
Abstract
Gene editing and therapy holds immense promise in addressing cardiovascular disorders by enabling precise modifications to the genetic groundworks of these conditions. This technology offers a targeted approach to correct or mitigate not only the genetic mutations that are responsible for inherited cardiovascular diseases, such as hypertrophic cardiomyopathy or familial hypercholesterolemia (FH), but also in controlling and regulating the genes in conditions that are age-related or that arise due to unnecessary gene activity, which often lack effective treatments. By editing the relevant genes, we aim to reduce disease severity, improve heart function, prevent future episodes and potentially improve the health span. Recent trials involving gene therapy and gene editing in cardiovascular disorders have revolutionized treatment strategies, offering hope for patients with genetic predispositions to heart and vessel-related ailments and advancing the pursuit for more personalized and effective therapies. In this review, we have consolidated the genetic mutations causing cardiovascular diseases (CVDs) followed by latest advancements in the gene editing technologies and their therapeutic implications along with involved ethical challenges and risk factors.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nucleic acids largely constitute the coding and non-coding genetic information and its role is central to the functioning of any eukaryotic cells. Nucleic acid damage is a major cause for the development of various CVDs the balance between damage and repair of nucleic acid plays a crucial role in cellular homeostasis in normal cells and interruption leads to severe diseases. Various extrinsic environmental or intrinsic genetic factors are the cause of this interruption in the maintenance of homeostasis. Genetics plays a crucial role in influencing the risk of various CVDs. Inherited CVDs include congenital heart diseases, arrhythmias, cardiomyopathy, and high blood cholesterol. A genetic variation in a single gene can have an impact on an individual’s risk of acquiring heart disease. A total of 2302 male and female offspring study participants (mothers < 65 years and fathers < 55 years) with a parental history of early CVD were included in the Framingham study and their CVD risk was assessed. Following 8 years of observation, CVD rose 75% in cases with a paternal history and about 60% in cases with a maternal history of early CVD. According to the Framingham study, the risk of CVD rose by almost 40% among people whose siblings also had the disease [54]. Advanced and emerging gene editing technologies lead to improved research models and therapy options. Molecular genetics plays a key role in predicting the genetic predisposition in CVDs. Gene editing is the potential cure for severe inherited CVDs. Somatic gene editing can permanently resolve the malfunctioning of a gene that leads to insufficient production of functional protein. In the past 30 years, gene editing techniques evolved from first-generation to third-generation starting from the use of homologous recombinant technology to the use of zinc fingers and recently clustered regularly interspaced short palindromic repeats/clustered regularly interspaced short palindromic repeat–associated proteins (CRISPR/Cas9). This aids in achieving better transfection efficiency, reducing off-target effects, and expanding application in gene therapy. This review discusses the recent development in gene editing and delivery techniques used in CVDs and the application of this technology to rectify and treat inherited and acquired diseases. This review also focuses on the ethical considerations and risk factors involved with these editing technologies.
DNA damage
The term “DNA Damage” encompasses any physical or expressible change in DNA either due to intrinsic or extrinsic factors, that leads to erratic expression/repression of genes. It also includes the accumulating epigenetic changes owing to the evolutionary nature of the gene concerning temporal and spatial switches. It not only includes nuclear DNA but also mitochondrial DNA and finally also takes into account RNA damage whether it is mRNA, rRNA/tRNA, or ncRNA. Damage caused by various factors is one of the major causes of CVDs both inherited and acquired. It can lead to the development and progression of atherosclerosis, heart failure, and arrhythmias. Accumulated genetic mutations and DNA lesions can disrupt or weaken the signaling pathways and cellular functions crucial for cardiovascular health. In addition to this, there are gradual changes in the expression of genes that are concomitant with age, lifestyle, and genetic makeup that accumulate over a long period that adversely affect the cardiac and vascular functions.
Growing body of evidence on the role of accumulating mutations (chronic DNA damage) promoting CHIP (clonal hematopoiesis of indeterminate potential) not only in cancers, but also in CVDs has kindled a lot of interest in studying the driver mutations and DNA damage beyond known mutation that are associated with CVDs. Clonal hematopoiesis, or CHIP is a state in which mutant hematopoietic lineage cells undergo a clonal expansion in the absence of an apparent hematological abnormality [17]. While the association between CHIP and hematological malignancies is expected, several large studies unexpectedly linked CHIP with CVDs such as Carotid artery diseases (CAD) and stroke, moreover, it was shown that CHIP carriers had a 1.5–2-fold heightened risk of developing CAD compared to non-carriers and nearly 4 times the risk of early-onset of myocardial infarction [39, 40, 81]. Clonal hematopoiesis increases the incidence of CAD and stroke, and worsens prognosis in the heart failure patients [16]. On the other hand, acute damage to the DNA has also been implicated in failing hearts, ischemic cardiomyopathy, and drug-induced cardiotoxicity. Experimental and clinical evidence indicates that DNA damage due to oxidative stress (e.g., ischemia–reperfusion injury) or after chemotherapy [74] is associated with heart failure [7, 8, 60]. Another study showed that there is progression of endothelial dysfunction and arteriosclerosis up to 15 months following initiation of anthracycline-based chemotherapy [1]. However, the precise effects of unrepaired DNA damage, and its role in the pathogenesis of heart failure, are still poorly understood.
In contrast to damaged DNA and its consequences on cardiovascular functions, aging heart and vessels experience gradual change in the gene regulation and expression. As we age, the cells experience change in gene regulation, leading to perturbation in the expression of genes responsible for maintaining cardiovascular health. Genes that regulate cardiac and vascular metabolism, calcium handling, energetics, proliferation (seen in a very small number of cardiomyocytes), response to increased demand, vasoconstriction and relaxation, often are observed to be deranged leading to decline in organ function [8, 12, 44, 95]. This decline can result in a range of issues, including weakened cardiac muscle, impaired blood vessel elasticity, and increased susceptibility to atherosclerosis. These age-related changes contribute to the higher incidence of heart disease and vascular dysfunction in older individuals. Understanding and potentially reversing these alterations in gene expression through emerging technologies like gene therapy and epigenetic modulation hold the promise of not only improving heart and vessel function in aging populations but also extending healthy lifespan.
Gene editing emerges as a promising tool to rectify these genetic anomalies, offering a precise means to repair or replace damaged DNA segments. Whether it is repairing malfunctioning genes responsible for regulating cholesterol metabolism, or cardiac muscle function, gene editing technologies like CRISPR-Cas9 and technologies that deliver the gene via targeted carriers hold the potential to address the underlying genetic and age-related cause of CVDs. By repairing or replacing damaged DNA sequences, we can not only halt disease progression but also potentially reverse the damage, offering hope for more effective treatments and even prevention in individuals with a genetic predisposition. In essence, correcting damaged DNA or aberrations in the gene expression is the key to unlocking a future where cardiac and vascular diseases are less prevalent and their devastating consequences minimized. This review discusses the latest advancements in systemic gene editing and gene therapy that currently are being tested or have shown promising result in clinical trials (Fig 1).
