
Abstract
Although there are many applications available for the analysis of chromatin immunoprecipitation with massively parallel DNA sequencing (ChIP-seq), users need some knowledge about the installation, alignment, and peak calling procedures prior to the analysis. Here, we present an easy-to-use application for ChIP-seq analysis called AutoChIP. With AutoChIP, installation of necessary programs, alignment of unmapped reads to a reference genome, and identification of genome-wide binding sites can be done in a single step with a large set of ChIP-seq data. Evaluation of the cocktail algorithm implemented in AutoChIP showed that it outperformed a single ChIP-seq tool in terms of the ratio of motif occurrences and the average height of normalized read density over the identified peaks. In addition, annotation of the identified peaks with the known gene and repeat elements information provides a comprehensive picture of the genome-wide binding sites of given proteins. Overall, AutoChIP provides a comprehensive platform to analyze a large set of ChIP-seq data in one step.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recent advances in genomic DNA sequencing, mainly driven by the next-generation sequencing (NGS) technique, have revolutionized ways to examine molecular events inside cells in several aspects. For example, chromatin immunoprecipitation with massively parallel DNA sequencing (ChIP-seq) generates molecular maps that pinpoint genome-wide binding positions of various proteins in many different cell types (Mouse et al. 2012; Kang et al. 2013). With these maps, researchers can distinguish target and non-target genes of transcription factors. Whole transcriptome shotgun sequencing (RNA-seq) can estimate the abundance of whole transcripts including protein-coding genes, non-coding RNAs, and small RNAs (Feuermann et al. 2013; Yamaji et al. 2013). As the number of NGS-based datasets increases, many tools have been developed to help turn sequenced short DNA fragments into biologically meaningful information. According to statistics from the OMICtools website (http://omictools.com/) (Henry et al. 2014), more than 2,000 tools are currently available for the analysis of NGS-based data. In case of ChIP-seq, several thousand datasets have been deposited in NCBIs gene expression omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) (Barrett et al. 2013).
In the era of NGS, however, there is no gold standard for analyzing a given NGS data set, although most of programs provide some statistics-based output. For instance, several studies have pointed out that the number of binding sites of a given ChIP-seq data set can vary depending on the algorithms and the parameters used for the analysis (Kang et al. 2013; Adomas et al. 2014). In addition, setting up a computer for the analysis can be challenging for novice users. Therefore, it is very difficult for users to select appropriate applications and establish a computing environment for the analysis. To address this issue, the present authors have developed AutoChIP, an automated analysis pipeline that can analyze a large number of ChIP-seq datasets simultaneously. Based on a graphical user interface (GUI), it installs required programs from websites automatically, generates an appropriate genome index for alignment and then processes several types of input such as FASTQ (unmapped reads) and BAM (mapped reads) files sequentially. To produces a list of high-confidence binding sites, which are defined as the peaks detected by all the algorithms as a cocktail strategy, AutoChIP utilizes the following popular peak calling programs; model-based analysis of ChIP-seq (MACS), hypergeometric optimization of motif enrichment (HOMER), and PeakRanger (Zhang et al. 2008; Heinz et al. 2010; Feng et al. 2011). Our evaluation demonstrated that the cocktail approach implemented in AutoChIP improves overall performance of peak finding in terms of the ratio of motif occurrences and the average density of ChIPed reads.
Materials and Methods
Development environment
AutoChIP was developed in Java programming language (JDK1.7) using Eclipse (Kepler version Java EE IDE). The GUI was made by using Swing and Windowbuilder. Since most applications implemented in AutoChIP are solely based on the Linux operating system, it can only run on a Linux operating system such as Ubuntu and Fedora. AutoChIP will download and install the following tools automatically: Samtools, twoBitToFa, and BEDTools for the manipulation of files and output (Li et al. 2009; Quinlan and Hall 2010); Bowtie2 and Subread for alignment (Langmead and Salzberg 2012; Liao et al. 2013); and MACS (version 1.4), HOMER, and PeakRanger for peak calling (Zhang et al. 2008; Heinz et al. 2010; Feng et al. 2011). To identify genome-wide binding sites, it runs all three peak calling programs and intersects the outputs.
ChIP-seq data sets used in the study
To assess the performance of AutoChIP, the following mouse and human ChIP-seq data were downloaded and analyzed by MACS, HOMER, PeakRanger, and AutoChIP; mouse STAT5A (GSM1005189) and STAT5B (GSM1005190) and their corresponding input (GSM1005193) in mammary gland tissues; human GATA3 (GSM1241752) and its corresponding input (GSM1241753) in MCF7 cell line; and human GATA3 (GSM1241754) and its corresponding input (GSM1241755) in T47D cell line (Adomas et al. 2014; Kang et al. 2014).
