, 13, 15 and 19); 10 members of group IIe have 3 conserved domains (domain 1, 2 and 9); and the 7 members of group III contain 3 conserved domains (domain 1, 2 and 14).
Overall, the conserved domains analysis along with the intron–exon distribution pattern among chickpea WRKYs seemed to be conserved in a group-specific manner, thus validating the grouping of CarWRKYs in the phylogenetic tree.
Comparative phylogenetic analysis of chickpea, M. truncatula and A. thaliana WRKYs
An unrooted neighbor-joining comparative phylogenetic tree was constructed from 238 protein sequences of CarWRKYs, MedtrWRKYs and AtWRKYs. The tree divided the WRKY protein into three major clusters of orthologous genes (MCOGI, II and III) (Fig. 3). These three major groups were subdivided into seven sub classes i.e. IN, IC, IIa, IIb, IIc, IId and IIe as reported by Eulgem et al. (2000). Among the three major groups, MCOG-II represents the largest major group of phylogenetic tree with 120 WRKYs distributed in five subgroups i.e. 22 in IIa, 13 in IIb, 40 in IIc, 27 in IId and 18 in IIe. MCOG-I is the second largest major group with 79 WRKYs distributed in two sub groups i.e. 57 in IN and 22 in IC. MCOG-III represents the smallest major group with 39 WRKYs from all the three species.

Comparative phylogenetic tree of WRKY proteins from chickpea, M. truncatula and A. thaliana. The deduced full-length amino acid sequences of CarWRKYs, MedtrWRKYs and AtWRKYs were aligned by Clustal Omega and an unrooted phylogenetic tree was constructed by Mega 7 using neighbor-joining method. Green rectangles, pink triangles and blue circles at the end of the nodes were used for the identification of the chickpea, M. truncatula and A. thaliana WRKY family members respectively. The numbers on the nodes represent percentage of bootstrap values from 1000 replicates. Various colours are used to distinguish different major clusters of orthologous genes (MCOG). (Color figure online)
Moreover, various orthologous gene paris were also identified among these three species in all the three major groups (Table S1). For instance, MCOG I contained 21 orthologous gene pairs, 14 among them were identified from sub-group IN; and 7 were identified in IC. Similarly, 31 orthologous gene pairs were identified in MCOG II, distributed in five subgroups as following i.e. 4 in IIa, 4 in IIb, 10 in IIc, 7 in IId and 6 in IIe. Only six pairs of orthologous genes were identified in MCOG III. It is interesting to note that the CarWRKYs shared more homology with MedtrWRKYs as compared to AtWRKYs. Overall, 57 orthologous gene pairs were identified between chickpea and M. truncatula. This sequence similarity of WRKY proteins among both these species is a result of the fact that both chickpea and M. truncatula are members of galegoid clade of Phaseoleae tribe of Fabaceae family. It has already been reported that the chickpea proteins shared a greater homology with M. truncatula as compared to A. thaliana (Varshney et al. 2013).
Duplication and selection pressure analyses of WRKY genes in chickpea
Gene duplications i.e. whole genome, segmental and tandem duplications play an important role in the expansion and evolution gene families. Whole genome duplications are mainly responsible for gene expansion and evolution in auto-polyploids (Mayrose et al. 2011), while segmental and tandem duplications have been known to play an important role in evolution of gene families in all the crop plants (Cannon et al. 2004). To study duplications and evolutionary events of CarWRKY genes in chickpea, 22 pairs of putative paralogous genes were recognized based on the chickpea WRKY phylogenetic tree. The paralogous genes located on the same chromosomes comes under the classification of tandem duplication while paralogous genes located on different chromosomes are considered to be segmentally duplicated (Panchy et al. 2016). A total of 21 CarWRKY gene pairs were identified to be segmentally duplicated whereas only one paralogous gene pair (CarWRKY62/CarWRKY 64 on chromosome 7) seemed to be evolved as a tandem duplication event (Table 3). Thus in chickpea, the evolution of WRKY genes seems to be predominantly associated with segmental duplications.