DNA damage causing inherited and acquired CVDs
Genetic mutations or anomalies in an individual’s genomic DNA are strongly associated with the onset of a category of medical conditions known as “inherited cardiovascular diseases” or “genetic cardiovascular disorders.” These conditions encompass a wide range of cardiovascular abnormalities that significantly impact both the structural and functional aspects of the heart and blood vessels. Foremost among these conditions is FH, a prevalent autosomal monogenic hereditary disorder characterized by elevated levels of cholesterol, low-density lipoprotein cholesterol (LDL-C), early xanthoma, and progressive atherosclerotic cardiovascular disease (ASCVD). The occurrence of this illness is associated with genetic abnormalities that regulate lipid metabolism, specifically involving genes such as LDLR, ApoB, and PCSK9 [79]. Another heritable disorder is Pulmonary Arterial Hypertension (PAH) which is marked by a group of unhealthy changes, such as the growth of vascular endothelial cells (ECs), the enlargement of smooth muscle cells, and the thickening of the adventitia. These changes lead to vascular remodeling and the blocking of precapillary pulmonary arteries. It has been found that loss-of-function mutations in the BMPR2 gene, which codes for the TGF-βII receptor, are the main cause of disease. These mutations cause heterodimers to form, which mess up the TGF-β pathway ligand binding and then change the activity of serine/threonine kinase [10, 11]. Further studies in this field revealed genes like BMPR1B, ACVRIJ, ENG, SMAD9, CAV1, and KCNK3 that are linked to the disease. It’s interesting to note that the effects of genes vary depending on the person who carries them, with rates of 14% for male carriers and 42% for female carriers [56]. BMPR2 mutations can be found in 50–80% of heritable PAH cases [88, 89]. Notably, it is important to note that hereditary and idiopathic PAH share a diagnosis of diminished BMPR2 expression and BMP signaling due to heterozygous deletion mutations. Another entity within the spectrum of inherited cardiovascular illness is Familial Thoracic Aortic Aneurysm and Dissection (FTAAD). When two or more affected members of the same family develop thoracic aortic aneurysms or dissections, but there are no other obvious abnormalities of the patient’s system from the aortic pathology, is termed as FTAAD [82]. The major cause of FTAAD is now thought to be a loss-of-function mutation in the gene ACTA2 that encodes alpha-smooth muscle actin. Mutations in genes like FBN1, TGFBR2, TGFB2, and TGFB3 that cause syndromes like Marfan syndrome may also induce FTAAD [27]. Marfan Syndrome is a connective tissue ailment that can cause abnormalities in many different organs. The extracellular matrix protein fibrillin-1 is encoded by the FBN1 gene, which most frequently experience autosomal dominant mutations [49]. Recently, Zeng et al. [98] discovered a person with Marfan syndrome who was heterozygous for the pathogenic T7498C mutation in the FBN1 gene and discovered human cells in culture with the same mutation. This finding highlights the genetic basis of Marfan syndrome. Cardiomyopathy as a hypernym, can be used to encompass a group of diseases/disorders (with differing aetiology) affecting the cardiac muscle which may become enlarged, thickened, or fibrotic in some cases resulting in impaired cardiac function. Some major types of cardiomyopathies include dilated cardiomyopathy, hypertrophic cardiomyopathy, and ischemic cardiomyopathy. Hypertrophy is a condition which involves extended period of increased workload on heart. This leads to hypertrophic enlargement of the walls in response to increased demand. Different types of pressure overload animal models confirm the involvement of DNA damage in hypertrophic condition. Nakada et al., reported that increased DNA double stranded breaks (DSBs) were found in Angiotensin II administered acutely hypertensive mice. This is indicated by a statistically increased γH2AX (Phosphorylated Histone H2A) and phosphorylated ATM, a primary kinase in DNA damage response (DDR). Transverse Aortic Constriction (TAC) induced pressure overload model leads to increased phosphorylated DNA dependent protein kinases (pDNA-PKcs) in cardiomyocyte nuclei, specifically inducing DSBs [71]. Accumulation of unrepaired DNA single strand breaks (SSB) in cardiomyocytes induce persistent DDR and subsequent NF-κβ pathway, resulting in increased expression of inflammatory cytokines. Some part of this pathway is understood to involve in the progression of pressure overload—induced heart failure [35].
In conclusion, there is a wide spectrum of hereditary cardiovascular illnesses, each with its own distinct genetic foundation and clinical implication. The clarification of these genetic narratives is a significant achievement in understanding and tackling these intricate medical conditions, therefore pushing forward the boundaries of clinical diagnosis, therapeutic treatments, and preventive strategies.
Acquired CVDs encompass a collection of heart and blood vessel ailments that manifest gradually because of many circumstances, such as individual choices pertaining to lifestyle, genetic predispositions, and effects from the surrounding environment. These disorders do not manifest at birth (congenital), but rather emerge or are acquired over the course of an individual’s lifespan. Atherosclerosis, a multifaceted pathological process, is one of the significant contributors to acquired CVDs. Atherosclerosis is the prime cause for the pathogenesis of CAD and cerebral and/or peripheral vascular disease [83]. Reported 50% of the deaths globally are responsible due to the clinical manifestation of the CAD and atherosclerosis—myocardial infarction and stroke [65]. From the literature studies it is very evident that somatic DNA damage is one of the major cause of CAD [2]. A fibrous cap with VSMCs, macrophage infiltration, and a defective EC layer indicate atherosclerotic plaque progression. The ‘necrotic core’—macrophage and VSMC foam cells, dead cells, extracellular lipid, and cellular debris is separated by fibrous cap. Cells in advanced lesions and circulating cells from aged and atherosclerotic patients have greater DNA damage, including DSB and DNA damage response activation, compared to controls. Severity of CAD in the patients correlates with the accumulation of oxidized purines including 8-oxoguanine followed by oxidative DNA damage [6]. In atherosclerosis, proliferation of smooth muscle cells take place and several literatures suggests that occurrence of alteration in the DNA level add up to the disease progression. Flouris et al. [18], reported the loss of heterozygosity in mismatch repair genes in atherosclerotic lesions lead to decreased fidelity of DNA repair in atherosclerotic tissues. It has also been reported by Hatzistamou et al. [33], that loss of heterozygosity and microsatellite instability occurs in human atherosclerotic plaque that leads to impaired DNA repair and thus subsequent accumulation of somatic mutation occurs. Telomere shortening has been reported in the VSMCs of human atherosclerotic plaque which can be the effect of chronic oxidative stress thus leading to VSMC senescence in atherosclerosis [63]. A genetic anomaly due to deficiency of apolipoproteins, apoA-I and apoC-III owing to an inversion in the DNA resulted in very low high density lipoproteins and occurrence of severe coronary atherosclerosis [63]. In conclusion, the development of acquired CVDs such as atherosclerosis, is driven by a multifaceted interaction between hereditary and environmental variables, which encompasses DNA damage and repair pathways. Understanding the genetic and molecular aspects is crucial to unravel the pathogenesis of these diseases and developing effective, preventive, and therapeutic strategies.
DNA damage in cells of cardiovascular system
Despite the multifactorial nature of the aetiology of CVDs described in the previous sections, ROS/RNS mediated DNA damage in cells is often a common and a key factor in the underlying mechanism of pathogenesis. Redox homeostasis in cells like VSMCs, cardiomyocytes and ECs are crucial due to their pivotal roles in the normal functioning of cardiovascular system. An imbalance resulting in excess ROS production in these cells leads to DNA damage. The effects of ROS/RNS damage when added with defects in DNA repair mechanisms leads to gradually accumulation of errors in DNA and ultimately results in and cell dysfunction and death. This often initiates (and later exacerbates) the onset of CVDs as discussed further in this section.
Vascular smooth muscle cells
Patients with atherosclerosis show both nuclear and mitochondrial DNA damage in VSMCs, macrophages and ECs. Gray et al., 2014 reported, both in vivo and in vitro studies indicate an increase in DSBs induced by oxidative stress in plaque VSMCs. Human plaque cultured VSMC display 2 fold increase in DNA damage markers and various proteins involved in multiple repair pathways; predominantly MRE11/RAD50/NBS1 complex, proteins involved in DSB repair [28]. In advanced lesions, increased DNA damage in VSMC reported to alter the plaque phenotype by inhibiting the fibrous cap area. However, upsurge in repair proteins contributes to growth in cap area and VSMC content. Human atherosclerosis demonstrates increased oxidative stress and accumulation of DNA breaks due to defective repair mechanism in VSMCs. A recent study stated that the defect in base excision repair (BER) is mainly due to failing enzyme 8-oxoguanine DNA glycosylase in atherosclerosis. Human plaque VSMCs have defective 8oxoBER, initiated due to decreased expression and acetylation of 8-oxo DNA glycosylase1 (OGG1) [87]. The activity and stability of OGG1 is regulated by sirtuin 1 deacetylase and P300 acetylation in VSMCs. Correcting the BER mechanism in VSMCs have significantly reduced the plaque size in extreme oxidative stress. This study indicates BER drives the accumulation of 8-oxoguanine and oxidative DNA damage in VSMCs promote atherosclerosis. Giuseppe Cafueri et al., 2012 explored that telomere length (TL) and oxidative DNA damage in endothelium and VSMCs plays an important role in abdominal aortic aneurysm (AAA). Nuclear staining for γ-H2AX histone revealed an increase in double strand break in VSMCs were detected in patients with AAA. The aortic segments collected from the AAA patients display shorter TL and several markers of oxidative stress in both endothelial cells and VSMCs. VSMCs and inflammatory cells are predominately present in the atherosclerotic plaques. In vitro studies have shown that plaque VSMCs have very slow cell proliferation rate, augmented population doubling time and early senescence compared to control VSMCs. Higher rate of apoptosis in plaque VSMCs ultimately primes to plaque rupture and breakdown [5].
