
Abstract
Solution-processable techniques, including spin-coating, roll-to-roll printing, and ink-jet printing, are suitable for large-scale manufacturing and flexible displays owing to their cost-effective and simple fabrication. These techniques are widely used in the fabrication of perovskite light-emitting diodes (PeLEDs) and organic light-emitting diodes (OLEDs). However, the energy level modulation between different functional layers, thickness control of the deposited layer, and interlayer mixing between the deposited and subsequent layers may affect the LED performance due to the poor solvent resistance of the deposited layer. Herein, we designed a novel poly[4,4'-(2-(4-((4-hexylphenyl)(phenyl)amino)phenyl)-9H-fluorene-9,9-diyl)bis(N,N-bis(4-butylphenyl)aniline)] (PFTPA-biTPA) as a hole transport layer (HTL) to modulate the hole injection. Subsequently, we introduced the bis(4-azido-2,3,5,6-tetrafluorobenzoate) (FPA) as a photo-crosslinker into the PFTPA-biTPA to prepare the photo-crosslinked HTL for both PeLED and OLED that inhibit attack from the processing solution and suppress the interlayer mixing between the deposited and subsequent layers. Both PeLEDs and OLEDs with photo-crosslinked PFTPA-biTPA exhibited maximum luminance of 175 and 4260 cd/m2 and maximum external quantum efficiency (EQE) of 4.16 and 10.86%, respectively, thus indicating superior properties to those without and with PFTPA-biTPA as the HTLs. Moreover, the photo-crosslinked PFTPA-biTPA showed significantly improved device stability compared to the reference devices having non-photo-crosslinked HTLs in PeLEDs.
Graphical abstract
Schematic illustration of crosslinked hole transport polymer with enhanced hole transport and solvent resistance

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Most light-emitting diodes (LEDs) use an efficient multilayer structure that can reduce each injection barrier to promote charge-carrier injection and improve the recombination of electrons and holes in the emissive layer (EML) [1,2,3,4,5,6,7,8]. The energy level matching and the conductivity of electrons and holes are important factors to balance the charge injection in multilayer devices. When charge injection is imbalanced, excess charges can accumulate at the interface between the EML and adjacent layer, and the accumulated holes and electrons can migrate to the electron transport layer (ETL) and hole transport layer (HTL), respectively. The emission in the transport layer causes leakage of major carriers, thereby degrading the LED performance [9, 10]. The larger hole injection barrier between the EML and HTL than the electron injection barrier between the EML and ETL results in charge imbalance in the perovskite light-emitting diodes (PeLEDs) and organic light-emitting diodes (OLEDs) due to the insufficient hole injection. Furthermore, the excitons can be quenched near the interface between the HTL and EML, resulting in luminescence quenching [11,12,13,14,15,16]. Therefore, it is important to obtain sufficient charge-carrier mobility and reduce the hole injection barrier for balancing the charge injection. Generally, the highest occupied molecular orbital (HOMO) level of perovskite in a PeLED is deeper than that of EML in an OLED which significantly limits the hole injection and makes the issue critical [17].
Multilayer devices are fabricated using either high-vacuum vapor deposition or solution-processing methods. The high-vacuum vapor deposition method is suitable only for thermally stable low-molecular-weight materials, and it is expensive as well as time consuming. Therefore, the solution-processing methods can be an attractive strategy to save money and time. However, the choice of solvents to fabricate multilayer devices is limited in solution-processing methods because each deposited layer can be redissolved by the processing solvent of the subsequent layer owing to the poor solvent resistance, thereby resulting in thickness variation and interlayer mixing [18, 19]. Therefore, crosslinkable materials have been developed, which can be crosslinked using thermal or UV irradiation to make the film insoluble in any solvent, thus exhibit excellent solvent resistance [20, 21]. Significant research has been conducted on the thermally crosslinkable charge transport materials in LEDs [22,23,24,25,26,27,28,29]. The thermal-crosslinking using specific functional groups such as styrene and oxetane required annealing at a high temperature (over 180 °C) for a long curing time that could damage layers under the crosslinked layer, thereby diminishing the device performance. In contrast, the photo-crosslinking with the molecules having allyl, acrylate, and azide groups as photo-crosslinkable functional groups was successfully performed at room temperature without causing thermal degradation of the deposited layer [30,31,32]. Particularly, azide-based photo-crosslinkers have attracted significant attention owing to their good chemical stabilities and photophysical properties [33, 34]. Investigations on the photo-crosslinkable charge transport layers are ongoing in OLEDs and quantum dot light-emitting diodes (QLEDs); however, these have not yet been extensively explored in PeLEDs. More research on the applicability of the photo-crosslinking method in PeLEDs is needed for the development and commercialization.
In this study, we synthesized a photo-crosslinkable poly[4,4′-(2-(4-((4-hexylphenyl)(phenyl) amino)phenyl)-9H-fluorene-9,9-diyl)bis(N,N-bis(4-butylphenyl)aniline)] (PFTPA-biTPA) to use as an HTL in the CsPbBr3 PeLEDs and OLEDs. Its HOMO level was calculated to be − 5.25 eV, which lies between that of poly(3,4-ethylenedioxythiophene):poly-styrene sulfonate (PEDOT:PSS, − 5.0 eV) and EMLs (− 6.2 eV for PeLEDs and − 5.5 eV for OLEDs), thereby facilitating better hole injection and charge balance in PeLEDs and OLEDs with the PFTPA-biTPA than those in PeLEDs and OLEDs without the PFTPA-biTPA [35]. Furthermore, we used bis(4-azido-2,3,5,6-tetrafluorobenzoate) (FPA) as a photo-crosslinker to evaluate the performance of the crosslinked PFTPA-biTPA as an HTL. FPA has also been used in previous studies to form an insoluble film and improve efficiency in red and green OLEDs [34, 36]. Herein, the crosslinked PFTPA-biTPA film showed higher solvent resistance and better external quantum efficiency (EQE) and operational stability than the non-crosslinked PFTPA-biTPA film. Both PeLEDs and OLEDs with the photo-crosslinked PFTPA-biTPA exhibited maximum luminance of 175 and 4260 cd/m2 and maximum EQE of 4.16 and 10.86%, respectively. The proposed photo-crosslinkable PFTPA-biTPA is a suitable HTL for both PeLEDs and OLEDs because of its improved robustness to redissolution from solvents used to deposit subsequent layers.
