
Abstract
Catechol 1, 2-dioxygenase (1, 2-CTD) plays an important role in the biodegradation of most aromatic pollutants (e.g. phenol) by cleaving the bond between the phenolic hydroxyl groups. In this study, we analysed microbial populations in petroleum-contaminated soil samples by the culture-dependent approach and chose one population as a sample location for metagenomic studies. This population showed the largest number of phenol-degrading isolates and the most phylogenetic diversity based on repetitive sequence-based PCR and 16S rDNA analyses. The isolates were assigned mainly to the bacterial genera Acinetobacter and Pseudomonas. To enable maximum recovery of 1, 2-CTD coding sequence from the genomic pool of bacterial populations, we extracted the substrate-induced RNA, reverse-transcribed it to the specific cDNA using primers designed for this purpose and constructed a metagenomic library. Alignment analysis of one recombinant sequence indicated a significant divergence, with the 1, 2-CTD gene of Pseudomonas aeruginosa DK2 as the closest species according to sequence similarity. This result indicates that such specifically designed primers are likely suitable candidates as biomarkers to construct 1, 2-CTD gene-targeted metagenomic libraries. By sequencing the larger clones of this metagenomic library, we hope to obtain novel and unpredicted 1, 2-CTD genes that can contribute to our knowledge of microbial diversity of these genes.
Similar content being viewed by others


Introduction
Access to high-quality microbial strains, both wild types and genetically engineered strains, is a major advantage for ex situ and in situ bioremediation strategies for the clean-up of contaminated environments (Mrozik and Piotrowska-Seget 2010). The potential for biodegradation by bacterial populations present in petroleum hydrocarbon-contaminated soils can be assessed by detecting their catabolic genes. These genes encode multi-component enzyme systems capable of removing aromatic pollutants from contaminated environments (Gibson and Parales 2000; Geng et al. 2006; Silva et al. 2013).
Despite the relatively broad diversity in aerobic bacterial aromatic compound degradation pathways, these catabolic pathways generally have a common feature, namely, catecholic intermediates. Catechol is a major ring cleavage substrate through which an exceptionally large number of peripheral pathways converge. This has led to extensive investigation of enzymes involved in catechol oxygenation reactions. In general, catechol is degraded either by catechol 1, 2-dioxygenase (1, 2-CTD; EC number 1.13.11.1) or catechol 2, 3 dioxygenase via the Ortho and Meta pathways, respectively (Chen et al. 2003; Uhlik et al. 2009). Catechol dioxygenases (CTD) belong to the aromatic dioxygenase family, a class of mononuclear non-heme iron intradiol-cleaving enzymes that catalyse the oxygenation of catecholates to aliphatic acids via the cleavage of aromatic rings (Kumar et al. 2005).
The study of gene diversity in nature can reveal the ecological, functional and evolutionary patterns of key genes (Rondon et al. 2000; Manickam et al. 2010). However, many studies have shown that a few variations in amino acids can essentially affect the structure and function of individual enzymes, resulting in changes in their substrate specificity and catalytic activities (Parales et al. 2000; Suenaga et al. 2002; Bagneris et al. 2005; Vardar and Wood 2005). The choice of degenerate primers designed from the conserved region of the targeted gene would enable sufficient coverage of a gene family (Sei et al. 1999; Tancsics et al. 2008; Morimoto and Fujii 2009). A degenerate PCR primer has several possible bases at certain positions in a sequence, and the degeneracy of the primer is the number of unique sequence combinations it contains (Linhart and Shamir 2005). Iwai et al. (2010) tested this approach on non-heme iron dioxygenase genes known to play important roles in pollutant biodegradation. Their results show a much greater diversity among genes potentially critical in nature’s carbon cycle than among those detected earlier.
The diversity of extradiol dioxygenases has also been analysed by Brennerova et al. (2009) and Suenaga et al. (2014), followed by the characterization of gene polymorphisms in contaminated environments. Their results indicated a broad diversity of catabolic genes expressing catechol 2, 3 dioxygenases, determined by pre-designed primers based on culture-dependent studies that targeted eight main groups of these genes for subsequent PCR screening of fosmid clones exhibiting extradiol dioxygenase activity (Brennerova et al. 2009).