To find out the expected period of divergence of the paralogous gene pairs, the synonymous (Ks) and non-synonymous (Ka) substitution rates were calculated. In chickpea, WRKY paralogous genes seemed to be duplicated from 55.80 to 313.50 million years ago (MYA) through segmental duplication with an average of 109.51 MYA. On the other hand, the tandemly duplicated gene pair seemed to be originated 74.30 MYA. The Ka/Ks ratios of all the 22 paralogous CarWRKYs were ranged from 0.20 to 0.84. It shows that the purifying selection might have operated on the codons during the evolution and expansion of paralogous WRKY genes in chickpea.
In silico expression analysis of CarWRKYs under salt, drought and cold stress
The expressions of WRKY genes were investigated from CTDB (Verma et al. 2015) and Garg et al. (2015) in root and shoot tissues under salt, drought and cold stress. A thorough investigation enabled us to identify the expression data of 43 CarWRKYs in both the tissues under all the three stress conditions. The heat map-based expression profiles of these CarWRKYs revealed their differential expression in root and shoot tissues under all the three abiotic stress (Fig. 4).

Heat map of 43 CarWRKY genes expression in chickpea. CarWRKY genes expression in root and shoot tissues under salt, drought and cold. The expression-based hierarchical clustering of genes was done to show various gene clusters. DS desiccation stress, SS salinity stress and CS cold stress
Under salt stress, 14 WRKY genes were downregulated in roots, while 9 WRKY genes were downregulated in shoots. Similarly, under drought stress, 15 WRKYs were downregulated in roots and shoots. Under cold stress, 20 WRKYs were downregulated in roots, while 15 WRKYs were downregulated in shoots. Interestingly, some genes exhibited similar expression trends under all the tissues and stresses. For example, CarWRKY59 and CarWRKY48 were always upregulated irrespective of the tissues and stresses applied. On the other hand, CarWRKY45 was always downregulated in all the tissues and under all the stresses. CarWRKY26 was only upregulated in shoots under salt stress, while it is downregulated under other stresses and tissues.
Some of the WRKY genes showed expression in a tissue- and/or stress-specific manner. For examples, the expression of CarWRKY47 was observed in shoot tissue under drought stress only, while no expression was found under salt and cold stress. Similarly, the transcripts of CarWRKY08 were only observed under cold stress in shoots, while its expression was completely absent in shoots under salt and drought stress. Combining all the three stresses, a total of 49 CarWRKYs were downregulated in roots as compared to 39 in shoots. On the other hand, the number of upregulated CarWRKY genes was greater in shoots (i.e. 90 genes) than in roots (i.e. 80 genes).
Discussion
Discovery, distribution and characterization of CarWRKY TFs
WRKY TF family is one of the most important families of regulatory proteins which are not only involved in the regulation of developmental processes in plants but also modulation of stress responses in plants (Rushton et al. 2010). Identification and characterization of WRKY family members in chickpea, a protein rich food legume, offer opportunities for the functional validation of these members against the abiotic stresses, which are a real threat to chickpea productivity.
Here we report the identification and characterization 70 genes encoding putative WRKY TFs in chickpea. When compared with other plant species, the number of WRKY genes in chickpea is greater than that in barley (45 WRKYs) (Mangelsen et al. 2008), castor bean (58 WRKYs) (Zou et al. 2016), cucumber (55 WRKYs) (Ling et al. 2011), rapeseed (43 WRKYs) (Yang et al. 2009), and grape wine (60 WRKYs) (Wang et al. 2014b). On the other hand, the number of WRKY genes in chickpea is less than that in tomato (78 WRKYs) (Chen et al. 2015), cotton (120 WRKYs) (Cai et al. 2014), maize (136 WRKYs) (Wei et al. 2012), soybean (131 WRKYs) (Yu et al. 2016) and rice (100 WRKYs) (Wu et al. 2005). From this comparison, it seems evident that the number of WRKY encoding genes is not proportional to the genome size of the respective plant species.