2 Results and discussion
The synthetic route of the synthesized PFTPA-biTPA is shown in Scheme 1. The detailed polymer synthesis procedure is described in the experimental section. Fluorene and triphenylamine building blocks were copolymerized to improve the hole transport ability and to meet HOMO energy level between PEDOT:PSS and EML. To further improve the hole transport ability, aryl amine groups were additionally introduced at the 9-position of fluorene (Figure S1 and Table S1) [37,38,39,40,41,42]. Moreover, alkyl groups were introduced not only to triphenylamine building block but also to the arylamines in 9-position of fluorene to give sufficient solubility to the hole-transporting polymer, and also increase the chemical crosslinking sites with the FPA unit [43]. The PFTPA-biTPA is polymerized by the Suzuki polycondensation reaction; subsequently, the synthesized polymer is dissolved in common organic solvents, such as toluene, chlorobenzene, and chloroform. The molecular weight of PFTPA-biTPA was measured by gel permission chromatography (GPC) using chloroform as an eluent. The number averaged molecular weight (Mn) and polydispersity index (PDI) of the PFTPA-biTPA were 87.8 kg/mol and 6.0, respectively. The 1H NMR, 13C NMR, and liquid chromatography quadrupole time-of-flight mass spectrometry (LC-QTOF-MS) of the synthesized polymer are shown in Figure S2–S16 and discussed in the experimental section.

Synthetic route of PFTPA-biTPA
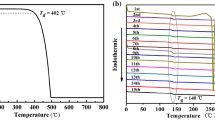
The thermogravimetric analysis (TGA) of the synthesized PFTPA-biTPA under a nitrogen atmosphere suggested high thermal stability (Figure S1(a)). The 5% weight loss of the PFTPA-biTPA was observed at a decomposition temperature (Td) of 462 ℃. The optical properties of the PFTPA-biTPA were investigated using the UV–visible absorption and photoluminescence (PL) spectra in 1 × 10–5 M chloroform solution and thin film states (Figure S1(b)). The PFTPA-biTPA shows the maximum absorption in solution and film states at 306 and 308 nm, respectively. The PL maximum of the PFTPA-biTPA in solution and film states is exhibited at 440 and 446 nm, respectively. The electrochemical properties of PFTPA-biTPA were studied using cyclic voltammetry (CV) in anhydrous acetonitrile (CH3CN) solution with 0.1 M tetrabutylammonium tetrafluoroborate as a supporting electrolyte (Figure S1(c)). The oxidation onset potential (Eox) of the PFTPA-biTPA film coated on the Pt working electrode was measured and the HOMO level was calculated to be − 5.25 eV. This is an intermediate energy level between PEDOT:PSS and EML; therefore, this is favorable for the hole injection from the anode to the EML in both PeLED and OLED. The lowest unoccupied molecular orbital (LUMO) level was calculated to be − 2.40 eV using a combination of the HOMO level from the oxidation onset and energy band gap (Egap) obtained from the absorption edge. The LUMO level of PFTPA-biTPA was also suitable for blocking the electron back-transport from the EML to the anode. The sufficient hole mobility was calculated to be 1.22 × 10–4 cm2 v−1 s−1 using space charge limited current (SCLC) (Figure S1(d)) [44, 45]. The results indicate that PFTPA-biTPA is suitable as the HTLs of LEDs.
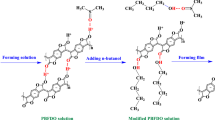
The process of photo-crosslinking PFTPA-biTPA was simple, and it is illustrated in Fig. 1(a) and the experimental section. The mixed solution of the PFTPA-biTPA and FPA (1.5 wt%) is spin coated and crosslinked using UV irradiation at 254 nm (84 mJ/cm2). Under UV irradiation, N2 separates from the terminal azide group (N = N = N) of FPA and a singlet nitrene is obtained. The lone pair electrons of the singlet nitrene cause C–H insertion reactions with carbon at the benzylic position of the alkyl side chains in the triphenylamine core, resulting in the crosslinking of the PFTPA-biTPA (Figure S17 and S18) [34, 36]. Fourier transform infrared (FTIR) spectroscopy was performed to confirm that crosslinking between the PFTPA-biTPA and FPA occurred through UV irradiation (Fig. 1(b)). The blending solution of the non-crosslinked and crosslinked PFTPA-biTPA:FPA was coated on a potassium bromide pellet. The intensity of the azide stretching peak decreases at 2127 cm−1 after crosslinking; thus, it indicates that N2 separates from the azide group present in FPA when crosslinking progresses [46].

a Molecular structure and crosslinking process of the PFTPA-biTPA with a crosslinker (FPA). b FTIR spectra of the PFTPA-biTPA. c UV–Vis absorption spectra of the PFTPA-biTPA film before and after rinsing with chlorobenzene (CB). d Thickness of the PFTPA-biTPA films before and after coating with octane. e Contact angles of the non-crosslinked and crosslinked PFTPA-biTPA. AFM images of f the non-crosslinked and g the crosslinked PFTPA-biTPA
Further analyses were carried out to investigate the performance of the crosslinked PFTPA-biTPA. The solvent resistance was confirmed by comparing the absorbance at the UV–Vis absorption maximum (306 nm and 394 nm for PFTPA-biTPA) before and after rinsing, as shown in Fig. 1c. The initial absorption spectra of the non-crosslinked and crosslinked PFTPA-biTPA films are not significantly different, indicating that the crosslinking using FPA does not affect the photophysical properties of the PFTPA-biTPA [33, 36]. The non-crosslinked and crosslinked films were rinsed in chlorobenzene (CB) for 1 min, which is used as a solvent for PFTPA-biTPA. The absorbance of the non-crosslinked film after rinsing shows poor solvent resistance of 13% at a wavelength of 306 nm, thereby suggesting that the non-crosslinked PFTPA-biTPA film can be easily redissolved by the solvents used in the upper layer during the solution processing. Furthermore, the crosslinked film has a residual ratio of approximately 93%, indicating that the solvent resistance can be enhanced through crosslinking. Therefore, a wide variety of solvents can be used on top of the crosslinked HTL to prevent redissolving and interlayer mixing during solution-processed LED fabrication [19, 32, 47]. These results were further confirmed by measuring the changes in the film thickness using processing solvent (Fig. 1d). The solvent octane was spin coated on the PFTPA-biTPA film as the perovskite layer and the thickness of the PFTPA-biTPA film was measured before and after coating with octane. Both non-crosslinked and crosslinked films exhibit the same thickness before coating with the solvents. After coating with octane, the thickness of the crosslinked film does not reduce significantly, whereas the non-crosslinked film shows a thickness reduction of approximately 50%. The absorption spectra and thickness analyses suggest that the crosslinked film could overcome the limitations on solvent selectivity used for device fabrication.