In addition to ring-hydroxylating oxygenase coding sequences, a great number of genes are being added to the list of gene markers required for isolating and identifying microorganisms which degrade aromatic compounds in soil and water. Among these are bacterial genes which direct recognition and chemotaxis towards these compounds (Grimm and Harwood 1999) and catabolic gene clusters (Khara et al. 2014). Application of the metagenomics approach for the discovery of local wild-type microorganisms capable of degrading aromatic contaminants is important in terms of estimating the possibility of applying autochthonous bioaugmentation as one of the feasible and environment-friendly procedures in bioremediation (Jorgensen 2008; Hosokawa et al. 2009; Mrozik and Piotrowska-Seget 2010).
Metagenomic approaches have been useful in retrieving genes involved in biodegradation from microbial communities in polluted areas (Eyers et al. 2004; Witzig et al. 2006; Cowan and Stafford 2007; Stenuit et al. 2008; Fernández-Arrojo et al. 2010; Silva et al. 2013). Metagenomics was first proposed by Handelsman et al. (1998), and today, in order to cover the extant diversity, gene-targeted metagenomics (GT-metagenomics) is currently a method of choice for PCR-based targeting (Warnecke and Hugenholtz 2007; Mocali and Bendetti 2010). This approach can not only provide insight into microbial gene diversity but also provide means to detect genes that could be important in environmental processes, as well as shedding light on their ecology, functional differences and evolutionary origins (Iwai et al. 2010, 2011). One of the most important factors in metagenomic analysis is the selection of an appropriate location, which is initially determined by culture-dependent methods, in order to obtain maximum versatility and variability of the target gene sequence and to reduce the costs (time, manpower, financial resources) during the process (Steele and Streit 2005; Uhlik et al. 2009; Prakash and Taylor 2012).
The aims of this study were to investigate the diversity and number of isolated bacteria involved in phenol degradation in soil samples collected at different locations (sampling stations) in an oil-contaminated area using repetitive extragenic palindromic sequence-based PCR (Rep-PCR) and 16S RNA sequence analyses and to design degenerate primers as biomarkers to amplify catechol-induced mRNA from the expressed cognate genes . The position and direction of metagenomics was also modelled to recover the 1, 2-CTD gene from a diverse bacterial population. We designed the degenerate primers based on sequences from the conserved regions of the 1, 2-CTD-encoding gene and covered the entire subfamily.
Material and methods
Sampling and media
For the isolation of phenol-degrading bacteria, we collected oil-contaminated soil samples from various locations (sampling stations) of the Tehran Oil Refinery (Tehran Province, Iran). Soil was horizontally sampled (50 g) at nine stations and transported in nylon bags immediately to the laboratory on ice. The soil temperature, pH value and relative humidity were also analysed according to the standard method of (Webster 2008) (results not shown). The synthetic phenol broth medium used was based on the mineral salts medium (MSM) [g/l: KH2PO4, 2.25; K2HPO4, 2.25; MgCl2.6H2O, 0.2; NaCl, 0.1; FeCl3.6H2O, 0.02; (NH4)2SO4, 1; bromothymol blue, 0.06; CaCl2, 0.01; pH 6.8–7]. These synthetic media were supplemented with phenol as the sole carbon and energy sources (MP) (Watanabe et al. 1998; Enan 2006) and with 15 g/l agar to obtain phenol agar medium. All media were sterilized at 121 °C for 15 min (all chemicals were obtained from Merck, Erlangen, Germany, including phenol which was chromatography grade purity 99.5 %).