Cardiomyocytes
Any damage to or deficiency of cardiomyocytes cause 50 percent of all the heart failure cases worldwide. Promoting endogenous cardiomyocyte proliferation or differentiation of stem cells into cardiomyocyte are used employed to recuperate the proper contractile function of heart. Oxidative DNA damages are associated with reduced proliferation of cardiomyocytes. Morphological changes in the cardiomyocyte leads to ventricular remodelling, hypertrophy, atrial fibrillation, and several other cardiac dysfunctions. Findings from the human cardiac tissue illustrate a decrease in the continuous turnover of the cardiomyocytes with age [37]. Lately, a population study was conducted to investigate the relationship between the oxidative DNA damage of cardiomyocytes and age of the human beings. Tissue specimens from different age groups were subjected to numerous DNA damage analyses. However, the reports do not indicate a positive correlation between the oxidative DNA damage in cardiomyocytes and increasing age of the human beings [37]. DNA damage studies are not enough to support the reduced proliferation of cardiomyocytes with age in the human beings. Further in depth studies are required to understand the underlying phenomenon of the reduced cardiomyocyte proliferation. Outcomes from several studies have reported an increase in the generation of ROS and accumulation of DNA damage in postnatal mice [19]. Hypoxia delays the cell cycle arrest in cardiomyocytes, while hyperoxia or ROS generating condition accelerates the process. Puente et al. [80], established a mouse model to study the effects of ROS and oxygen on cardiomyocyte proliferation. The study highlights a physiological phenomenon where in a transition from birth to breathing atmospheric oxygen arrests the cell-cycle of mature aerobic cardiomyocytes. Moreover, a transient shift from glycolytic to oxidative metabolism in postnatal mouse and zebra fish heart shows an increase in the generation of ROS, which arrests the cardiomyocytes cell cycle through activation of DNA damage response.
Endothelial cells
Endothelium is a dynamic interface between circulating cells in the blood and vascular smooth muscle cells and basement membrane and acts as the key regulator of vascular homeostasis. It plays a wide range of roles including inhibition of inflammatory responses, regulation of the growth and migration of vascular smooth muscle cells, prevention of low density lipid (LDL) oxidation and maintenance of balance between vasodilation and vasoconstriction [24, 41]. Endothelial dysfunction is one of the major factors in progression/ exacerbation of Atherosclerosis as well as an important preclinical marker of Atherosclerosis [31, 57]. It has also been implicated in other diseases like hypertension [91]. Oxidative stress plays a critical role in endothelial dysfunction and senescence by decreasing bioavailability of ·NO, affecting cell signalling and increasing endothelium permeability. ROS mediated oxidative stress has been shown to alter the redox potential, cause DNA damage, downregulation of RNA transcripts and inhibition of protein synthesis in HUVEC mitochondria causing dysfunction (also nuclear DNA damage) [3]. High 8-oxo-dG (DNA damage marker) immunoreactivity has also been observed in endothelial cells in plaque [62, 66]. In endothelial progenitor cells from patients with coronary artery disease, reduced telomerase activity and higher oxidative DNA damage as compared to control healthy patients [85]. Increase in ROS in hypertensive rats has been shown to increase endothelial permeability which leads to inflammatory reaction aggravating the vascular damage [48, 91]. Increase in mitochondrial oxidative stress also causes eNOS uncoupling leading to reduction in NO bioavailability [94]. Sirtuin 6 (SIRT6) is a NAD+ -dependent deacetylase expressed in response to oxidative stress (which often results in double-strand breaks (DSBs)) and activates PARP-1 mediated DSB repair via. homologous recombination and non-homologous end joining [61]. It participates in repression of inflammation and DNA damage and regulation of ageing. It has been found to be highly expressed in endothelial cells (HUVECs) and have cytoprotective role against genomic and telomeric DNA damage. RNAi silencing of SIRT6 results in premature senescence and reduced cell proliferation [11]. SIRT6 expression levels has been found be decreased in atherosclerotic lesions from human patients as well as in ApoE-/- mouse models. It’s overexpression in endothelial results in decreased monocyte adhesion via. decreased VCAM-1 expression and reduced pro-atherogenic gene expression like TNFSF4 (tumor necrosis factor superfamily member 4) [97].
Reparable damage and potentially rectifiable disorders
Unlike mutation driven myeloproliferative disorders (cancers, leukemia) and non-malignant diseases (α- and β-hemoglobinopathies, sickle cell anemia, bone marrow failure syndrome and rare blood disorders such as Primary Immune Deficiency disease, Inborn Errors of Metabolism), the disorders of other solid organs (e.g., heart and liver) are not amenable to damage repair strategies per se, owing to the difficulty in replacing all or most faulty cells of that organ. Currently, among the treatment options for many forms of cancers other than leukemia, chimeric antigen receptors T cells (CAR-T cells) that includes the deactivation of disease modifier genes and the delivery of chimeric antigen receptors (CAR) in the production of CAR cells as adoptive immunotherapy, has been actively tested. However, in case of non-malignant congenital or acquired anomalies that arise due to DNA damage or sporadic somatic mutations and developmental aberrations, the treatment options are limited to overcoming the deficiency by drugs, gene delivery, endogenous gene therapy, removal/resection and surgical course of action to correct the abnormality, and many a times no permanent treatment option than palliative long-term care. Non-malignant but devastating CVDs, such as atherosclerosis, ischemic cardiomyopathy, peripheral artery disease, venous thromboembolism, aortic stenosis, and pulmonary hypertension have recently been associated with driver mutation causing CHIP. Converging on the most likely candidate genes for CHIP highlighted the role of three genes, namely, DNA methyltransferase 3 alpha (DNMTA3A), Ten-eleven translocation-2 (TET2), Additional Sex Combs Like Transcriptional Regulator 1 (ASXL1), and Janus Kinase 2 (JAK2). In fact, the risk of developing coronary disease is 2-times higher in patients with the most common CHIP mutations in these genes whereas having a JAK2 mutation conferred a 12-fold relative risk of incident CAD [97]. An important aspect of CHIP diagnosis and prognosis is its clone size. Currently, CHIP is considered present if the variant allele frequencies (VAF) reach 2% or more, corresponding to 4% of circulating cells, presuming heterozygosity [22, 97]. These observations lead to a proposition that the outcomes of these risk conferring CHIP mutation may be mitigated by reducing the number of mutations carrying cells or by replenishing with the normal cells without driver mutation. However, the bottleneck in this approach is the problem in early identification and selection of these cells limiting its feasibility and utility. Notwithstanding the issues related to early identification, in principle, exogenous gene therapy coupled with precise gene editing may rectify the mutation that confer the risk of developing cardiovascular pathologies.