As shown in Fig. 1e, the crosslinked film demonstrates a significantly higher contact angle of 99.7° with water than the non-crosslinked film (44.5°), suggesting that the crosslinking process increases the hydrophobicity of the film. Therefore, the adjacent perovskite layer can be prevented from being degraded by moisture to some extent [48,49,50]. Changes in the surface texture are observed in the AFM images of the PFTPA-biTPA films as shown in Fig. 1f and g. The root-mean-square (RMS) values are calculated to be 0.768 and 0.603 nm for the non-crosslinked and crosslinked films, respectively. The dense and uniform crosslinked film suggests improved morphology, which is beneficial for improving the hole transport performance and reducing leakage current. Moreover, the interface quality in contact with the perovskite nanocrystals (NCs) layer could be improved, which is closely related to the interfacial and long operational stability [51]. These results confirmed effective crosslinking of the PFTPA-biTPA film and good solvent resistance. Furthermore, the insolubility of the crosslinked HTL could improve the interfaces and reduce defects, thereby improving the device performance.
In general, the photo-crosslinkable PFTPA-biTPA can improve the device performance of LED by improving the robustness of HTL. To evaluate the device performance of PeLEDs in the presence or absence of crosslinking of the PFTPA-biTPA, the device was fabricated in the structure shown in Fig. 2a. The multilayered PeLED comprises ITO/PEDOT:PSS/PFTPA-biTPA/CsPbBr3 NCs/TPBi/LiF/Al. The PFTPA-biTPA with HOMO level of − 5.25 eV is introduced between PEDOT:PSS and CsPbBr3 NCs, as shown in the energy level diagrams (Fig. 2b), to facilitate hole injection. Moreover, effective electron blocking is also possible with a high LUMO level (− 2.40 eV) [52, 53].

a Device structure of PeLED. b Energy level diagram of PeLED. c–f Device performance of PeLED. c J–V–L, d EQE, e EL spectra at the peak of EQE, f Operational stability
Perovskite CsPbBr3 NCs were synthesized through the hot-injection synthesis and purification process (see experimental section for details). The cubic phase structure of CsPbBr3 was identified by the X-ray diffraction (XRD) shown in Figure S20(a). The diffraction peaks located at 13.2°, 21.4°, 30.5°, 34.1°, 37.5°, and 43.6° can be assigned to (100), (110), (200), (210), (211), and (220) planes, respectively; thereby, confirming the cubic phase of the CsPbBr3 NCs [54]. The UV–Vis absorption and PL spectra of the CsPbBr3 NCs solution are obtained with the absorption and PL peaks at 500 and 512 nm, as shown in Figure S20(b). The full-width half-maximum (FWHM) in the PL spectrum is approximately 24 nm, thereby indicating a high-purity green emission. The results indicate the excellent properties of the crystalline NCs that were used to fabricate PeLEDs in this study.
The current density and luminance-driving voltage (J–V–L), external quantum efficiency (EQE) versus current density, and electroluminescence (EL) spectra of devices using the pristine PEDOT:PSS (without PFTPA-biTPA) and non-crosslinked and crosslinked PFTPA-biTPA are shown in Fig. 2c–e and Table 1. As shown in the J–V–L characteristic curves of PeLEDs (Fig. 2c), the device using PFTPA-biTPA shows significantly reduced leakage current and lower turn-on voltage of 3.0 V than the device using only the PEDOT:PSS, indicating better electrical contact and more efficient hole injection between the NCs layer and HTL [32]. The device with crosslinked HTL exhibits a lower turn-on voltage of 2.7 V than the device with non-crosslinked HTL. It indicates that the film with crosslinked HTL has a smoother surface, therefore facilitates better contact between the NCs and HTL through crosslinking. The PLQYs of perovskites with HTLs were significantly reduced, which results from exciton quenching due to charge transfer at the interface between the perovskites and HTL, as shown in Figure S21.. In particular, PLQY (12.3%) of perovskite on PFTPA-biTPA:FPA is lower than that on PEDOT:PSS (15.9%). This means that the energy barrier decreases between the PFTPA-biTPA:FPA and perovskite, which makes it more effective to inject hole. On the other hand, PLQY of perovskite with non-crosslinked PFTPA-biTPA shows 20.3% due to the intermixed emission of HTL and perovskite. However, the device with the crosslinked HTL shows lower current density than the device with the non-crosslinked HTL at a high applied voltage, suggesting that the FPA crosslinker is an insulating compound [51]. This phenomenon is demonstrated in Figure S22 and Table S2, where the device performance is presented with respect to the amount of FPA used in crosslinking. The J–V curve shows that the current density decreases with the increasing amount of FPA. Nevertheless, when the amount of FPA increases from 0.3 to 1.5%, the degree of crosslinking increases; consequently, the luminance and EQE values increase. Furthermore, the device performance decreases when 2% FPA is added, indicating that the excess amount of insulating compound significantly lowers the charge injection. Thus, the device using crosslinked HTL with 1.5% of FPA shows slightly improved device performance with the maximum luminance of 175 cd m−2 and the peak EQE of 4.16%.
Figure 2e is the electroluminescence (EL) spectrum of the device using the PFTPA-biTPA. The device with only PEDOT:PSS had too low luminance value to determine the EL spectrum. The EL peaks of the devices using the non-crosslinked and crosslinked PFTPA-biTPA have similar wavelengths, indicating the standard spectrum for CsPbBr3 NCs [55]. Notably, the insolubility of HTL can improve the interface and reduce defects. The FWHMs of the EL peaks for the non-crosslinked and crosslinked HTLs were 22 nm and 20 nm, respectively. It indicates that the interface between the crosslinked HTL and NCs layer has better contact, which was also confirmed by the AFM images (Fig. 1g). Moreover, the shoulder EL peak at about 440 nm indicates that the use of the non-crosslinked HTLs causes the HTLs to be heterogeneously redissolved using the solvent of NCs solution, resulting in an imbalance of carrier injection, thereby causing the recombination at the HTL than the EML (The inset of Fig. 2e) [56, 57]. Consequently, when the half-lifetime (T50) is measured at an initial luminance of ~ 20 cd m−2 (Fig. 2f), the device using the rigid crosslinked HTL shows more than nine times improved operational stability than the device using the non-crosslinked HTL. It suggests that crosslinking of the HTLs in solution-processed multilayer PeLEDs can be the key factor in solving the stability concern, which is the biggest issue in the present.