Motif analysis and peak annotation
To predict over-represented motifs in peaks, MEME-ChIP (http://meme.nbcr.net/meme/cgi-bin/meme-chip.cgi) was used with the default setting (Machanick and Bailey 2011). The absence or presence of the predicted motifs within a 200 bp flanking sequence was assessed by the FIMO tool (http://meme.nbcr.net/meme/cgi-bin/fimo.cgi) (Grant et al. 2011). Distributions of peaks were estimated according to gene and repetitive element annotation (mm9 for mouse and hg19 for human) using HOMER.
Program availability
AutoChIP can be downloaded at https://sites.google.com/site/kangklab/.
Results and Discussion
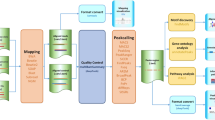
AutoChIP workflow
AutoChIP automatically installs required programs upon its first run. For the analysis, it accepts FASTQ (unmapped reads) and BAM (mapped reads) files as inputs and performs one of the following analyses: Indexing, Alignment, or Align-ChIP (Fig. 1). In the indexing tab, users can generate a genome index for alignment using either Bowtie2 or Subread (Fig. 1a) (Langmead and Salzberg 2012; Liao et al. 2013). This is a necessary step when input files are provided in the FASTQ format and there is no existing genome index. Human, mouse, and drosophila genomes can be downloaded from the designated AutoChIP server. In the alignment tab, FASTQ files can be aligned to an indexed genome (Fig. 1b). Multiple files can be processed sequentially. The Align-ChIP tab provides a fully automated mode for the ChIP-seq analysis when FASTQ files and an indexed genome are provided (Fig. 1c). The main algorithm to detect binding sites of proteins of interest is a cocktail approach that generates a list of high-confidence peaks by intersecting the outputs from MACS, HOMER, and PeakRanger (Zhang et al. 2008; Heinz et al. 2010; Feng et al. 2011). We chose those peak calling tools due to the following reasons: (1) MACS is one of the most widely used peak calling programs for ChIP-seq analysis; (2) HOMER is a versatile tool that can analyze different types of NGS-based data including ChIP-seq, RNA-seq, and MNase-seq; and (3) PeakRanger was used to process a large set of ChIP-seq data produced by the modENCODE consortium. Additionally, AutoChIP can annotate the identified peaks by means of HOMER with appropriate gene and repetitive element information. All the functions are provided in GUI mode; therefore, there is no need for users to learn how to install and execute each program.

Three main functions of AutoChIP. a If mapped files (BAM) were not provided, input files (FASTQ) should be aligned to a reference genome before peak calling. The genome should be indexed prior to the alignment. To generate an index for the given genome, the Indexing tab provides a function that generates an index of the genome by using either Bowtie2 (default) or Subread. The following reference genomes can be automatically downloaded from https://sites.google.com/site/kangklab/: hg19 (Human), mm9 and mm10 (Mouse), and dm3 (Drosophila). b The alignment tab provides a function that can align unmapped reads (FASTQ) to the indexed genome. Multiple files can be processed sequentially. If an indexed genome and mapped files (BAM) were provided, the analysis of peak calling, and annotation of the identified peaks can be conducted. c If an indexed genome and unmapped files (FASTQ) were provided, the alignment, peak calling, and annotation steps can be conducted sequentially in the Align-ChIP tab
Inconsistency between peak calling applications
To identify genome-wide binding sites from given ChIP-seq datasets, available peak calling programs including MACS, HOMER, and PeakRanger use different strategies despite being based on statistical methods such as false discovery rate (FDR) (Zhang et al. 2008; Heinz et al. 2010; Feng et al. 2011). Due to the differences in strategy, several studies reported that the number of genome-wide binding sites identified can vary by up to several thousand (Malone et al. 2011; Kang et al. 2013). We confirmed the inconsistency by reanalyzing available mouse STAT5A (GSE40930, mammary gland tissues) (Kang et al. 2014) and human GATA3 (GSE51274, MCF7 and T47D breast cancer cell lines) (Adomas et al. 2014) ChIP-seq data with MACS, HOMER, and PeakRanger. Up to several thousand peaks were differentially identified between peak calling programs (Fig. 2). For example, 33495 STAT5A binding sites were identified by all three tools, while 8987, 2891, and 1703 STAT5A peaks were uniquely detected by MACS, HOMER, and PeakRanger, respectively. Similarly, 3751, 6031, and 429 GATA3 peaks were identified in MCF7 cells only by each respective program. Totals of 33495 STAT5A, 9486 STAT5B, 18296 GATA3 (MCF7), and 12513 GATA3 (T47D) peaks were identified by all three applications. The results demonstrated that current ChIP-seq analysis tools still have room for improvement.