The phylogenetic tree-based classification of chickpea followed the same trend as in other crop species. The characterization of WRKYs with respect to intron/exon distribution and conserved domains revealed the conservation of gene structure as well as domains among the members of a same group in the phylogenetic tree. The number of introns in CarWRKY genes ranged from 1 to 7, which is in accordance with the gene structure of most WRKY genes in other plant species like rubber plant (Li et al. 2015). In some species like rice, intron-less WRKY genes were also reported (Xie et al. 2005; Ross et al. 2007), which might be result of intron loss events during evolution. On the other hand, WRKY genes with 6 or 7 introns were absent in cassava (Wei et al. 2016). The structural diversity between exons and introns is also considered as helpful tool for phylogenetic grouping of these genes, as a supporting evidence. This diversity is an important part of the evolution, diversification and neo-functionalization of gene families (Shiu and Bleecker 2003; Wang et al. 2014a; Han et al. 2016).
Intron size was variable within and between each WRKY sub-family. This could be due to the consequence of chromosomal rearrangements like fusions, inversions and duplications (Li et al. 2016). However, the size of the exon was conserved within a cluster of closely related CarWRKY genes. Similar results have been reported recently for WRKY genes in different plants e.g. cassava, common bean and cacao (Wei et al. 2016; Wang et al. 2016; de Almeida et al. 2017). Moreover, it has been shown previously that divergence in exon is less common with the same pace of duplications in orthologs as compared to paralogs (Xu et al. 2010).
Comparative phylogenetic, functional and expression analyses
WRKY proteins in chickpea were primarily divided in three main groups by the phylogenetic analysis i.e. MCOG-I, II and III, while MCOG-II was further classified into five sub-groups (MCOG-IIa, b, c, d and e). These results are in-consistency with the findings of earlier studies e.g. by Eulgem et al. (2000) and Wei et al. (2016) in Arabidopsis and cassava respectively. In chickpea, most of the CarWRKYs were present in MCOG-II as in the case of potato 52 StWRKYs were recorded (Zhang et al. 2017).
The comparative phylogenetic tree can be used to infer the gene function of CarWRKYs, because the CarWRKYs were present in an orthologous relationship with MedtrWRKYs and AtWRKYs. The orthologous gene pairs usually retain their function after the speciation (Blanc and Wolfe 2004). Being a model plant, extensive efforts have been made to functionally characterize the genes of A. thaliana. Therefore, most of the AtWRKYs have been functionally characterized.
For instance, in group IIa, AtWRKY58 (ortholog of CarWRKY67) has been shown to negative regulate systematic acquired resistance in A. thaliana (Eulgem 2006). Wilt disease symptoms caused by Ralstonia solanacearum are also influenced by the ortholog of CarWRKY32 i.e. AtWRKY27 (Mukhtar et al. 2008). In Arabidopsis, the level of mRNA has been increased in radicules and roots by AtWRKY65 by lowering the expression levels in plant leaves (Mangelsen et al. 2008). The ortholog of CarWRKY25 i.e. AtWRKY44 has been observed to control the trichome development in Arabidopsis (Johnson et al. 2002). AtWRKY02 (an ortholog of CarWRKY38) is a vital WRKY family member which is involved in the regulation of seed germination and post germination growth (Jiang and Yu 2009).
In MCOG-IId, AtWRKY72 which is an ortholog of CarWRKY60, has been involved in defense system against pathogen or nematode (Dong et al. 2003; Yu et al. 2001). In MCOG-IIe, the ortholog of CarWRKY05 in A. thaliana i.e. AtWRKY40 was up-regulated under salt treatment at the transcriptional level (Zou et al. 2016). An ortholog of CarWRKY12 (AtWRKY57) can uplift the level of ABA which might be helpful to enhance the drought tolerance under abiotic stresses (Huang et al. 2015). In MCOG IIc, AtWRKY12 (ortholog of CarWRKY69) works as a negative regulator during the formation and development of secondary cell wall in Arabidopsis (Qin et al. 2015). Transcriptomic data-based expression analysis of most of orthologous CarWRKYs revealed their differential expression in root and shoot tissues under abiotic stresses. The identification of AtWRKY orthologs in chickpea can help in functional validation of their functions in chickpea. The candidate WRKYs in chickpea can then be employed for functional genomics followed by their potential use in stress breeding programs.