Isolation and selection of phenol-degrading bacteria
An aliquot (1 g) of each soil sample was inoculated into phenol broth medium and after 7 days of culture, 5 ml of this medium was sub-cultured into fresh phenol broth media. Following four successive sub-cultures into phenol broth media, drops from the broth cultures were transferred onto solid phenol medium, and colonies growing on this medium were subsequently isolated and purified. The phenotypic characteristics of the isolates were also investigated by basic biochemical tests (results not shown; Boone et al. 2005). The growth rate of the isolates was routinely assessed indirectly by making turbidity measurements at 600 nm using a UV-visible spectrophotometer (model UV-1601PC; Shimadzu, Kyoto, Japan) every 12 h, up to 72 h. Experiments were carried out in 250-ml flasks, each containing 50 ml of MP (500 mg phenol l−1), and the test media was inoculated with 100 μl of a 24-h culture grown in nutrient broth. Cultures were then incubated with shaking at 30 °C. Isolates were also inoculated into basal MP without the substrate, which served as the control experiment. Cell diversity and pH were also measured in the presence of 1000 and 2000 mg l−1 phenol (as the sole carbon and energy sources) over a 72-h period.
Molecular studies on phenol-degrading bacteria
Chromosomal DNA was extracted from the bacterial isolates using the SET buffer method, and the quality and quantity of the extracted DNA were checked by measuring the UV absorption (Sambrook and Russel 2001). Pure DNA was used as the template in PCR reactions to study genetic diversity and identify bacterial isolates. Genomic fingerprints of the isolates were obtained by Rep-PCR performed with primers REP1R-I (5′-CGICGICATCIGGC-3′) and REP2-I (5′-ICGICTTATCIGGCCTAC-3′) (Louws et al. 1994). The PCR reaction mixture contained 75 ng template DNA, 20 mM of each forward and reverse primer, 0.1 mM dNTPs, 1 mM Mg Cl2, 1× Taq Polymerase buffer, 1.25 U Taq Polymerase (CinnaGen, Tehran, Iran) and ultrapure water to a volume of 25 μl. The PCR amplification regimen was 1 cycle of 5 min at 94 °C, 30 cycles of 1 min at 94 °C, 1 min at 45 °C and 1 min at 72 °C, with a final extension cycle at 72 °C for 10 min. This was followed by final incubation at 4 °C after which 10 μl of each amplified PCR product was separated by electrophoresis on a 1.5 % agarose gel in 0.5× TBE buffer (Liang et al. 2010).
The 16S rRNA gene of the isolates were amplified from the genomic DNA by PCR using universal primers 27 F (5′-GTTTGATCCTGGCTCAG-3′) and 1492R (5′-TACGGTTACCTTGTTACGACTT-3′) (Heuer et al. 1997). The reaction mixture (25 μl) contained 75 ng of DNA extract, 0.5 μM of primers, 1× PCR buffer, 0.2 mM of each dNTP, 1.6 mM MgCl2, 1.25U Taq DNA polymerase and ultrapure water. The PCR amplification regimen was 1 cycle of 5 min at 95 °C, 30 cycles of 1 min at 94 °C, 1 min at 61 °C and 1 min at 72 °C, with an additional cycle of 5 min at 72 °C. The PCR products were then sequenced, and the phylogenic tree was drawn using MEGA 5 software (Tamura et al. 2011).