Reanalysis of published STAT5 and GATA3 ChIP-seq with MACS, HOMER, and PeakRanger. Different numbers of STAT5A, STAT5B and GATA3 (MCF7 and T47D cell lines) binding sites were identified by MACS, HOMER, and PeakRanger. The published data were downloaded from the Gene Expression Omnibus (GEO accession number GSE40930 and GSE51274) (Adomas et al. 2014; Kang et al. 2014)
Performance evaluation of the cocktail approach implemented in AutoChIP
AutoChIP takes advantage of each algorithm by intersecting their outputs and provides a list of high-confidence peaks, which are defined as the peaks identified by all the algorithms, as a cocktail approach. To assess the cocktail approach, motif frequency (absence or presence of a given motif per peak) was calculated. Since DNA binding proteins recognize specific DNA sequences (motifs), STAT5 and GATA3 binding motifs were first predicted by using MEME-ChIP, with the peaks identified by AutoChIP (Machanick and Bailey 2011). The known STAT5 and GATA3 motifs were significantly identified (Fig. 3a). With the top motifs showing the lowest E-value, motif frequency (p value < 0.0001 for detecting motifs) in T47D cells was estimated using the FIMO tool with the identified peaks (Grant et al. 2011): 33495 (AutoChIP), 50266 (MACS), 40476 (HOMER), and 40696 (PeakRanger) STAT5A peaks; 9486 (AutoChIP), 15866 (MACS), 14825 (HOMER), and 12129 (PeakRanger) STAT5B peaks; 18296 (AutoChIP), 30222 (MACS), 32186 (HOMER), and 20151 (PeakRanger) GATA3 peaks in MCF7 cells; and 12513 (AutoChIP), 32279 (MACS), 29128 (HOMER), and 13678 (PeakRanger) GATA3 peaks. The result shows that the cocktail approach by AutoChIP outperformed the single peak calling tool in all cases according to the percentage of peaks containing at least one top motif (Fig. 3b). This tendency was maintained when applied to degenerated motifs defined by increasing the p value cutoff from 0.001 to 0.1 (Fig. 3c). In addition, the normalized read densities of the peaks identified by AutoChIP in STAT5A, STAT5B, and GATA3 (MCF7 and T47D) ChIP-seq are higher than those identified by the single method (Fig. 3d). The results demonstrated that the cocktail approach implemented in AutoChIP identified high-quality peaks in terms of the ratio of motif occurrence and the binding strength of the given proteins to the sites.

Comparison of AutoChIP to MACS, HOMER, and PeakRanger. a Top motifs (the lowest E-value) predicted in the common peaks of the given ChIP-seq data by MEME-ChIP are shown (Machanick and Bailey 2011). These were used for further analysis. b The percentage of the common peaks containing at least one motif was calculated with different p value thresholds for detecting motifs in the peaks. c The percentages of the motifs in the peaks identified by HOMER, MACS, PeakRanger, and AutoChIP were shown as bar graphs. d The average of normalized read density (reads per million per nucleotide, RPM) on the AutoChIP and marginal peaks is shown. The marginal peaks were defined as the peaks identified that were unique to a single program
Annotation of the identified peaks with the information of genes and repetitive elements
AutoChIP provides several advantages to users. First, all the required programs are automatically installed and necessary procedures for peak calling such as alignment are conducted in one step. Second, additional analyses can be performed after the peak calling. For example, annotation of the identified peaks can easily be executed with the Perl script (annotatePeaks.pl) provided by HOMER. Annotation analysis of the identified peaks by AutoChIP showed that the majority of STAT5A, STAT5B, and GATA3 peaks were located in intergenic and intron regions (Fig. 4a; Table S1). The result is consistent with the known feature of STAT5 and GATA3 as enhancer binding proteins (Ranganath et al. 1998; Gonsky et al. 2004). Additionally, the relationship between STAT5 (or GATA3) binding and repetitive elements was assessed by using the Perl script (analyzeRepeat.pl) from HOMER. Interestingly, 2.95 and 0.80 % of the known repetitive elements coincided with STAT5A and STAT5B in mouse mammary glands, respectively (Fig. 4b; Table S2). In addition, 1.44 % (MCF7) and 0.98 % (T47D) of promoter regions of the repetitive elements were bound by GATA3. However, the importance of these bindings related to the activity of repetitive elements needs to be validated in near future.