The PFTPA-biTPA was also introduced into an OLED as an HTL to investigate the device performance using the PFTPA-biTPA and photo-crosslinking method (Fig. 3 and Table 2). The device structure of the OLED comprises ITO/PEDOT:PSS/PFTPA-biTPA/Ir(mphmq)2tmd:TCTA:TPBi/TPBi/LiF/Al (Fig. 3a), which is basically arranged in the same layers as those of PeLEDs except EML. Ir(mphmq)2(tmd) was used as the red phosphorescent dopant; the energy level diagram and molecular structures of the materials used as the EML of the OLED are shown in Figure S23. The device using the crosslinked PFTPA-biTPA shows better device performance than devices using the pristine PEDOT:PSS and non-crosslinked PFTPA-biTPA (Fig. 3b and c). As shown in the J–V curve, the leakage current at low applied voltage is reduced using the device with crosslinked PFTPA-biTPA, which is similar to the results of the PeLED. In addition, the crosslinked PFTPA-biTPA exhibits the highest EQE and maximum luminance of 10.86% and 4,260 cd m−2, respectively. Similar to the results of the PeLED, the photo-crosslinked PFTPA-biTPA as an HTL with improved solvent resistance improved the hole injection, transport capabilities, and device performance of OLED [33].

a Device structure of OLED. b–d Device performance of OLED. b J–V–L, c EQE, d EL spectra at the peak of EQE
3 Conclusion
We demonstrated the effect of photo-crosslinkable PFTPA-biTPA as an HTL of solution-processed PeLEDs. The crosslinkable PFTPA-biTPA was designed for efficient crosslinking reaction by attaching the alkyl chains as crosslinking sites. The crosslinked PFTPA-biTPA film exhibited more than 90% solvent resistance and improved surface morphology and interface contact compared to the non-crosslinked PFTPA-biTPA film. The device based on the crosslinked PFTPA-biTPA with FPA had the luminance and the peak EQE of 175 cd m−2 and 4.16%, respectively, in PeLEDs. A significant improvement in operational stability was observed in the crosslinked HTL. We also introduced the PFTPA-biTPA as an HTL in OLEDs to demonstrate the various possibilities of the photo-crosslinking method and PFTPA-biTPA. The device exhibited the luminance and peak EQE of 4260 cd m−2 and 10.86%, respectively. This photo-crosslinking strategy with the PFTPA-biTPA in various solution-processed LEDs can be the key factor in resolving the stability concern and enhancing the device performance.
4 Experimental section
4.1 Materials
Fluorene, 4-butylbromobeznene, 4-hexylbromobenzene, diphenylamine, chromium(VI) oxide, sodium tert-butoxide, aniline, bromine, potassium acetate, zinc chloride, sulfuric acid, hydrochloric acid, acetic anhydride, triphenylphosphine, tetrabutylammonium bromide, N-bromosuccinimide, hexanoyl chloride, triethylsilane, trifluoroaceticacid, tris(dibenzylideneacetone)dipalladium, tri-tert-butylphosphine, tetrakis(triphenylphosphine) palladium, bis(pinacolato)diboron were purchased from Sigma Aldrich, Alfa Aesar, and Tokyo chemical industry without further purification. Tris[2-(p-tolyl)pyridine]iridium(III) (Ir(mppy)3), iridium(III) bis[4-methyl-2-(3,5-dimethylphenyl)quinolinato-N,C2′] tetramethylheptadionate (Ir(mphmq)2(tmd)), and FPA were synthesized according to reported methods [36]. Cesium carbonate (99.9%), 1-octadecene (ODE, 90% tech.), oleylamine (OLA, 70% tech.), oleic acid (OA, 90% tech.), methylacetate (MeOAc; anhydrous 99.5%), octane (anhydrous, ≥ 99%), toluene (anhydrous 99.8%), hexane (anhydrous 95%), didodecyldimethylammonium bromide (DDAB 98%), and chlorobenzene (99.8%) were purchased from Sigma Aldrich. Lead bromide (PbBr2; 99.999%) was purchased from Alfa Aesar. Indium tin oxide-based transparent conductive electrode (~ 4.5 Ω/sq) purchased from AMG. Aqueous solution of poly(3,4-ethylenedioxythiophene) doped with poly (styrenesulfonic acid) (PEDOT:PSS) (Clevios AI 4083) purchased from Heraeus. Poly[bis(4-phenyl) (4-butylphenyl) amine] (poly-TPD) and 2,2′,2″-(1,3,5-benzinetriyl)tris(1-phenyl-1-H-benzimidazole) (TPBi; 99.9%) were purchased from OSM. LiF (99.9%) and Al (99.9%) were purchased form iTASCO.
4.2 Synthesis of PFTPA-biTPA
4.2.1 Synthesis of 2,7-dibromo-9H-fluorene (1)
Fluorene (2.0 g, 12.0 mmol) was dissolved in dichloromethane (60 ml), and bromine (4.2 g, 26.4 mmol) was added dropwise in 0 °C. Subsequently, the mixture was allowed to warm to room temperature at 30 min, and the mixture was refluxed 12 h. And the reaction mixture was cooled to 0 °C and quenched sodium hydroxide solution. After quenching process, the mixture was extracted with dichloromethane and dried over magnesium sulfate. After removing the solvent under reduced pressure, the crude solid was purified using chloroform and methanol to give a pure product. (3.7 g 94.2%) 1H NMR (CDCl3, 400 MHz, ppm): δ 7.66 (s, 2H), 7.60 (d J = 7.8 Hz, 2H), 7.51 (d, = 7.8 Hz 2H), 3.85 (s, 2H) 13C NMR (CDCl3, 100 MHz, ppm) δ 145.0, 139.9, 130.3, 128.5, 121.3, 121.1, 36.7.