Design of degenerate primers and PCR conditions
The nucleic acid sequences were introduced to the GeneFisher server (GeneFisher ver. 2.0; BiBiServ, Bielefeld University) to be aligned by DIALIGN or ClustalW subsequent to which degenerate primers were designed (Giegerich et al. 1996). The primer pairs’ INDO-S-F (5′-CRCGHACCATYGAMGG −3′) and INDO-S-R (5′-TTKATYTKSGTRGKTCAG-3′) were selected following alignment of several conserved DNA sequences of the 1, 2-CTD gene. PCR amplification under these conditions should result in an amplicon in the size range of 400–430 bp. In another approach, the amino acid sequences were multialigned, and conserved regions of EGPLYV and PAHIHY were determined at the – and C-terminal sites, respectively, in the catechol intradiol dioxygenase family. The degenerate primer pairs’ INDO-U-F (5′-GARGGTCCWYTRTAYGTKG-3′) and INDO-U-R (5′ARTGYAYRTGKGCTGGRC-3′) were designed by the GeneFisher server and BioToolKit software (BioToolKit 300; Primer Degenerate ver. 3.4, Chang Bioscience, Inc.) based on the codon usage method to produce amplicons in the size range of 360–380 bp. The melting temperature (Tm) for degenerate primer oligos was calculated using an enhanced nearest neighbour (NN) approach (SantaLucia 1998). The optimal annealing temperature was determined by setting a gradient of 57 °C to 63 °C. The annealing temperature of the INDO-S/INDO-U primer pairs was determined to be 58 °C/60 °C. The following cycling parameters were selected: 1 cycle of 5 min at 95 °C, 30 cycles of 1 min at 94 °C, 1 min at 58 °C or 60 °C and 1 min at 72 °C, with an additional 5-min cycle at 72 °C.
Touchdown PCR was carried out using a cycling programme in which the annealing temperature was gradually reduced (e.g. 1–2 °C /every second cycle). The initial annealing temperature was several degrees above the estimated Tm of the primers (65 °C), and then it was gradually decreased (10 cycles) until it reached the calculated annealing temperature of the primers or some degrees below (52 °C). Amplification was continued using this annealing temperature (20 cycles).
Soil sampling and metagenomic RNA extraction
The equipment was treated with ethanol, bleach and sterile water to prevent contamination between sampling runs (Kakirde et al. 2010). Following sampling, it was critical to place samples on ice and process them as quickly as possible. The samples were immediately inoculated into mineral medium containing 500 mg l−1 phenol and incubated at 30 °C for 7 days. This step was carried out in triplicate. For the extraction of environmental RNA, the pre-culture was prepared by incubating microorganisms in 5 ml of PMS medium at 30 °C overnight. This culture was inoculated into 50 ml of PMS medium and grown for 12 h at 30 °C, to an absorbance of 0.45–0.60 at 600 nm. The cells were centrifuged at 3400 g for 10 min at 4 °C and washed with 300 μl saline. The pellet was re-suspended in 300 μl of 1 mg lysozyme ml−1 Tris-EDTA buffer (TE; 10 mM Tris, 1 mM EDTA, pH 8.0) and sonicated for 15 s (Folsom et al. 2010). Total RNA was isolated using the RNX-Plus kit (CinnaGen, Tehran, Iran). RNA concentrations and purity were determined by measuring the absorbance at 260, 280 and 230 nm using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE).
Synthesis of GT-metagenomic cDNA and library construction
The reverse transcription (RT) reaction was carried out using the Expand Reverse Transcriptase kit (Roche, Mannheim, Germany) and the INDO-U-R primer. The cDNA was then used as template in PCR analyses using INDO-U-R and Oligo dG primers after a pre-denaturation stage at 94 °C for 3 min, 30 cycles. The cDNA fragments were gel-purified with a DNA Gel Extraction kit (Fermentase, Pittsburgh, PA). The metagenomic library was constructed by TA cloning of PCR amplified community cDNA, one of the most widely used techniques in bacterial community analysis. To this end, the cDNA fragments were subjected to agarose gel electrophoresis, following which 1.5- to 3-kbp fragments were extracted from the gel. Finally, the fragments were subjected to overnight ligation with 50 ng of a TA vector (pGEMR-T Vector Systems; Promega, Madison, WI) at 4 °C (Newton and Graham 1996). The resultant product was transformed to competent Escherichia coli (DH 5α) cells by heat shock, and the recombinant plasmid carrying the insert was then purified for further subcloning and characterization. One recombinant colony was selected randomly, and plasmid DNA was purified by the alkaline lyses method followed by sequencing (Microgen Inc., Seoul, South Korea).
Results
The location and sampling methods were determined by investigating the variability and diversity of the targeted population at each sampling station. The geographical locations of the sampling stations are shown in Fig. 1. Following enrichment and purification of the samples, 68 isolates were screened on medium containing 100 mg l−1 of phenol. A total of 22 isolates were obtained from station 6.