Annotation of the common peaks. The distribution of the identified peaks by AutoChIP relative to genes and repetitive elements was estimated
Conclusion
Owing to the NGS technique, various molecular events can be captured and visualized by means of bioinformatic approaches. Among them, ChIP-seq has been widely used to detect genome-wide binding sites of proteins. Currently, more than 30 peak calling programs and several thousand ChIP-seq datasets have been reported. Incorporating the available data into an ongoing study can give rise to new biological insights. However, it is a daunting task for novice users to install programs and use them to detect genome-wide binding sites of proteins of interest. In addition, false positive peaks might be identified along with true positive peaks, regardless of statistical methods, due to problematic genomic regions, sequencing bias, inadequate statistical power, and insufficient sequencing depth. Through a series of analyses, we showed that the cocktail approach implemented in AutoChIP outperformed a single peak calling method. Using AutoChIP, all necessary steps including the installation of required programs, genome indexing, alignment, peak calling, and annotation of identified peaks can be done in one step. The easy-to-use GUI will help novice users to analyze their own and available ChIP-seq datasets. Understanding of genome-wide protein binding networks could be facilitated by using AutoChIP along with various other NGS-based methods such as RNA-seq and MNase-seq.
References
Adomas AB, Grimm SA, Malone C, Takaku M, Sims JK, Wade PA (2014) Breast tumor specific mutation in GATA3 affects physiological mechanisms regulating transcription factor turnover. BMC Cancer 14:278
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M et al (2013) NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res 41:D991–D995
Feng X, Grossman R, Stein L (2011) PeakRanger: a cloud-enabled peak caller for ChIP-seq data. BMC Bioinformatics 12:139
Feuermann Y, Kang K, Gavrilova O, Haetscher N, Jang SJ, Yoo KH, Jiang C, Gonzalez FJ, Robinson GW, Hennighausen L (2013) MiR-193b and miR-365-1 are not required for the development and function of brown fat in the mouse. RNA Biol 10:1807–1814
Gonsky R, Deem RL, Bream J, Young HA, Targan SR (2004) Enhancer role of STAT5 in CD2 activation of IFN- gene expression. J Immunol 173:6241–6247
Grant CE, Bailey TL, Noble WS (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics 27:1017–1018
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38:576–589
Henry VJ, Bandrowski AE, Pepin AS, Gonzalez BJ, Desfeux A (2014) OMICtools: an informative directory for multi-omic data analysis. Database (Oxford) 2014. doi:10.1093/database/bau069
Kang K, Robinson GW, Hennighausen L (2013) Comprehensive meta-analysis of signal transducers and activators of transcription (STAT) genomic binding patterns discerns cell-specific cis-regulatory modules. BMC Genom 14:4
Kang K, Yamaji D, Yoo KH, Robinson GW, Hennighausen L (2014) Mammary-specific gene activation is defined by progressive recruitment of STAT5 during pregnancy and the establishment of H3K4me3 marks. Mol Cell Biol 34:464–473
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Liao Y, Smyth GK, Shi W (2013) The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41:e108. doi:10.1093/nar/gkt214
Machanick P, Bailey TL (2011) MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27:1696–1697
Malone BM, Tan F, Bridges SM, Peng Z (2011) Comparison of four ChIP-Seq analytical algorithms using rice endosperm H3K27 trimethylation profiling data. PLoS One 6:e25260. doi:10.1371/journal.pone.0025260
Mouse EC, Stamatoyannopoulos JA, Snyder M, Hardison R, Ren B, Gingeras T, Gilbert DM, Groudine M, Bender M, Kaul R et al (2012) An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol 13:418
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842
Ranganath S, Ouyang W, Bhattarcharya D, Sha WC, Grupe A, Peltz G, Murphy KM (1998) GATA-3-dependent enhancer activity in IL-4 gene regulation. J Immunol 161:3822–3826
Yamaji D, Kang K, Robinson GW, Hennighausen L (2013) Sequential activation of genetic programs in mouse mammary epithelium during pregnancy depends on STAT5A/B concentration. Nucleic Acids Res 41:1622–1636
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol 9:R137
Acknowledgments
We thank members of the Kang laboratory for valuable comments.
Conflict of interest
The authors state that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kim, T., Lee, W., Han, K. et al. An automated analysis pipeline for a large set of ChIP-seq data: AutoChIP. Genes Genom 37, 305–311 (2015). https://doi.org/10.1007/s13258-014-0260-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-014-0260-3