4.2.2 Synthesis of 2,7-dibromo 9H- fluoren-9-one (2)
Compound 1 (3.2 g, 9.8 mmol) was dissolved in acetic anhydride (140 ml) and chromium trioxide (3.0 g, 29.4 mmol) was added dropwise in 0 °C. Subsequently, the mixture was allowed to warm to room temperature for 3 h, the reaction mixture was cooled to ice bath and quenched water. After quenching process, the mixture was filtered by reduced pressure and the crude product was purified by washing with water and isopropyl alcohol to give a product. (2.8 g, 86.2%) 1H NMR (CDCl3, 300 MHz, ppm): δ 7.75 (d, J = 1.8 Hz, 2H), 7.60 (dd J = 8.1 Hz, J = 1.8 Hz 2H), 7.51 (d = 7.8 Hz 2H). 13C NMR (CDCl3, 100 MHz, ppm) δ 191.1, 142.4, 137.6, 135.4, 128.0, 123.5, 122.0.
4.2.3 Synthesis of 4-butyl-N-(4-butylphenyl)-N phenylaniline (3)
Aniline (1.6 g, 17.2 mmol) and tris(dibenzylideneacetone)dipalladium (0.3 g, 3.4 mmol) and sodium tert-butoxide (5.0 g, 51.6 mmol) and 4-bromobutylbenzene (9.2 g, 36.6 mmol), tri-tert-butylphosphine (0.2 g, 10.2 mmol) were dissolved in toluene under N2 atmosphere and refluxed for overnight. After cooling to room temperature, the mixture was extracted with ethyl acetate and dried over magnesium sulfate. The crude product was purified by silica gel column chromatography (dichloromethane/hexane, 1:8 v/v) to obtain product. (4.7 g, 77.1%) 1H NMR (CDCl3, 400 MHz, ppm): δ 7.27–7.23 (m, 2H), 7.12–7.03 (m, 10H), 7.00–6.97 (m, 1H), 2.64 (t, J = 7.6 Hz, 4H), 1.69–1.62 (m, 4H), 1.48–1.32 (m, 4H), 1.04 (t, J = 7.6 Hz 6H). 13C NMR (CDCl3, 100 MHz, ppm) δ 148.3, 145.6, 137.4, 129.1, 129.0, 124.4, 123.1, 121.8. 35.1, 33.8, 22.5, 14.1. MS HRMS (ESI): m/z calcd. for C26H31N [M + H] + 358.2529, found 358.2524.
4.2.4 Synthesis of 4,4′-(2,7-dibromo-9H-fluorene-9,9-diyl) bis(N,N-bis(4-butylphenyl)aniline) (4)
Compound 2 (1.4 g, 4.2 mmol), compound 3 (4.5 g, 12.6 mmol) ,and zinc chloride were dissolved in small amount of dichloromethane. Subsequently, HCl gas injected into the mixture for 2 h and stirred overnight at room temperature. And the reaction mixture was cooled to ice bath and quenched NaOH solution. After quenching process, the mixture extracted with dichloromethane and dried over magnesium sulfate. The crude product was purified by silica gel column chromatography (dichloromethane/hexane, 1:7 v/v) and crude solid was purified using hexane to give a pure product. (2.9 g, 68.1%) 1H NMR (CDCl3, 400 MHz, ppm): δ 7.56 (d, J = 8.0 Hz, 2H), 7.51 (d, J = 1.6 Hz, 2H) 7.47 (dd, J = 8.4 Hz, 1.6 Hz) 7.06–6.94 (m, 20H), 6.87 (d, J = 8.8 Hz, 4H), 2.66 (t, J = 7.6 Hz, 8H), 1.62–1.54 (m, 8H), 1.39–1.33 (m, 8H), 0.94 (t, J = 7.2 Hz, 12H). 13C NMR (CDCl3, 100 MHz, ppm) δ 153.7, 147.2, 145.2, 138.0, 137.8, 136.7, 130.7, 129.4, 129.2, 128.6, 124.9, 121.7, 121.6, 121.5, 64.6, 35.1, 33.7, 22.4, 14.0. MS HRMS (ESI): m/z calcd. for C65H65Br2N2 [M + H] + 1033.3666, found 1033.3681.
4.2.5 Synthesis of 4-hexyl-N,N-diphenylaniline (5)
Diphenylamine (3.0 g, 17.7 mmol) and tris(dibenzylideneacetone)dipalladium (0.3 g, 3.4 mmol) and sodium tert-butoxide (5.1 g, 53.1 mmol) and 4-bromohexylbenzene (5.1 g, 21.2 mmol), tri-tert-butyl phosphine (0.2 g, 10.2 mmol) were dissolved in toluene under N2 atmosphere and refluxed for overnight. After cooling to room temperature, the mixture was extracted with ethyl acetate and dried over magnesium sulfate. The crude product was purified by silica gel column chromatography (dichloromethane/hexane, 1:10 v/v) to obtain product (4.8 g, 82.1%) 1H NMR (CDCl3, 400 MHz, ppm): δ 7.32–7.28 (m, 4H), 7.17–7.03 (m, 10H), 2.66 (t = 7.6 Hz, 2H), 1.77–1.68 (m, 2H) 1.47–1.36 (m, 6H), 1.01–0.97 (m, 3H). 13C NMR (CDCl3, 100 MHz, ppm): δ 148.2, 145.5, 138.0, 129.4, 129.3, 124.9, 123.9, 122.4, 35.6, 31.9, 31.7, 29.3, 22.8, 14.3. MS HRMS (ESI): m/z calcd. for C24H27N [M + H] + 330.2216, found 330.2217.
4.2.6 Synthesis of 4-bromo-N-(4-bromophenyl)-N-(4-hexylphenyl)aniline (6)
Compound 5 (3.0 g, 9.1 mmol) was dissolved in chloroform, and then N-bromosuccinimide (3.7 g, 20.9 mmol) was added dropwise at 0 °C. Subsequently, the mixture was stirred in 0 °C for 3 h. After, the mixture extracted with dichloromethane and dried over magnesium sulfate. The crude product was purified by silica gel column chromatography (dichloromethane/hexane, 1:10 v/v) to obtain product (3.4 g, 75.2%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.36 (d, J = 8.8 Hz, 4H), 7.13 (d, J = 8.8 Hz, 2H), 6.97 (d, J = 8.8 Hz, 2H), 6.94 (d, J = 8.8 Hz, 4H), 2.63 (t, J = 7.6 Hz, 2H), 1.67–1.61 (m, 2H), 1.41–1.34 (m, 6H), 0.97–0.92 (m, 3H). 13C NMR (CDCl3, 100 MHz, ppm): δ 146.9, 144.5, 139.1, 132.4, 129.7, 125.2, 125.1, 115.1, 35.6, 31.9, 31.6, 29.2, 22.8, 14.3. MS HRMS (ESI): m/z calcd. for C24H25Br2 [M + H] + 486.0427, found 486.0415.