Gene divergence and selection pressure analysis
Gene duplication events i.e. whole genome, tandem and segmental duplications are considered as key mechanisms involved in the expansion and complexities of gene families (Liu et al. 2011). Segmental duplications seemed to be more common as compared to tandem duplication for the expansion of WRKY family in chickpea, oil palm, soybean and tomato (Xiao et al. 2017; Guo and Qiu 2013; Cai et al. 2013). On the other hands, tandem duplications have been reported to play a major role for the expansion of WRKY family in potato and sweet orange (Zhang et al. 2017; da Silva et al. 2017). It is also interesting to note that all the paralogous gene pairs in chickpea seemed to have gone through the strong purifying selection. Purifying selection has also been reported as a key player during the evolution of WRKY genes in A. thaliana, M. truncatula, and Gossypium species (Ding et al. 2015; Wang et al. 2011; Song et al. 2014). The purifying selection usually selectively removes the deleterious alleles over time (Biswas and Akey 2006), suggesting the conservation of key nucleotide sequences in WRKYs to perform important roles for the survival of plants.
Conclusions
In summary, the present study identified 70 non-redundant WRKY encoding genes in an important cool season pulse-legume, chickpea. Their classification; characterization with respect to gene structure and conserved domains; and comparative phylogenetic analyses suggests the conservation of WRKY classes among the studies plant species. The paralogous genes were suggested to expand as a result of segmental duplications, while the codons went through the purifying selection. Most of the genes were observed to be up-regulated under stress environments, suggesting their role in the mediation of stress responses in chickpea. Overall, these analyses will help to accelerate the functional characterization of WRKY TFs especially in response to biotic and abiotic stress. The candidate CarWRKY genes can then be employed in stress breeding program.
References
Acharjee S, Sarmah BK (2013) Biotechnologically generating ‘super chickpea’ for food and nutritional security. Plant Sci 207:108–116
Ali L, Madrid E, Varshney R, Azam S, Millan T, Rubio J, Gil J (2014) Mapping and identification of a Cicer arietinum NSP2 gene involved in nodulation pathway. Theor Appl Genet 127:481–488
Ali MA et al (2018) Transcription factors WRKY11 and WRKY17 are involved in abiotic stress responses in Arabidopsis. J Plant Physiol 226:12–21
Bailey TL et al (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37:W202–W208
Biswas S, Akey JM (2006) Genomic insights into positive selection. Trends Genet 22:437–446
Blanc G, Wolfe KH (2004) Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 16:1679–1691
Brand LH, Fischer NM, Harter K, Kohlbacher O, Wanke D (2013) Elucidating the evolutionary conserved DNA-binding specificities of WRKY transcription factors by molecular dynamics and in vitro binding assays. Nucleic Acids Res 41:9764–9778
Cai X et al (2013) Genome-wide analysis of plant-specific Dof transcription factor family in tomato. J Integr Plant Biol 55:552–566
Cai C, Niu E, Du H, Zhao L, Feng Y, Guo W (2014) Genome-wide analysis of the WRKY transcription factor gene family in Gossypium raimondii and the expression of orthologs in cultivated tetraploid cotton. Crop J 2:87–101
Cannon SB, Mitra A, Baumgarten A, Young ND, May G (2004) The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol 4:10
Chen L, Yang Y, Liu C, Zheng Y, Xu M, Wu N, Sheng J, Shen L (2015) Characterization of WRKY transcription factors in Solanum lycopersicum reveals collinearity and their expression patterns under cold treatment. Biochem Biophys Res Commun 464:962–968