Geographical location of the sampling stations. Symbols + numbers Sampling stations, filled triangle sampling station 6. DD Decimal degrees
The isolates were subsequently cultured on medium containing a higher amount of phenol (500 mg l−1), resulting in the growth of only 28 isolates in the medium. Among these, 12 isolates from station 6 were renamed as GSN8, GSN10, GSN11, GSN13, GSN17, GSN18, GSN19, GSN21, GSN22, GSN25; GSN26 and GSN28. Figure 2 shows the growth kinetics and variations in cell density of 21 bacterial isolates cultured in broth containing 500 mg phenol l−1 at 600 nm over a 72-h period. The GSN15 and GSN11 isolates showed minimum and maximum growth, respectively, based on absorbency measurements, and all other isolates fell within this range. Cell density and pH were also measured in medium containing 1000 and 2000 mg phenol l−1 after 72 h as indices of growth capabilities. Comparison between measurements of the original medium at time of initial inoculum (pH 7) and those as later time-points showed that the absorbance increased from >0.05 to 1.54 (wavelength 600 nm) (Table 1).

Time course of growth of 21 bacterial species in culture broth according to spectrophotometric measurement of absorbency. The GSN15 (blue line, filled circles) and GSN11 (red line, filled triangles) represent minimum and maximum growth, respectively; the growth profiles of all other isolates (in grey, no symbol) fall in between this maximum–minimum range
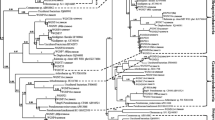
PCR products were separated by agarose gel electrophoresis, revealing species-specific patterns (Fig. 3). These patterns were analysed by NTSYSpc 2.02e and a dendrogram was generated (Fig. 4). The isolates with similar patterns (i.e. belonging to the same species) clustered together, and 16 various isolates with >50 % coefficient of similarity were selected for further study. The most variable isolates were obtained from sampling station 6, which demonstrates the potential of this location for further investigation of biodiversity. Identification was achieved by 16S rDNA sequencing for a limited number of isolates from each group within the cluster. The phylogenic relationship was generated based on the 16S rDNA sequences of the phenol-degrading strains (Fig. 5).

Repetitive extragenic palindromic sequence-based PCR (Rep-PCR) genomic fingerprints of 25 isolates by the Rep-primer set. Lanes: 1 GSN8, 2 GSN6, 3 GSN1, 4 GSN2, 5 GSN22, 6 GSN4, 7 GSN23, 8 GSN3, 9 GSN20, 10 GSN14, 11 GSN10, 12 GSN15, 13 GSN26, 14 GSN16, 15 GSN9, 16 GSN12, 17 GSN24, 18 GSN28, 19 GSN21, 20 GSN25, 21 GSN5, 22 GSN11, 23 GSN13, 24 GSN7, 25 GSN17, M 100-bp marker

Dendrogram obtained by Rep-PCR fingerprints of the phenol-degrading bacteria isolated. The dendrogram is based on Dice's coefficient of similarity with the unweighed pair group method with arithmetic average clustering algorithm (UPGMA)

Phylogenetic analysis based on the 16S rDNA sequences of the phenol-degrading isolates. Neighbour-joining model is employed for the tree construction. Bootstrap values obtained with 1000 repetitions are indicated as percentages at the nodes and >50 % similarity in sequences are shown
PCR products in the size range of 400–430 bp were obtained by INDO-S primers only for isolates GSN9, GSN12, GSN4, GSN11, GSN22, and GSN23 (results not shown). However, as expected, primer pair INDO-Us amplified fragments of 360–380 bp in all isolates (Fig. 6). Most of the nonspecific bands were eliminated by Touchdown-PCR; Fig. 7a shows the desired bond amplified by Touchdown-gradient PCR in the selected GSN28 isolate. To recover the 1, 2-CTD gene from catechol intradiol dioxygenases, gene expression was induced by the specific substrate, catechol. The amplified cDNA of the 1, 2-CTD gene shown in Fig. 7b is from the selected GSN28 isolate.