4.2.7 Synthesis of 4-hexyl-N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline (7)
Compound 6 (1.7 g, 3.5 mmol), [1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium (II) (127.6 mg, 0.1 mmol), potassium acetate (2.1 g, 21.0 mmol), and bis(pinacolato)diboron (3.5 g, 14.0 mmol) were dissolved in 1,4-dioxane under N2 atmosphere and refluxed for overnight. After cooling to room temperature, the mixture was extracted with dichloromethane and dried over magnesium sulfate. The crude product was purified by silica gel column chromatography (dichloromethane/hexane, 1:2 v/v) to obtain product, crude solid was purified using hexane to give a pure product (1.5 g, 73.8%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.68 (d, J = 8.4 Hz, 4H), 7.10–7.01 (m, 8H), 2.60 (t, J = 7.6 Hz, 2H), 1.64–1.58 (m, 2H), 1.38–1.30 (m, 30H), 0.91–0.88 (m, 3H). 13C NMR (CDCl3, 100 MHz, ppm): δ 150.4, 144.7, 139.2, 136.0, 129.5, 126.0, 122.6, 83.7, 35.6, 31.9, 31.6, 29.2, 25.0, 22.8, 14.3. MS HRMS (ESI): m/z calcd. for C36H49B2NO4 [M + H] + 582.3920, found 582.3948.
4.2.8 Polymerization of PFTPA-biTPA
The desired polymer was synthesized by Suzuki polycondensation. To a mixture of 4-hexyl-N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline (356.0 mg, 0.3 mmol), 4,4'-(2,7-dibromo-9H-fluorene-9,9-diyl)bis(N,N-bis(4-butylphenyl)aniline) (200.0 mg, 0.3 mmol) tetrakis(triphenylphosphine) palladium(0) (27.0 mg), anhydrous toluene (8 ml), K2CO3 2 M solution (8 ml) were added under Ar atmosphere. The solution was stirred at 110 °C for 24 h. To terminate polymerization, a small amount of bromobenzene was added into the solution. After 1 h, a small amount of phenylboronic acid was added into the solution and the reaction mixture was stirred for an additional hour. Subsequently, the solution was poured into a flask containing 200 ml of methanol. The crude polymer was purified by Soxhlet extraction with acetone, methanol, and hexane, sequentially. The crude polymer was collected, dissolved in chloroform, and re-precipitated using methanol. The yield of the polymer was 385.0 mg (91.2%), Mn = 87.8 kg/mol, polydispersity index (PDI) = 6.0, temperature (Td) = 393 °C.
4.3 Synthesis of CsPbBr3 nanocrystals
4.3.1 Cesium-oleate solution
Cs2CO3 (0.407 g), OAc (1.25 ml), and ODE (20 ml) were put into 3-neck flask and degassed under vacuum at 100 °C for 1 h. After degassing, N2 gas was filled into flask and then the temperature was increased to 140 °C.
4.3.2 CsPbBr3 NCs
PbBr2 (0.g), OAc (2.5 ml), OLA (2.5 ml), and ODE (25 ml) were put into 3-neck flask and degassed under vacuum at 100 °C for 1 h. After degassing, N2 gas was filled into flask and then the temperature was increased to 180 °C. When the temperature reached 180 °C, cesium-oleate solution (2 ml) was swiftly injected into the flask. After 10 s, heating mantle was removed and the flask was immersed in an ice bath. The crude solution was collected into centrifuge tubes and centrifuged at 8000 rpm for 10 min. After that, supernatant was discarded and the remaining NCs was dispersed into 2 ml toluene into each tube. MeOAc (6 ml) was added and the NCs were purified by centrifuging at 8,000 rpm for 3 min. The purified NCs was dispersed in 5 ml hexane and filtered with 0.2 μm PTFE syringe filter (Whatman) and stored.
4.3.3 Ligand exchange
The purified NCs, capped with OA and OLA, were dispersed in toluene (8 mg ml−1). OA (100 μl) and DDAB solution (0.05 M in toluene, 200 μl) were added into NCs solution and stirred for 30 min. After ligand exchange, MeOAc (2 ml) was added in exchanged NCs (1 ml) and centrifuged at 8000 rpm for 3 min. The NCs were dispersed in octane for LED fabrication.
4.4 Device fabrication
4.4.1 PeLED
ITO was washed by acetone and isopropanol. After drying in an oven, the ITO substrates were treated with O2 plasma for 10 min. The PEDOT:PSS (AI 4083) was spin coated on ITO at 4,500 rpm for 40 s and annealed at 140 °C for 10 min. The substrates were transferred to an N2-filled glove box. PFTPA-biTPA solution (4 mg ml−1) in chlorobenzene was spin coated on PEDOT:PSS layer at 4,000 rpm for 40 s and annealed at 120 °C for 5 min for non-crosslinked device. To crosslink PFTPA-biTPA, PFTPA-biTPA solution mixed with various amount of FPA was spin coated as in the preceding condition and the film coated with PFTPA-biTPA:FPA was irradiated with UV (254 nm, 84 mJ/cm2) for 10 s. The CsPbBr3 NCs (5 mg ml−1) was spin coated on PFTPA-biTPA layer at 2000 rpm for 30 s. Finally, TPBi (70 nm), LiF (2 nm), and Al (100 nm) were sequentially deposited at ~ 10–6 torr by the thermal evaporation. The area of the electrode defines a 0.135cm2 emission area of the device.