da Silva EG, Ito TM, de Souza SGH (2017) ‘In silico’genome-wide identification and phylogenetic analysis of the WRKY transcription factor family in sweet orange (‘Citrus sinensis’). Aust J Crop Sci 11:716
de Almeida DSM et al (2017) Genome-wide identification and characterization of cacao WRKY transcription factors and analysis of their expression in response to witches’ broom disease. PLoS ONE 12:e0187346
Ding M, Chen J, Jiang Y, Lin L, Cao Y, Wang M, Zhang Y, Rong J, Ye W (2015) Genome-wide investigation and transcriptome analysis of the WRKY gene family in Gossypium. Mol Genet Genom 290:151–171
Dong J, Chen C, Chen Z (2003) Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Mol Biol 51:21–37
Eulgem T (2006) Dissecting the WRKY web of plant defense regulators. PLoS Pathog 2:e126
Eulgem T, Rushton PJ, Robatzek S, Somssich IE (2000) The WRKY superfamily of plant transcription factors. Trends Plant Sci 5:199–206
Garg R, Bhattacharjee A, Jain M (2015) Genome-scale transcriptomic insights into molecular aspects of abiotic stress responses in chickpea. Plant Mol Biol Rep 33:388–400
Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. In: Walker JM (ed) Proteomics protocols handbook. Springer, New York, pp 571–607
Giacomelli JI, Weigel D, Chan RL, Manavella PA (2012) Role of recently evolved miRNA regulation of sunflower HaWRKY6 in response to temperature damage. New Phytol 195:766–773
Guo Y, Qiu L-J (2013) Genome-wide analysis of the Dof transcription factor gene family reveals soybean-specific duplicable and functional characteristics. PLoS ONE 8:e76809
Han Y, Ding T, Su B, Jiang H (2016) Genome-wide identification, characterization and expression analysis of the chalcone synthase family in maize. Int J Mol Sci 17:161
Hu B, Jin J, Guo A-Y, Zhang H, Luo J, Gao G (2014) GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31:1296–1297
Huang X, Li K, Xu X, Yao Z, Jin C, Zhang S (2015) Genome-wide analysis of WRKY transcription factors in white pear (Pyrus bretschneideri) reveals evolution and patterns under drought stress. BMC Genom 16:1104
Ishiguro S, Nakamura K (1994) Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5′ upstream regions of genes coding for sporamin and β-amylase from sweet potato. Mol Gen Genet MGG 244:563–571
Jain M, Misra G, Patel RK, Priya P, Jhanwar S, Khan AW, Shah N, Singh VK, Garg R, Jeena G (2013) A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J 74:715–729
Jiang Y, Deyholos MK (2009) Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol Biol 69:91–105
Jiang W, Yu D (2009) Arabidopsis WRKY2 transcription factor mediates seed germination and postgermination arrest of development by abscisic acid. BMC Plant Biol 9:96
Jin J, Tian F, Yang D-C, Meng Y-Q, Kong L, Luo J, Gao G (2016) PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw982
Jing Z, Liu Z (2018) Genome-wide identification of WRKY transcription factors in kiwifruit (Actinidia spp.) and analysis of WRKY expression in responses to biotic and abiotic stresses. Gene Genom 40:429–446
Johnson CS, Kolevski B, Smyth DR (2002) TRANSPARENT TESTA GLABRA2, a trichome and seed coat development gene of Arabidopsis, encodes a WRKY transcription factor. Plant Cell 14:1359–1375
Knudsen B, Knudsen T, Flensborg M, Sandmann H, Heltzen M, Andersen A, Dickenson M, Bardram J, Steffensen P, Mønsted S (2011) Clc sequence viewer. A/S Cb, version 6
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Li W, Cheng J, Wu Z, Qin C, Tan S, Tang X, Cui J, Zhang L, Hu K (2015) An InDel-based linkage map of hot pepper (Capsicum annuum). Mol Breed 35:32
Li M-Y, Xu Z-S, Tian C, Huang Y, Wang F, Xiong A-S (2016) Genomic identification of WRKY transcription factors in carrot (Daucus carota) and analysis of evolution and homologous groups for plants. Sci Rep 6:23101