Gene editing in CVDs
The area of cardiovascular biology has reaped advantages from the advancement of gene editing technologies over the past 3 decades. These include homologous mediated gene editing in mice comprising of knockout, knock-in, conditional and inducible gene targeting using the Cre-LoxP recombination system [15, 50]. Maneuvering these techniques facilitated in generation and evaluation of thousands of mice model uncovering the mechanistic pathways of embryonic heart development followed by growth of the postnatal heart and remodeling of the adult heart. Despite, the conventional gene targeting in mice encountered difficulties involving low targeting efficiency in mouse embryonic stem cells, the extensive procedure of blastocyst injection and transmission of germline and the high cost of generating these mice limited the application of embryonic stem cell-based mouse genetics. Nevertheless, genome-editing technologies are continuously progressing and improving for the advancement of biomedical research. The beginning of genome-editing era started with homologous gene recombination technology employed for knockout and knock-in gene mutations. Subsequent second-generation gene editing technologies like ZFNs and TALENs were invented that depended on the usage of nucleic acid binding proteins and endonucleases [10]. Superseding this, the third generation of gene editing technologies involved the CRISPR/Cas system based on protein-nucleic acid complexes. The CRISPR genome editing innovation has steered a new era of cardiovascular research to counter the difficulties in gene editing with potentialities for genetic-correction of disease. The discovery of yeast rare endonucleases that generate site-specific DSBs increased the proficiency of targeted gene integration [84]. Advancement of this field ushered programmable nucleases that can generate site-specific DNA DSBs. Genome engineering has become convenient due to the discovery of RNA-guided CRISPR-Cas nuclease system [47]. The CRISPR-Cas system unraveled itself as a breakthrough in gene targeting technique transforming biomedical research. Furthermore, advancement in the new CRISPR-Cas systems enhanced gene targeting specificities increasing the scope for genome editing strategies and beyond, viz., gene regulation, epigenetic modification, and chromatin imaging. With the advent of CRISPR-Cas technology, it became pertinent to find the best method for therapeutic in-vivo delivery along with its effectivity and safety. Studies reported the delivery of CRISPR-Cas tools in the form of DNA, mRNA or ribonucleoprotein complexes (RNP) [51]. Among the various methods, gene therapy by AAVs were found to be robust by delivering editor-encoding DNA and targeting to the specific tissue [64, 93]. Yet packaging the AAV particles with Cas9 protein has been a major challenge in delivering this system in vivo [68]. Nevertheless, in CVDs like Duchenne muscular dystrophy (DMD) leading to lethal cardiac and skeletal muscle degeneration are caused due to X-linked dystrophin gene (DMD). Corrections of the DMD mutations has been achieved by genome editing with CRISPR/Cas9 in iPSCs from multiple patients and restoring dystrophin protein expression in derived cardiomyocytes [55].
CRISPR/Cas9 system
CRISPR/Cas originated in the chromosome of bacteria and archaea as an adaptive immune mechanism against viruses, bacteriophages, plasmids, and foreign nucleic acids. CRISPR present in the host chromosome that integrates new spacers of short fragments of foreign DNA into the CRISPR repeat-spacer array. This equips the host with a genetic memory of a previous infection, preventing it from the succeeding intrusion of the same intruder. The Cas was named due to its proximal location and functional interdependence with CRISPR. Short mature CRISPR-RNAs (crRNAs) are generated following transcription of the CRISPR array [42]. Due to this genetic memory from previous infection, the 5’ end of this crRNAs has the spacer fragment that complements with a sequence from the foreign genetic material and the 3’ end contains a part of the CRISPR repeat sequence [42]. Upon subsequent infection, the hybridization of the crRNA with the foreign genetic material takes place, activating a sequence specific disruption of the invading genetic fragment by Cas nucleases. Furthermore, there is a short-conserved sequence motif (2–5 bp), known as the protospacer adjacent motif (PAM) adjoining the crRNA-targeted sequence on the invading nucleic acid. In most of the CRISPR-Cas complexes, PAM is crucial for selection and disruption of the DNA [43].
The latest classification of the CRISPR-Cas loci categorizes the CRISPR system into six different types (I-VI) [7]. The class II CRISPR-Cas system employs a single endonuclease Cas9 that recognizes dsDNA and cleaves each strand with separate nuclease domain. Gene editing technology of the eukaryotic cells substantially employs this CRISPR-Cas9 system [21]. In the class II CRISPR-Cas9 mediated DNA degradation process, a unique dual-RNA hybrid structure forms due to the base-pairing of another small non-coding RNA, the trans-activating crRNA (tracrRNA) with the repeat sequence of the crRNA. This dual-RNA hybrid structure guides the Cas9 to degrade any DNA containing a 20-nucletide (nt) complementary target sequence adjacent to the PAM [76]. Single-guide RNA (sgRNA) is synthesized by incorporating the crRNA and tracrRNA into a single RNA chimera streamlining into a two-component system—Cas9 and sgRNA. Once the sgRNA base pairs with target DNA sequence, the Cas9 generates DSBs near to the PAM sequence located in a proper distance from the specific target sequence on the sgRNA. Different Cas9 proteins, naturally occurring or engineered, offers identification of diverse PAM sequence extending various options for determining sites specific for gene editing as per requirement [76].
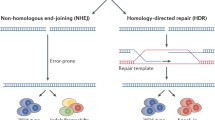
The DSB occurred due to the cleavage of the Cas9 being repaired either by an error prone nonhomologous end joining (NHEJ) mechanism leading to short arbitrary insertion and/or deletion (INDELs) or by highly accurate homology directed repair (HDR) with exact genome modification using a homologous repair template at the DSB site [43]. The critical advantage of the CRISPR-Cas9 genome editing technique lies in the identification of the target DNA by 20nt sgRNA rather than engineering specific protein domains recognizing the target DNA used in gene editing by conventional ZFNs or TALENs.
Cas9 has been engineered into nCas9 (Cas9 nickase) with a single amino acid mutation in one of the nuclease domains and only cleaves the target strand without any DSBs creating low INDELs and efficient HDR. Base editing and prime editing are the advanced gene editing technology that uses engineered nCas9 nuclease precisely altering a single base without DSB or all kinds of point mutations involving nucleotide transitions and transversions [42, 43]. Dead Cas9 (dCas9), another engineered catalytically dead Cas9 with mutations in both the nuclease domains implemented in techniques beyond gene editing to fuse with transcription activation or repression of the sgRNA-targeted genes.
Implication of CRISPR/Cas9 in CVDs
Application of CRISPR/Cas9 is implemented in correction of mutation by introducing INDELs followed by NHEJ repair causing a frameshift of the mutant allele removing the disease-causing mutant allele. However, only dominant-negative mutations will be corrected by this strategy if it is possible to target the mutant allele and not the wild type allele [53]. A missense mutation (H530R) in the PRKAG2 gene [26], encoding the protein kinase AMP-activated noncatalytic subunit gamma 2 causes an autosomal dominant form of Wolff-Parkinson-White syndrome [25]. Paroxysmal supraventricular tachycardia is the underlying cause of this syndrome affecting ~ 10% of sudden cardiac deaths in young patients [4]. A sgRNA targeting the mutant allele of the PRKAG2 gene was designed that differed from the wild type allele by a single nucleotide. The H530R/ + mouse model replicated PRKAG2 cardiac syndromes in patients. Significant improvement of cardiac problems was accomplished by 20% reduction of PRKAG2 mRNA after systemic delivery of the AAV9-sgRNA and AAV9-Cas9 to neonatal pups [96]. Due to one nucleotide difference between the mutant and the WT allele, editing efficiency and specificity were relatively low. Catecholaminergic polymorphic ventricular tachycardia is caused by autosomal dominant mutations in the RYR2 gene [96]. Mice heterozygous for the Ryr2 mutation received a single subcutaneous injection of the AAV9-SaCas9 system degrading the disease-causing allele [78]. This somatic genome editing approach has the potential to treat the lethal autosomal dominant inherited cardiac disorders. DMD occurs due to mutations in the dystrophin gene on the X chromosome affecting 1 in 3500 boys [92]. DMD patients die by the age of 25 mostly due to breathing complications and cardiomyopathy [75]. CRISPR/Cas9 has been successfully employed in correcting the mutations in the Dmd genes of the mdx mice, a model for DMD [99]. In nutshell, genome editing was done by injection of the sgRNA, Cas9 and HDR template into the mdx mouse zygotes to rectify the disease-causing gene mutation in the germ line [99]. Since, genome editing in the germ line produced genetically corrected animals with a varied range of mosaicism (2–100%), therefore, the scientists were able to compare the percentage of genomic correction required to achieve normal muscular structure and function.