DNA fragments of different lengths (360–380 bp) were amplified by the INDO-U primer pair. Lanes 1–7 GSN3, GSN8, GSN15, GSN20, GSN23, GSN4, GSN16; lanes 8–14 GSN9, GSN13, GSN12, GSN5, GSN21, GSN28, GSN22; rightmost lane 100-bp DNA marker

Molecular analysis of GSN28 isolate. a 360-bp fragment was produced by Touchdown-gradient PCR using the INDO-U-F and INDO-U-R primer pair in the GSN28 isolate. b The specific cDNA amplified by reverse transcription-PCR by the INDO-U-R and Oligo dG primers. Lane M 100-bp DNA marker
The sequence analysis indicated 93 % similarity to the 1, 2-CTD of Pseudomonas stutzeri DSM4166; the gene sequence was submitted as GenBank accession number KF921490.
Station 6 was chosen as the sample location for metagenomic studies. Microbial cells were recovered from contaminated soil prior to lysis, which resulted in the reduction or removal of inhibitors such as humic acid, phenolic polymers, polysaccharides and nucleases (Malik et al. 2008; Delmont et al. 2011a, b). In addition, the lysis treatment meant that there were fewer non-specific bacterial genomes for further analysis.
After early enrichment of soil microorganisms, specific cDNA fragments were produced by RT-PCR of environmental bacterial RNA by the INDO-U-R and Oligo dG primers (Fig. 8a). GT-metagenomic cDNA , purified from the agarose gel (Fig. 8b) and showing relative homogeneity in size, was used for to construct the GT-metagenomic library.

Total environmental cDNA amplification: a The 500-bp environmental cDNA of catechol 1, 2-dioxygenase was amplified by reverse transcription-PCR. b Specific environmental cDNA was extracted from the agarose gel. Lane M 100 bp DNA marker
One recombinant plasmid was randomly selected, and sequenced. The sequence was subsequently aligned with 466 nucleotides of the 1, 2-CTD gene of Pseudomonas aeruginosa DK2 by BLAST and was deposited in GenBank under accession number KF921491.
Discussion
In this study, degenerate primers were designed as a marker for GT-metagenomics selection of 1, 2-CTD sequences in microbial populations isolated from oil-contaminated environments. To this end, the use of culture-dependent approaches assisted in the initial recovery of phenol-degrading microorganisms. Rep-PCR, which is a technique ideally suitable for the rapid grouping of microorganisms, was performed to detect the more diverse bacterial populations; this was followed by 16 s rDNA sequence analysis to identify the bacterial isolates. Lisek et al. (2014) have shown Rep-PCR can efficiently differentiate isolates within clusters. Several of the microorganisms which were able to degrade phenolic compounds in this study were assigned to the bacterial genera Acinetobacter and Pseudomonas. Previous research has shown the capacities of immobilized Acinetobacter cells to remove phenol from wastewater (Jiang et al. 2013). Our ongoing study also indicates that one of the Acinetobacter sp. identified here is capable of both biofilm formation as well as phenol degradation in a moving bed biofilm bioreactor (unpublished data).