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Ling J et al (2011) Genome-wide analysis of WRKY gene family in Cucumis sativus. BMC Genom 12:471
Liu H, Yang W, Liu D, Han Y, Zhang A, Li S (2011) Ectopic expression of a grapevine transcription factor VvWRKY11 contributes to osmotic stress tolerance in Arabidopsis. Mol Biol Rep 38:417–427
Mangelsen E, Kilian J, Berendzen KW, Kolukisaoglu ÜH, Harter K, Jansson C, Wanke D (2008) Phylogenetic and comparative gene expression analysis of barley (Hordeum vulgare) WRKY transcription factor family reveals putatively retained functions between monocots and dicots. BMC Genom 9:194
Mayrose I, Zhan SH, Rothfels CJ, Magnuson-Ford K, Barker MS, Rieseberg LH, Otto SP (2011) Recently formed polyploid plants diversify at lower rates. Science 333:1257–1257
Mukhtar MS, Deslandes L, Auriac MC, Marco Y, Somssich IE (2008) The Arabidopsis transcription factor WRKY27 influences wilt disease symptom development caused by Ralstonia solanacearum. Plant J 56:935–947
Panchy N, Lehti-Shiu M, Shiu S-H (2016) Evolution of gene duplication in plants. Plant Physiol 171:2294–2316
Pandey SP, Somssich IE (2009) The role of WRKY transcription factors in plant immunity. Plant Physiol 150:1648–1655
Qin Y, Tian Y, Liu X (2015) A wheat salinity-induced WRKY transcription factor TaWRKY93 confers multiple abiotic stress tolerance in Arabidopsis thaliana. Biochem. Biophys Res Commun 464:428–433
Qiu Y, Yu D (2009) Over-expression of the stress-induced OsWRKY45 enhances disease resistance and drought tolerance in Arabidopsis. Environ Exp Bot 65:35–47
Rinerson CI, Rabara RC, Tripathi P, Shen QJ, Rushton PJ (2015) The evolution of WRKY transcription factors. BMC Plant Biol 15:66
Ross CA, Liu Y, Shen QJ (2007) The WRKY gene family in rice (Oryza sativa). J Integr Plant Biol 49:827–842
Rushton PJ, Somssich IE, Ringler P, Shen QJ (2010) WRKY transcription factors. Trends Plant Sci 15:247–258
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sani AS et al (2017) Genetic diversity, population structure, and genetic correlation with climatic variation in chickpea (Cicer arietinum) landraces from Pakistan. Plant Genome 11:170067
Schluttenhofer C, Yuan L (2015) Regulation of specialized metabolism by WRKY transcription factors. Plant Physiol 167:295–306
Shiu S-H, Bleecker AB (2003) Expansion of the receptor-like kinase/Pelle gene family and receptor-like proteins in Arabidopsis. Plant Physiol 132:530–543
Sievers F et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539
Song H, Nan Z (2014) Genome-wide identification and analysis of WRKY transcription factors in Medicago truncatula. Hereditas 36:152–168
Song H, Wang P, Nan Z, Wang X (2014) The WRKY transcription factor genes in lotus japonicus. Int J Genom. https://doi.org/10.1155/2014/420128
Sturn A, Quackenbush J, Trajanoski Z (2002) Genesis: cluster analysis of microarray data. Bioinformatics 18:207–208
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Thudi M et al (2016) Whole genome re-sequencing reveals genome-wide variations among parental lines of 16 mapping populations in chickpea (Cicer arietinum L.). BMC Plant Biol 16:10
Varshney RK et al (2013) Achievements and prospects of genomics-assisted breeding in three legume crops of the semi-arid tropics. Biotechnol Adv 31:1120–1134
Verma M, Kumar V, Patel RK, Garg R, Jain M (2015) CTDB: an integrated chickpea transcriptome database for functional and applied genomics. PLoS ONE 10:e0136880
Vom Endt D, Kijne JW, Memelink J (2002) Transcription factors controlling plant secondary metabolism: what regulates the regulators? Phytochemistry 61:107–114