A pathogenic 4-bp deletion in the MYBPC3 gene causes a dominant negative hypertrophic cardiomyopathy (HCM). Human sperm carrying this MYBPC3 mutation was coinjected with CRISPR/Cas9 into normal oocytes [59]. The homologous WT maternal gene functioned as the template DNA to repair the DSBs of the mutant paternal allele instead of a synthetic DNA template. This study showed the potential of gene editing to rectify genetic mutations in human embryos by taking care of safety and ethical concerns. Apart from these, CRISPR/Cas9 has been extensively utilized to model heart diseases in mice and generate specific mouse models with the diseased phenotype. Furthermore, iPSCs derived cardiomyocytes are targeted by CRISPR/Cas9 gene editing technology to mutate the genes and study their phenotype involved in causing several cardiomyopathies and then correcting the mutations in the patient derived iPSCs [45]. Calmodulinopathies cause severe genetic arrhythmia with long-QT syndrome (LQTS) due to mutations in the CALM1, CALM2, or CALM3 gene. Hampered Ca2+/calmodulin (CaM)-dependent inactivation of L-type Ca2+ channels are the primary reason of this kind of LQTS. The patient derived iPSCs deived cardiomyocytes revealed defective L-type Ca2+ channels due to long action potentials, impeding Ca2+ properties and diminished Ca2+/CaM-dependent inactivation of these channels. The mutant genes were selectively suppressed by involving CRISPR without interfering with the wild-type counter parts. After CRISPR intervention and suppression of CALM2 expression, D130G-CALM2 cardiyomyocytes derived from patient iPSCs functionally improved with normalized action potential duration. This advancement of gene editing strategies by CRISPR can be applied to knockdown any of the mutated CALM genes as a therapeutic tool for calmodulinopathy [23]. Two different groups has targeted to introduce loss-of-function mutation in PCSK9 gene in mouse liver by packaging the CRISPR/Cas9 tool in either adenovirus (AdV) or AAVs [45]. Both the groups observed > 40–50% PCSK9 gene modification with 3–4 days or 1 week of in vivo administration with lowered serum cholesterol levels [14]. All these reports along with progressing genome-wide association studies (GWAS) studies have led to a general scientific consensus that combining CRISPR gene editing and GWAS will play important role of development in the human personalized medicine [90].
Gene therapy in CVDs
The burden of cardiovascular maladies on humankind is steadily rising year-on-year. Several reports indicate genetic mutations as the root cause of over a hundred such afflictions. Advancements in genetic engineering have prompted scientists and clinicians to explore the potential of gene therapy in treating CVDs. However, due to multiple complexities and concerns associated with the gene editing techniques, the therapies have been limited to conditions involving single gene mutation. Grossman and group in 1995 conducted the first gene therapy using recombinant retroviruses carrying genetically altered human LDLR to treat FH. However, the outcome was not successful with only three out of five subjects showing a small reduction in LDL-c (6–25%) while liver biopsies showed hepatocytes expressing normal LDLR. This shows the poor transfection efficiency of retrovirus [29]. With the advancement in the gene therapy delivery system, Zhao et al. 2020 used the CRISPR/Cas9 system to produce a homozygous E208X mutant of the LDLR through HDR in mouse-fertilized eggs. Later, dual AAV8 systems were used to deliver liver-specific cas9 and sgRNA to repair the mutation, and the Sanger sequencing showed 6.7% gene repair. This gene-editing treatment decreased atherosclerotic lesions, lipid accumulation, macrophage infiltration, and plaque fibrosis, which depicts high transfection efficiency compared to the retroviral systems [100]. Lately, using CRISPR base editing technology, Musunuru and his group showed near complete knockout of PCSK9 in the liver of cynomolgus monkeys. They showed single administration of lipid nanoparticles leads to a 90% reduction of serum PCSK9 levels and a 60% drop of LDL-c, which was stable for 8 months post-treatment [69]. One such example of base editing was used to correct the pathogenic FBN1 gene involved in Marfan syndrome. In this study human zygotes created by sperms collected from the Marfan syndrome patients were microinjected with BE3 (altered in syndrome) and gRNA, showing that out of eight, seven had successful mutation repaired compared to 50% of the control embryos [98]. Nevertheless, achieving such editing efficiency using CRISPR/Cas nuclease and base editors’ system does not omit the risks of off-target effects. In-depth account of gene therapy against inherited and acquired CVDs is explained excellently in recently published reviews [10] [9].
Mesenchymal stem/ stromal cells are one of the most widely used cell-based therapies with more than 800 clinical trials being conducted to fully exploit their benefits in treating a diverse range of diseases [58]. With an enhanced ability to express functional genes, genetically modified mesenchymal stem cells proffer high desirable transfection. They also have improved homing, migration ability to target organ and enhanced proliferation and anti-apoptotic survival after transfection [13]. For better homing and targeted delivery under inflammatory conditions, the MSCs were genetically modified to overexpress the SDF1 receptor, which helped in myoangionesis and improved cardiac remodeling under ischemic conditions [52]. Similarly enhanced expression of GCP2/CXCL6 in human derived mesenchymal stem cells, improved cardiac function and reduced infarct size in mouse myocardial infraction model [46]. Recently, a study reported the cardioprotective nature of mesenchymal stem cells in mouse pressure overload induced heart failure. Circulating adiponectin in the animal body stimulates the exosome production of mesenchymal cells which have organ protective properties [72].
Another step that has been taken in the direction of gene therapy in heart failure is to augment the cardiomyocytes with the genes that show decline in the expression over advancing age. One such gene is SERCA2a [30, 70]. CUPID and AGENT-HF trials were conducted to deliver the SERCA2a with the help of AAV vectors, however both the trials could not show any substantial differences between the treatment and placebo [38]. The reasons behind the similar results in both the groups was partly thought to be unsuccessful delivery of transgene in the myocardium as the transgene was not detected in the samples later when the tissue was available. The absence of transgene from the treated samples was believed to be due to poor tropism of AAVs for the human cardiomyocytes. However, there were no safety concerns in any of the trials comprising small number of patients.
Ethical concerns
Germline gene editing raise ethical concerns because changes, whether intentional or not, are inherited by future generations. Genetic testing, informed permission, privacy, and responsible genetic information usage are ethical issues in inherited CVDs [32]. Ethical considerations include encouraging individuals with a genetic condition to inform their family members about potential risks and seek testing or counseling [77]. Respect for individual autonomy is essential and they should have the right to make decisions about their own genetic testing, treatment, reproductive choices. Continuous education is essential for healthcare professionals to provide the best care and guidance to patients with inherited CVDs [32]. Mosaicism is also a significant cause of concern. A study found high levels of mosaicism in a bovine embryo model after germline editing using the Cas9 system, confirming its potential in human embryos if unregulated [34]. Ma et al. discovered that adding CRISPR before cell division for editing shorter genes mainly avoid mosaicism with no off-target consequences [59]. However, longer genes may have more off-target effects. It is unknown if these off-target effects are automatically detected and remedied in the human embryo. Thus, research is needed to validate the absence of off-target effects during editing of the human genome and advance the development of novel techniques such as base editing. It has been argued that germline gene editing is unnecessary if we can identify healthy embryos using pre-implantation genetic diagnostic approaches. However, this does not apply for polygenic diseases [20].
There has been use of germline gene editing for non-therapeutic objectives. China has created newborns using CRISPR-Cas9, and this started a global debate and highlighted the need for stringent regulations and international agreement to address these difficulties and limit the use of this technology to certain medical procedures [36]. According to the National Academy of Medicine, efforts for safe and secure gene therapy should be made as there are those who for moral or religious reasons, choose to forgo germline gene editing [73]. Germline gene editing and in vitro fertilization may only benefit wealthy individuals due to their high cost. This worry could be put to rest with the help of insurance and government health care policies should consider this.