4.4.2 OLED
The PEDOT:PSS (AI 4083) was spin coated onto cleaned ITO at 2,000 rpm for 30 s. PFTPA-biTPA and PFTPA-biTPA:FPA (1.5wt%) were deposited onto the PEDOT: PSS layer. And then coated film was irradiated with UV irradiation. The emissive layer Ir(III) complex in the composition ratio of TCTA:TPBi:Ir(mphmq)2tmd (= 50:50:5wt%) was spin coated and then annealed at 110 °C for 30 min to remove the residual solvent. Finally, TPBi, LiF, and Al were deposited as in the PeLED fabrication.
4.5 Characterization
1H and 13C NMR spectra were recorded on a Bruker, Avance Neo 400 400 MHz spectrometer in CDCl3. UV–Visible absorption spectra were carried out with a Shimadzu UV-3600 spectrophotometer, whereas photoluminescence (PL) spectra were measured on a Hitachi F-7000 spectrophotometer. Cyclic voltammetry (CV) measurements were carried out with a CHI 600D system in acetonitrile solutions containing 0.1 M tetrabutylammonium tetrafluoroborate (TBABF4) as the supporting electrolyte, Ag/AgNO3 as the reference electrode, a platinum wire as the counter electrode, and a platinum working electrode. An Alliance e2695 measurement unit was used to measure molecular weight. Molecular weight was measured using gel-permeation chromatography (with polystyrene standard and chloroform as the eluent, with a Waters Alliance e2695 instrument). The surface morphologies were measured by Park Systems XE-100 as atomic force microscopy (AFM) instrument operated using non-contact scanning mode. XRD patterns were measured using an X’Pert-MPD diffractometer (Philips, Netherlands) employing CuKα radiation. The current density–voltage–luminance (I–V–L) characteristics were measured and electroluminescence (EL) spectra of the PeLEDs and OLEDs were recorded using a Keithley 2400 source measurement unit, CS-2000 and CS-1000 spectrophotometer to evaluate the device performances.
References
B. Ma, F. Lauterwasser, L. Deng, C.S. Zonte, B.J. Kim, J.M. Fréchet, C. Borek, M.E. Thompson, Chem. Mater. 19, 4827 (2007)
S. Park, J.T. Lim, W.-Y. Jin, H. Lee, B.-H. Kwon, N.S. Cho, J.-H. Han, J.-W. Kang, S. Yoo, J.-I. Lee, ACS Photonics 4, 1114 (2017)
Y. Wang, W. Wang, Z. Huang, H. Wang, J. Zhao, J. Yu, D. Ma, J. Mater. Chem. C 6, 7042 (2018)
L. Qian, Y. Zheng, J. Xue, P.H. Holloway, Nat. Photonics 5, 543 (2011)
S.D. Stranks, H.J. Snaith, Nat. Nanotechnol. 10, 391 (2015)
J. Wang, N. Wang, Y. Jin, J. Si, Z.K. Tan, H. Du, L. Cheng, X. Dai, S. Bai, H. He, Z. Ye, M.L. Lai, R.H. Friend, W. Huang, Adv. Mater. 27, 2311 (2015)
J. Li, X. Shan, S.G.R. Bade, T. Geske, Q. Jiang, X. Yang, Z. Yu, J. Phys. Chem. Lett. 7, 4059 (2016)
S.-P. Wang, C.-K. Chang, S.-H. Yang, C.-Y. Chang, Y.-C. Chao, Mater. Res. Express 5, 015037 (2018)
C. Li, N. Wang, A. Guerrero, Y. Zhong, H. Long, Y. Miao, J. Bisquert, J. Wang, S. Huettner, J. Phys. Chem. Lett. 10, 6857 (2019)
Y. Shynkarenko, M.I. Bodnarchuk, C. Bernasconi, Y. Berezovska, V. Verteletskyi, S.T. Ochsenbein, M.V. Kovalenko, ACS Energy Lett. 4, 2703 (2019)
N. Kirkwood, B. Singh, P. Mulvaney, Adv. Mater. Interfaces 3, 1600868 (2016)
Y. Zou, M. Ban, Y. Yang, S. Bai, C. Wu, Y. Han, T. Wu, Y. Tan, Q. Huang, X. Gao, T. Song, Q. Zhang, B. Sun, ACS Appl. Mater. Interfaces 10, 24320 (2018)
Y. Liu, T. Wu, Y. Liu, T. Song, B. Sun, APL Mater. 7, 021102 (2019)
Y.S. Shin, Y.J. Yoon, J. Heo, S. Song, J.W. Kim, S.Y. Park, H.W. Cho, G.H. Kim, J.Y. Kim, ACS Appl. Mater. Interfaces 12, 35740 (2020)
C. Zou, Y. Liu, D.S. Ginger, L.Y. Lin, ACS Nano 14, 6076 (2020)
Z. Yu, W.H. Jeong, K. Kang, H. Song, X. Shen, H. Ahn, S.W. Lee, X. Fan, J.W. Jang, S.R. Ha, J.W. Min, J.H. Park, J. Han, E.D. Jung, M.H. Song, D.W. Chang, W.B. Im, S.H. Park, H. Choi, B.R. Lee, J. Mater. Chem. A 10, 13928 (2022)
Y.H. Kim, H. Cho, J.H. Heo, T.S. Kim, N. Myoung, C.L. Lee, S.H. Im, T.W. Lee, Adv. Mater. 27, 1248 (2015)
C.-Y. Lin, Y.-C. Lin, W.-Y. Hung, K.-T. Wong, R.C. Kwong, S.C. Xia, Y.-H. Chen, C.-I. Wu, J. Mater. Chem. 19, 3618 (2009)
R. Li, L. Cai, Y. Zou, H. Xu, Y. Tan, Y. Wang, J. Li, X. Wang, Y. Li, Y. Qin, D. Liang, T. Song, B. Sun, ACS Appl. Mater. Interfaces 12, 36681 (2020)
W. Sun, L. Xie, X. Guo, W. Su, Q. Zhang, ACS Appl. Mater. Interfaces 12, 58369 (2020)