Voorrips R (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wang QQ, Liu F, Chen XS, Ma XJ, Zeng HQ, Yang ZM (2010) Transcriptome profiling of early developing cotton fiber by deep-sequencing reveals significantly differential expression of genes in a fuzzless/lintless mutant. Genomics 96:369–376
Wang Q, Wang M, Zhang X, Hao B, Kaushik S, Pan Y (2011) WRKY gene family evolution in Arabidopsis thaliana. Genetica 139:973
Wang H-L, Yang J, Boykin LM, Zhao Q-Y, Wang Y-J, Liu S-S, Wang X-W (2014a) Developing conversed microsatellite markers and their implications in evolutionary analysis of the Bemisia tabaci complex. Sci Rep 4:6351
Wang L et al (2014b) Genome-wide identification of WRKY family genes and their response to cold stress in Vitis vinifera. BMC Plant Biol 14:103
Wang N, Xia E-H, Gao L-Z (2016) Genome-wide analysis of WRKY family of transcription factors in common bean, Phaseolus vulgaris: chromosomal localization, structure, evolution and expression divergence. Plant Gene 5:22–30
Wei K-F, Chen J, Chen Y-F, Wu L-J, Xie D-X (2012) Molecular phylogenetic and expression analysis of the complete WRKY transcription factor family in maize. DNA Res 19:153–164
Wei Y et al (2016) Genome-wide identification and expression analysis of the WRKY gene family in cassava. Front Plant Sci 7:25
Wu K-L, Guo Z-J, Wang H-H, Li J (2005) The WRKY family of transcription factors in rice and Arabidopsis and their origins. DNA Res 12:9–26
Xiao Y et al (2017) Genome-wide identification of WRKY genes and their expression profiles under different abiotic stresses in Elaeis guineensis. PLoS ONE 12:e0189224
Xie Z, Zhang Z-L, Zou X, Huang J, Ruas P, Thompson D, Shen QJ (2005) Annotations and functional analyses of the rice WRKY gene superfamily reveal positive and negative regulators of abscisic acid signaling in aleurone cells. Plant Physiol 137:176–189
Xu X, Breen G, Chen C-K, Huang Y-S, Wu Y-Y, Asherson P (2010) Association study between a polymorphism at the 3′-untranslated region of CLOCK gene and attention deficit hyperactivity disorder. Behav Brain Funct 6:481
Yang B, Jiang Y, Rahman MH, Deyholos MK, Kav NN (2009) Identification and expression analysis of WRKY transcription factor genes in canola (Brassica napus L.) in response to fungal pathogens and hormone treatments. BMC Plant Biol 9:68
Yu D, Chen C, Chen Z (2001) Evidence for an important role of WRKY DNA binding proteins in the regulation of NPR1 gene expression. Plant Cell 13:1527–1540
Yu Y, Wang N, Hu R, Xiang F (2016) Genome-wide identification of soybean WRKY transcription factors in response to salt stress. Springer Plus 5:920
Zhang Y, Wang L (2005) The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evol Biol 5:1
Zhang C et al (2017) Genome-wide identification of the potato WRKY transcription factor family. PLoS ONE 12:e0181573
Zhang M et al (2018) Transcriptome-wide identification and screening of WRKY factors involved in the regulation of taxol biosynthesis in Taxus chinensis. Sci Rep 8:5197
Zou Z, Yang L, Wang D, Huang Q, Mo Y, Xie G (2016) Gene structures, evolution and transcriptional profiling of the WRKY gene family in castor bean (Ricinus communis L.). PLoS ONE 11:e0148243
Acknowledgements
The authors acknowledge the Punjab Agricultural Research Board (Government of Punjab), Lahore, Pakistan for funding through Project PARB-938, as well as Centre for Advanced Studies in Agriculture and Food Security (CAS-AFS).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Waqas, M., Azhar, M.T., Rana, I.A. et al. Genome-wide identification and expression analyses of WRKY transcription factor family members from chickpea (Cicer arietinum L.) reveal their role in abiotic stress-responses. Genes Genom 41, 467–481 (2019). https://doi.org/10.1007/s13258-018-00780-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-018-00780-9