Challenges of gene editing technologies in treating CVDs
The genome editing field is rapidly evolving, with the cardiovascular field experiencing significant advances. However, challenges remain in the therapeutic application of CRISPR-Cas9 in cardiovascular research, including delivery and balancing efficacy and safety. The possibility for off-target effects is one of the main technical concerns of CRISPR-Cas9 gene editing. Inadvertent cleavage of DNA at regions that are like but not identical to the target sequence can occur when the Cas9 enzyme is used to cut DNA at specified loci. Off-target effects can cause detrimental genetic changes in the setting of CVDs. To reduce unintended consequences, scientists are always attempting to increase CRISPR-Cas9’s specificity. A major challenge in gene editing involves getting the necessary components, such as Cas9 and gRNA, to the cells that need editing in the cardiovascular system. Reaching targeted cardiac or vascular areas without causing injury or activating an immune response requires effective and safe delivery systems [67]. For gene editing to be used effectively in treating cardiovascular illnesses, advancements in delivery systems are required. Making changes to an individual’s DNA that can be passed on to future generations through germline editing is highly controversial and ethically problematic component of gene editing technology. The broader ethical considerations surrounding germline editing need to be addressed within the context of cardiovascular research due to the potential utilization of gene editing for the assessment and prevention of genetic risks associated with CVDs, which has a tenuous but real connection to cardiovascular illnesses. Concerns about the immune response and safety have been raised in relation to the use of Cas9 proteins and viral vectors in gene editing. These reactions could reduce the efficiency of gene editing or cause unwanted side effects [86]. The dynamics of the immune response must be fully understood, and techniques developed to limit hazards, before gene editing technologies may be used safely in cardiovascular applications. Many CVDs, such as CAD, are complicated multigene disorders impacted by a wide range of genetic and environmental factors. It is possible that trying to stop a disease’s progression by focusing on a single gene won’t be enough. To properly treat such illnesses, researchers must find and change numerous genes or pathways all at once. It is extremely difficult to evaluate the long-term safety and effectiveness of gene editing therapy for cardiovascular illnesses. Careful planning and long-term follow-up of trial participants are essential for detecting and treating any unanticipated adverse events or complications that may arise.
Future directions
Genome editing techniques like CRISPR/Cas9, base editing, and prime editing have great potential for future applications in various fields. However, further understanding and advances are needed to improve technical approaches, characterize, and optimize base and prime editing technologies, and address delivery challenges. The immunogenicity of CRISPR/Cas9 proteins also needs to be addressed. Concerns about the ethics of human genome editing have increased, highlighting the need for specific regulatory systems. Optimized genome editors with increased target specificity, low off-target effects, small size, flexible PAM availability, and easy accessibility are ideal for clinical applications. Ex vivo genome editing is likely the first possible clinical application, but direct in vivo editing of post-mitotic tissues like the heart is still far away. The potential of genome editing is great and should be further researched to benefit humankind and potentially treat diseases previously untreatable [86].
Abbreviations
- AAV:
-
Adeno associated virus
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- Cas:
-
CRISPR associated protein
- CVDs:
-
Cardiovascular diseases
- iPSC:
-
Induced pluripotent stem cells
- FH:
-
Familial hypercholesterolemia
- PAH:
-
Pulmonary arterial hypertension
- FTAAD:
-
Familial thoracic aortic aneurysm and dissection
- ZFNs:
-
Zinc-finger nucleases
- PAM:
-
Protospacer adjacent motif
- HCM:
-
Hypertrophic cardiomyopathy
- sgRNA:
-
Single-guide RNA
- crRNA:
-
CRISPR-RNAs
- tracrRNA:
-
trans-activating CrRNA
- TALENS:
-
Transcription activator–like effector nucleases
- VSMCs:
-
Vascular smooth muscle cells
References
Anastasiou M, Oikonomou E, Theofilis P, et al. Prolonged impact of anti-cancer therapy on endothelial function and arterial stiffness in breast cancer patients. Vasc Pharmacol. 2023;152:107195. https://doi.org/10.1016/j.vph.2023.107195.
Andreassi MG. Coronary atherosclerosis and somatic mutations: an overview of the contributive factors for oxidative DNA damage. Mutat Res Rev Mutat Res. 2003;543:67–86.
Ballinger SW, Patterson C, Yan C, et al. Mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ Res. 2000;86:960–7.
Basso C, Corrado D, Rossi L, Thiene G. Ventricular preexcitation in children and young adults: atrial myocarditis as a possible trigger of sudden death. Circulation. 2001;103:269–75. https://doi.org/10.1161/01.CIR.103.2.269.
Bennett MR, Evan GI, Schwartz SM. Apoptosis of human vascular smooth muscle cells derived from normal vessels and coronary atherosclerotic plaques. J Clin Invest. 1995;95:2266–74. https://doi.org/10.1172/JCI117917.
Botto N, Masetti S, Petrozzi L, et al. Elevated levels of oxidative DNA damage in patients with coronary artery disease. Coron Artery Dis. 2002;13:269–74. https://doi.org/10.1097/00019501-200208000-00004.
Brouns SJJ, Jore MM, Lundgren M, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–4. https://doi.org/10.1126/SCIENCE.1159689.
Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation. 2013;128:388–400. https://doi.org/10.1161/CIRCULATIONAHA.113.001878.
Cannatà A, Ali H, Sinagra G, Giacca M. Gene therapy for the heart lessons learned and future perspectives. Circ Res. 2020. https://doi.org/10.1161/CIRCRESAHA.120.315855.
Cao G, Xuan X, Zhang R, et al. Gene Therapy for Cardiovascular Disease: Basic Research and Clinical Prospects. Front Cardiovasc Med. 2021;8:760140. https://doi.org/10.3389/FCVM.2021.760140.
Cardus A, Uryga AK, Walters G, Erusalimsky JD. SIRT6 protects human endothelial cells from DNA damage, telomere dysfunction, and senescence. Cardiovasc Res. 2013;97:571–9. https://doi.org/10.1093/cvr/cvs352.
Cui M, Wang Z, Bassel-Duby R, Olson EN. Genetic and epigenetic regulation of cardiomyocytes in development, regeneration and disease. Development. 2018. https://doi.org/10.1242/DEV.171983.
Devetzi M, Goulielmaki M, Khoury N, et al. Genetically-modified stem cells in treatment of human diseases: tissue kallikrein (KLK1)-based targeted therapy (Review). Int J Mol Med. 2018;41:1177–86. https://doi.org/10.3892/IJMM.2018.3361.
Ding Q, Strong A, Patel KM, et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res. 2014;115:488–92. https://doi.org/10.1161/CIRCRESAHA.115.304351/-/DC1.
Doetschman T, Azhar M. Cardiac-specific inducible and conditional gene targeting in mice. Circ Res. 2012;110:1498–512. https://doi.org/10.1161/CIRCRESAHA.112.265066.
Dorsheimer L, Assmus B, Rasper T, et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. 2019;4:32. https://doi.org/10.1001/JAMACARDIO.2018.3965.
Evans MA, Sano S, Walsh K. Cardiovascular disease, aging, and clonal hematopoiesis. Annu Rev Pathol. 2019. https://doi.org/10.1146/annurev-pathmechdis.
Flouris GA, Arvanitis DA, Parissis JT, et al. Loss of heterozygosity in DNA mismatch repair genes in human atherosclerotic plaques. Mol Cell Biol Res Commun. 2001;4:62–5. https://doi.org/10.1006/mcbr.2000.0255.
Foglia MJ, Poss KD. Building and re-building the heart by cardiomyocyte proliferation. Development. 2016;143(5):729–40. https://doi.org/10.1242/dev.132910.
Fu Y, Foden JA, Khayter C, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–6. https://doi.org/10.1038/nbt.2623.
Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012. https://doi.org/10.1073/PNAS.1208507109/-/DCSUPPLEMENTAL/PNAS.201208507SI.PDF.
Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–87. https://doi.org/10.1056/NEJMOA1409405.
German DM, Mitalipov S, Mishra A, Kaul S. Therapeutic genome editing in cardiovascular diseases. JACC Basic Transl Sci. 2019;4:122–31. https://doi.org/10.1016/j.jacbts.2018.11.004.
Gimbrone MA Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:6072–8. https://doi.org/10.1002/cncr.27633.Percutaneous.
Gollob MH, Green MS, Tang AS-L, et al. Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med. 2001;344:1823–31. https://doi.org/10.1056/NEJM200106143442403.
Gollob MH, Green MS, Tang ASL, Roberts R. PRKAG2 cardiac syndrome: familial ventricular preexcitation, conduction system disease, and cardiac hypertrophy. Curr Opin Cardiol. 2002;17:229–34. https://doi.org/10.1097/00001573-200205000-00004.