J. Wang, H. Liu, S. Wu, Y. Jia, H. Yu, X. Li, S. Wang, Chem. Eng. J. 391, 123479 (2020)
J. Zhao, J.A. Bardecker, A.M. Munro, M.S. Liu, Y. Niu, I.-K. Ding, J. Luo, B. Chen, A.K.-Y. Jen, D.S. Ginger, Nano. Lett. 6, 463 (2006)
S.K. Ahn, T. Ban, P. Sakthivel, S.-H. Jin, Y.-S. Gal, J.H. Lee, Macromol. Res. 20, 459 (2012)
C.A. Zuniga, J. Abdallah, W. Haske, Y. Zhang, I. Coropceanu, S. Barlow, B. Kippelen, S.R. Marder, Adv. Mater. 25, 1739 (2013)
C.F. Huang, M.L. Keshtov, F.C. Chen, ACS Appl. Mater. Interfaces 8, 27006 (2016)
F.R.P. Limberg, T. Schneider, S. Höfle, F. Reisbeck, S. Janietz, A. Colsmann, H. Krüger, Adv. Func. Mater. 26, 8505 (2016)
Z. Zhu, D. Zhao, C.-C. Chueh, X. Shi, Z. Li, A.K.Y. Jen, Joule 2, 168 (2018)
C.C. Lin, S.Y. Yeh, W.L. Huang, Y.X. Xu, Y.S. Huang, T.H. Yeh, C.H. Tien, L.C. Chen, Z.L. Tseng, Polym. (Basel) 12, 2243 (2020)
K.W. Tsai, M.K. Hung, Y.H. Mao, S.A. Chen, Adv. Func. Mater. 29, 1901025 (2019)
J. Lee, H. Han, J. Lee, S.C. Yoon, C. Lee, J. Mater. Chem. C 2, 1474 (2014)
J. Park, C. Lee, J. Jung, H. Kang, K.-H. Kim, B. Ma, B.J. Kim, Adv. Func. Mater. 24, 7588 (2014)
Y. Zou, Y. Liu, M. Ban, Q. Huang, T. Sun, Q. Zhang, T. Song, B. Sun, Nanoscale Horiz 2, 156 (2017)
R.Q. Png, P.J. Chia, J.C. Tang, B. Liu, S. Sivaramakrishnan, M. Zhou, S.H. Khong, H.S. Chan, J.H. Burroughes, L.L. Chua, R.H. Friend, P.K. Ho, Nat. Mater. 9, 152 (2010)
G.W. Preston, A.J. Wilson, Chem. Soc. Rev. 42, 3289 (2013)
S.-H. Jeong, S. Ahn, T.-W. Lee, Macromol. Res. 27, 2 (2018)
J.Y. Park, J.H. Jang, J.H. Lee, D.H. Hwang, J. Nanosci. Nanotechnol. 20, 4661 (2020)
C. He, Q. He, X. Yang, G. Wu, C. Yang, F. Bai, Z. Shuai, L. Wang, Y. Li, J. Phys. Chem. C 111, 8661 (2007)
Z. Ge, T. Hayakawa, S. Ando, M. Ueda, T. Akiike, H. Miyamoto, T. Kajita, M.-A. Kakimoto, Adv. Func. Mater. 18, 584 (2008)
W. Xu, B. Peng, J. Chen, M. Liang, F. Cai, J. Phys. Chem. C 112, 874 (2008)
H.T. Lin, C.L. Huang, G.S. Liou, ACS Appl. Mater. Interfaces 11, 11684 (2019)
J.Y. Kim, G. Kwak, Y.C. Choi, D.-H. Kim, Y.S. Han, J. Ind. Eng. Chem. 73, 175 (2019)
H. Hong, M.Y. Jo, Y.E. Ha, J.H. Kim, Macromol. Res. 21, 321 (2013)
M. Schock, S. Brase, Molecules 25, 1009 (2020)
J. Li, L. Xu, T. Wang, J. Song, J. Chen, J. Xue, Y. Dong, B. Cai, Q. Shan, B. Han, H. Zeng, Adv. Mater. 29, 1603885 (2017)
S. Kang, R. Jillella, J. Jeong, Y.I. Park, Y.J. Pu, J. Park, ACS Appl. Mater. Interfaces 12, 51756 (2020)
P. Tao, Y. Li, A. Rungta, A. Viswanath, J. Gao, B.C. Benicewicz, R.W. Siegel, L.S. Schadler, J. Mater. Chem. 21, 18623 (2011)
H. Ha, Y.J. Shim, D.H. Lee, E.Y. Park, I.H. Lee, S.K. Yoon, M.C. Suh, ACS Appl. Mater. Interfaces 13, 21954 (2021)
Z. Yu, S.R. Ha, J.H. Park, E.D. Jung, W.H. Jeong, S. Kim, M.H. Song, J.-M. Kim, S. Lee, S.H. Park, H. Choi, B.R. Lee, J. Power Sources 478, 228810 (2020)
D. Fan, R. Zhang, Y. Li, C. Shan, W. Li, Y. Wang, F. Xu, H. Fan, Z. Sun, X. Li, M. Zhao, A.K.K. Kyaw, G. Li, J. Wang, W. Huang, Front. Chem. 9, 746365 (2021)
Y. Xia, G. Yan, J. Lin, Nanomaterials (Basel) 11, 3119 (2021)
Y.-Q.-Q. Yi, J. Yang, L. Xie, Y. Liu, W. Su, Z. Cui, Chem. Eng. J. 439, 135702 (2022)
H. Shen, W. Cao, N.T. Shewmon, C. Yang, L.S. Li, J. Xue, Nano. Lett. 15, 1211 (2015)
E. Yoon, K.Y. Jang, J. Park, T.W. Lee, Adv. Mater. Interfaces 8, 2001712 (2020)
G. Nedelcu, L. Protesescu, S. Yakunin, M.I. Bodnarchuk, M.J. Grotevent, M.V. Kovalenko, Nano. Lett. 15, 5635 (2015)
H. Yu, G. Tian, W. Xu, S. Wang, H. Zhang, J. Niu, X. Chen, Front. Chem. 6, 381 (2018)
Q. Zheng, F. You, J. Xu, J. Xiong, X. Xue, P. Cai, X. Zhang, H. Wang, B. Wei, L. Wang, Org. Electron. 46, 7 (2017)
Z.P. Yang, H.C. Su, Adv. Func. Mater. 30, 1906788 (2020)
Acknowledgements
This work was supported by a Research Grant of Pukyong National University(2021).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Park, J.Y., Jang, J.W., Shen, X. et al. Fluorene- and arylamine-based photo-crosslinkable hole transporting polymer for solution-processed perovskite and organic light-emitting diodes. Macromol. Res. 31, 721–732 (2023). https://doi.org/10.1007/s13233-023-00151-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13233-023-00151-8