Gong J, Zhou D, Jiang L, et al. In vitro lineage-specific differentiation of vascular smooth muscle cells in response to SMAD3 deficiency: implications for SMAD3-related thoracic aortic aneurysm. Arterioscler Thromb Vasc Biol. 2020;40:1651–63. https://doi.org/10.1161/ATVBAHA.120.313033.
Gray K, Kumar S, Figg N, et al. Effects of DNA damage in smooth muscle cells in atherosclerosis. Circ Res. 2014;116(5):816–26. https://doi.org/10.1161/CIRCRESAHA.116.304921.
Grossman M, Rader DJ, Muller DWM, et al. A pilot study of ex vivo gene therapy for homozygous familial hypercholesterolaemia. Nat Med. 1995;111(1):1148–54. https://doi.org/10.1038/nm1195-1148.
Gwathmey JK, Yerevanian A, Hajjar RJ. Cardiac gene therapy with SERCA2a: from bench to bedside. J Mol Cell Cardiol. 2010;50(5):803–12. https://doi.org/10.1016/j.yjmcc.2010.11.011.
Halcox JPJ. Endothelial dysfunction. Prim Auton Nerv Syst. 2012;12:319–24. https://doi.org/10.1016/B978-0-12-386525-0.00066-4.
Hall AE, Burton H. Legal and ethical implications of inherited cardiac disease in clinical practice within the UK. J Med Ethics. 2010;36:762–6. https://doi.org/10.1136/jme.2009.034108.
Hatzistamou J, Kiaris H, Ergazaki M, Spandidos DA. Loss of heterozygosity and microsatellite instability in human atherosclerotic plaques. Biochem Biophys Res Commun. 1996;225:186–90. https://doi.org/10.1006/bbrc.1996.1151.
Hennig SL, Owen JR, Lin JC, et al. Evaluation of mutation rates, mosaicism and off target mutations when injecting Cas9 mRNA or protein for genome editing of bovine embryos. Sci Rep. 2020;10(1):22309. https://doi.org/10.1038/s41598-020-78264-8.
Higo T, Naito AT, Sumida T, et al. DNA single-strand break-induced DNA damage response causes heart failure. Nat Commun. 2017;8:1–13. https://doi.org/10.1038/ncomms15104.
How to respond to CRISPR babies. Nature. 2018;564:5. https://doi.org/10.1038/d41586-018-07634-0
Huang Y, Hong H, Li M, et al. Age-dependent oxidative DNA damage does not correlate with reduced proliferation of cardiomyocytes in humans. PLoS ONE. 2017;12(1):1–13. https://doi.org/10.1371/journal.pone.0170351.
Ishikawa K, Weber T, Hajjar RJ. Human cardiac gene therapy. Circ Res. 2018;123:601–13. https://doi.org/10.1161/CIRCRESAHA.118.311587.
Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;26:2488–98. https://doi.org/10.1056/NEJMoa1408617.
Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–21. https://doi.org/10.1056/NEJMOA1701719.
Jean D, Peter G. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III27–32. https://doi.org/10.1161/01.CIR.0000131515.03336.f8.
Jiang F, Doudna JA. CRISPR–Cas9 structures and mechanisms. Annu Rev Biophys. 2017;46:505–29. https://doi.org/10.1146/ANNUREV-BIOPHYS-062215-010822.
Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21. https://doi.org/10.1126/SCIENCE.1225829.
Khan M, Völkers M, Wende AR. Editorial: metabolic regulation of cardiac and vascular cell function: physiological and pathophysiological implications. Front Physiol. 2022;13: 849869. https://doi.org/10.3389/FPHYS.2022.849869.
Khouzam JPS, Tivakaran VS. CRISPR-Cas9 applications in cardiovascular disease. Curr Probl Cardiol. 2021;46: 100652. https://doi.org/10.1016/J.CPCARDIOL.2020.100652.
Kim SW, Lee DW, Yu LH, et al. Mesenchymal stem cells overexpressing GCP-2 improve heart function through enhanced angiogenic properties in a myocardial infarction model. Cardiovasc Res. 2012;95:495–506. https://doi.org/10.1093/CVR/CVS224.
Knott GJ, Doudna JA. CRISPR-Cas guides the future of genetic engineering. Science. 2018;361:866–9. https://doi.org/10.1126/SCIENCE.AAT5011.
Kurose I, Wolf R, Cerwinka W, Granger DN. Microvascular responses to ischemia/reperfusion in normotensive and hypertensive rats. Hypertension. 1999;34:212–6. https://doi.org/10.1161/01.HYP.34.2.212.
Landis BJ, Veldtman GR, Ware SM. Genotype-phenotype correlations in Marfan syndrome. Heart. 2017;103:1750–2. https://doi.org/10.1136/heartjnl-2017-311513.
Lewandoski M. Conditional control of gene expression in the mouse. Nature Rev Genet. 2002;2(10):743–55.
Li ZH, Wang J, Xu JP, et al. (2023) Recent advances in CRISPR-based genome editing technology and its applications in cardiovascular research. Mil Med Res. 2023;101(10):1–20. https://doi.org/10.1186/S40779-023-00447-X.
Li Q, Zhang A, Tao C, et al. The role of SDF-1-CXCR4/CXCR7 axis in biological behaviors of adipose tissue-derived mesenchymal stem cells in vitro. Biochem Biophys Res Commun. 2013;441:675–80. https://doi.org/10.1016/J.BBRC.2013.10.071.
Liu N, Olson EN. CRISPR modeling and correction of cardiovascular disease. Circ Res. 2022;130:1827–50. https://doi.org/10.1161/CIRCRESAHA.122.320496.
Lloyd-Jones DM, Nam BH, D’Agostino RB, et al. Parental cardiovascular disease as a risk factor for cardiovascular disease in middle-aged adults: a prospective study of parents and offspring. JAMA. 2004;291:2204–11. https://doi.org/10.1001/JAMA.291.18.2204.
Long C, Li H, Tiburcy M, et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci Adv. 2018. https://doi.org/10.1126/SCIADV.AAP9004/SUPPL_FILE/AAP9004_SM.PDF.
Long L, Ormiston ML, Yang X, et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat Med. 2015;21:777–85. https://doi.org/10.1038/nm.3877.
Ludmer PL, Selwyn AP, Shook TL, et al. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med. 1986;315:1046–51. https://doi.org/10.1056/NEJM198610233151702.
Luo R, Lu Y, Liu J, et al. Enhancement of the efficacy of mesenchymal stem cells in the treatment of ischemic diseases. Biomed Pharmacother. 2019;109:2022–34. https://doi.org/10.1016/J.BIOPHA.2018.11.068.
Ma H, Marti-Gutierrez N, Park SW, Wu J, Lee Y, Suzuki K, Koski A, Ji D, Hayama T, Ahmed R, et al. Correction of a pathogenic gene mutation in human embryos. Nature. 2017;548:413–9. https://doi.org/10.1038/nature23305.
Machado RD, Aldred MA, James V, et al. Mutations of the TGF-β type II receptorBMPR2 in pulmonary arterial hypertension. Hum Mutat. 2006;27:121–32. https://doi.org/10.1002/humu.20285.
Mao Z, Hine C, Tian X, et al. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011;332:1443–6. https://doi.org/10.1126/science.1202723.
Martinet W, Knaapen MWM, De Meyer GRY, et al. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation. 2002;106:927–32. https://doi.org/10.1161/01.CIR.0000026393.47805.21.
Matthews C, Gorenne I, Scott S, et al. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res. 2006;99:156–64. https://doi.org/10.1161/01.RES.0000233315.38086.bc.
Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, et al. Current clinical applications of in vivo gene therapy with AAVs. Mol Ther. 2021;29:464–88. https://doi.org/10.1016/J.YMTHE.2020.12.007.
Mensah GA, Moran AE, Roth GA, Narula J. The global burden of cardiovascular diseases, 1990–2010. Glob Heart. 2014;9:183–4. https://doi.org/10.1016/j.gheart.2014.01.008.
Mercer J, Mahmoudi M, Bennett M. DNA damage, p53, apoptosis and vascular disease. Mutat Res Fundam Mol Mech Mutagen. 2007;621:75–86. https://doi.org/10.1016/j.mrfmmm.2007.02.011.
Moore OM, Ho KS, Copeland JS, et al. Genome editing and cardiac arrhythmias. Cells. 2023. https://doi.org/10.3390/cells12101363.
Motta BM, Pramstaller PP, Hicks AA, Rossini A. The impact of CRISPR/Cas9 technology on cardiac research: from disease modelling to therapeutic approaches. Stem Cells Int. 2017. https://doi.org/10.1155/2017/8960236.
Musunuru K, Chadwick AC, Mizoguchi T, et al. (2021) In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature. 2021;5937859(593):429–34. https://doi.org/10.1038/s41586-021-03534-y.
Naim C, Yerevanian A, Hajjar RJ. Gene therapy for heart failure: Where do we stand? Curr Cardiol Rep. 2013;15:1–10. https://doi.org/10.1007/s11886-012-0333-3.
Nakada Y, Nhi Nguyen NU, Xiao F, et al. DNA damage response mediates pressure overload-induced cardiomyocyte hypertrophy. Circulation. 2019;139:1237–9. https://doi.org/10.1161/CIRCULATIONAHA.118.034822.
Nakamura Y, Kita S, Tanaka Y, et al. Adiponectin stimulates exosome release to enhance mesenchymal stem-cell-driven therapy of heart failure in mice. Mol Ther. 2020;28:2203–19. https://doi.org/10.1016/j.ymthe.2020.06.026.
National Academies of Sciences, Engineering, and Medicine. Human genome editing: science, ethics, and governance. Washington: The National Academies Press; 2017. https://doi.org/10.17226/24623.
Octavia Y, Tocchetti CG, Gabrielson KL, et al. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012;52(6):1213–25. https://doi.org/10.1016/j.yjmcc.2012.03.006.
Olson EN. Toward the correction of muscular dystrophy by gene editing. Proc Natl Acad Sci USA. 2021. https://doi.org/10.1073/PNAS.2004840117.
Van Der Oost J, Westra ER, Jackson RN, Wiedenheft B. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat Rev Microbiol. 2014;12:479–92. https://doi.org/10.1038/NRMICRO3279.
Ormond KE, Borensztein MJ, Hallquist MLG, et al. Defining the critical components of informed consent for genetic testing. J Pers Med. 2021. https://doi.org/10.3390/jpm11121304.
Pan X, Philippen L, Lahiri SK, et al. In Vivo Ryr2 editing corrects catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2018;123:953–63. https://doi.org/10.1161/CIRCRESAHA.118.313369.
Paquette M, Baass A. A novel cause of familial hypercholesterolemia: PCSK9 gene duplication. Can J Cardiol. 2018;34:1259–60. https://doi.org/10.1016/j.cjca.2018.08.027.
Puente BN, Kimura W, Muralidhar SA, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–79. https://doi.org/10.1016/j.cell.2014.03.032.
Reiner AP, Bhattacharya R, Zekavat SM, et al. Clonal hematopoiesis is associated with higher risk of stroke. Stroke. 2022;53:788–97. https://doi.org/10.1161/STROKEAHA.121.037388.
Renard M, Francis C, Ghosh R, et al. Clinical validity of genes for heritable thoracic aortic aneurysm and dissection. J Am Coll Cardiol. 2018;72:605–15. https://doi.org/10.1016/j.jacc.2018.04.089.
Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–9. https://doi.org/10.1038/362801a0.
Rouet P, Smih F, Jasin M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol. 1994;14:8096–106.
Satoh M, Ishikawa Y, Takahashi Y, et al. Association between oxidative DNA damage and telomere shortening in circulating endothelial progenitor cells obtained from metabolic syndrome patients with coronary artery disease. Atherosclerosis. 2008;198:347–53. https://doi.org/10.1016/j.atherosclerosis.2007.09.040.
Schreurs J, Sacchetto C, Colpaert RMW, et al. Recent advances in crispr/cas9-based genome editing tools for cardiac diseases. Int J Mol Sci. 2021;22:10985. https://doi.org/10.3390/ijms222010985.
Shah A, Gray K, Figg N, et al. Defective base excision repair of oxidative DNA damage in vascular smooth muscle cells promotes atherosclerosis. Circulation. 2018;138:1446–62. https://doi.org/10.1161/CIRCULATIONAHA.117.033249.
Shukla PC, Singh KK, Yanagawa B, et al. DNA damage repair and cardiovascular diseases. Can J Cardiol. 2010;26:13A-16A. https://doi.org/10.1016/S0828-282X(10)71055-2.
Tian W, Jiang X, Sung YK, et al. Phenotypically silent bone morphogenetic protein receptor 2 mutations predispose rats to inflammation-induced pulmonary arterial hypertension by enhancing the risk for neointimal transformation. Circulation. 2019;140:1409–25. https://doi.org/10.1161/CIRCULATIONAHA.119.040629.
Torres-Ruiz R, Rodriguez-Perales S. CRISPR-Cas9 technology: applications and human disease modelling. Brief Funct Genomics. 2017;16:4–12. https://doi.org/10.1093/BFGP/ELW025.
Touyz RM. Oxidative stress and vascular damage in hypertension. Curr Hypertens Rep. 2000;2(1):98–105.
Verhaart IEC, Aartsma-Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol. 2019;15(7):373–86.
Wang D, Zhang F, Gao G. CRISPR-based therapeutic genome editing: strategies and in vivo delivery by AAV vectors. Cell. 2020;181:136–50. https://doi.org/10.1016/J.CELL.2020.03.023.
Wenzel P, Schuhmacher S, Kienhöfer J, et al. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc Res. 2008;80:280–9. https://doi.org/10.1093/cvr/cvn182.
Wolf CM. Hypertrophic cardiomyopathy: genetics and clinical perspectives. Cardiovasc Diagn Ther. 2019;9:S388. https://doi.org/10.21037/CDT.2019.02.01.
Xie C, Zhang YP, Song L, Luo J, Qi W, Hu J, Lu D, Yang Z, Zhang J, Xiao J, Zhou B, Du JL. Genome editing with CRISPR/Cas9 in postnatal mice corrects PRKAG2 cardiac syndrome. Cell Res. 2016;26(10):1099–111.
Xu S, Yin M, Koroleva M, et al. SIRT6 protects against endothelial dysfunction and atherosclerosis in mice. Aging (Albany NY). 2016;8:1064–82. https://doi.org/10.18632/aging.100975.
Zeng Y, Li J, Li G, et al. Correction of the marfan syndrome pathogenic FBN1 mutation by base editing in human cells and heterozygous embryos. Mol Ther. 2018;26:2631–7. https://doi.org/10.1016/J.YMTHE.2018.08.007.
Zhang Y, Li H, Min YL, et al. Enhanced CRISPR-Cas9 correction of Duchenne muscular dystrophy in mice by a self-complementary AAV delivery system. Sci Adv. 2020;6:6812. https://doi.org/10.1126/SCIADV.AAY6812/SUPPL_FILE/AAY6812_SM.PDF.
Zhao H, Li Y, He L, et al. In vivo AAV-CRISPR/Cas9–mediated gene editing ameliorates atherosclerosis in familial hypercholesterolemia. Circulation. 2020;141:67–79. https://doi.org/10.1161/CIRCULATIONAHA.119.042476.
Funding
This work did not receive any funding.
Author information
Authors and Affiliations
Contributions
DS, RC, PS, and DG—wrote the manuscript. PCS—ideation and finalization of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Financial interests: RC and SD received no financial support for this work. Non-financial interests: The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Corresponding Editor: Nishant Chakravorty; Reviewers: Syamantak Majumder, Amit Kumar Pandey.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Duddu, S., Chakrabarti, R., Sharma, P. et al. Gene editing and therapy in acquired and inherited cardiovascular disorders. Nucleus 67, 237–250 (2024). https://doi.org/10.1007/s13237-024-00480-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13237-024-00480-8