The metabolic pathways and genes involved in the degradation of aromatic compounds have been characterized in many bacterial strains, predominantly Proteobacteria and Actinobacteria (Brennerova et al. 2009; Silva et al. 2013). To investigate the potential of novel catabolic genes from unculturable microorganisms in these diverse populations, we analysed the requirements for degenerate primer design and tested their functionality to model the direction of GT-metagenomics. The particular region of the gene selected for amplification, coverage percentage of the target gene family and the specificity of primer sets are criteria that need to be taken into consideration when designing PCR primers to indicate gene populations in a community. Since we designed the INDO-S by aligning a limited number of sequences, PCR products were obtained for only some isolates. However, the INDO-U primer pairs were designed using the amino acid sequence of the gene family and subsequently amplified various expected fragments in all isolates. The catechol intradiol dioxygenase family can be divided into several subgroups in accordance with their substrate specificity for catechol, chlorocatechols and hydroxyquinols. These substances contain conserved regions PAHIHY and EGPLYV, from which primer pair INDO-U was designed. The 1, 2-CTD gene is expressed by a specific substrate, catechol, and therefore, following induction, RNA amplification into cDNA by RT-PCR using the INDO-U-R and Oligo dG primers allowed the construction of a GT-metagenomic library for a specific functional activity. The relative abundance of targeted microbial taxa was monitored during the enrichment process and during construction of the metagenomic library. Analysis of one recombinant sequence from the library, aligned with that of Pseudomonas aeruginosa DK2, indicated one gap and 60 mismatches, with a 87 % similarity between query and subject sequences. It seems likely that designed primers are appropriate biomarkers to construct the 1, 2-CTD gene-targeted metagenomic libraries. By sequencing the larger clones of this metagenomic library, we hope to obtain novel and unpredicted 1, 2-CTD genes which can contribute to our knowledge of the biodegradion capacity and pathways of microbial populations present in contaminated environments.
We believe that the coupling of classical enrichment processes to modern metagenomic technologies is an efficient approach for the isolation of a large number of novel biomolecules and target genes from soil samples. This strategy has been instrumental in the recovery of several hundreds of industrially important genes that have been utilized in the construction of novel gene banks (Daniel 2004, 2005; Lorenz and Eck 2005; Handelsman 2007; Fernández-Arrojo et al. 2010; Hirsch et al. 2010; Silva et al. 2013). These catalytic gene banks or the corresponding clones can serve as starting material for the development of novel processes as well as the bioremediation of organic compounds.
References
Bagneris C, Cammack R, Mason R (2005) Subtle difference between benzene and toluene dioxygenases of Pseudomonas putida. Appl Environ Microbiol 71:1570–1580
Boone DR, Castenholz RW, Garrity GM, Brenner DJ, Krieg NR, Staley JT (2005) Bergey's manual of systematic bacteriology. Springer Science & Business Media, New York
Brennerova M, Josefiova J, Brenner V, Pieper D (2009) Metagenomics reveals diversity and abundance of meta-cleavage pathways in microbial communities from soil highly contaminated with jet fuel under air-sparging bioremediation. Environ Microbiol 11:2216–2227
Chen Y, Liu H, Chen H (2003) Characterization of phenol biodegradation by Comamonas testosteroni zd4-1 and Pseudomonas aeruginosa zd4-3. Biomed Environ Sci 16:163–172
Committee on Metagenomics: Challenges and Functional Applications (2007) The new science of metagenomics: revealing the secrets of our microbial planet, 1st edn. National Academies Press, Washington D.C.
Cowan D, Stafford W (2007) Metagenomic methods for the identification of active micro-organisms and genes in biodegradation processes. Univ West Cape Res Repository 3:1–19
Daniel R (2004) The soil metagenome: a rich resource for the discovery of novel natural products. Curr Opin Biotechnol 15:199–204
Daniel R (2005) The metagenomics of soil. Nat Rev Microbiol 3:470–478
Delmont T, Clark I, Simonet P, Vogel T (2011a) Metagenomic comparison of direct and indirect soil DNA extraction approaches. J Microbiol Methods 86:397–400
Delmont T, Robe P, Cecillon S, Clark I, Constancias F (2011b) Accessing the soil metagenome for studies of microbial diversity. Appl Environ Microbiol 77:1315–1324
Enan R (2006) Isolation and molecular characterization of phenol degrading Gram-positive bacteria from oil contaminated soil. Res J Microbiol 4:307–318
Eyers L, Schuler L, Stenuit B, Agathos N, Fantroussi S (2004) Environmental genomics: exploring the unmined richness of microbes to degrade xenobiotics. Appl Microbiol Biotechnol 66:123–130
Fernández-Arrojo L, Guazzaroni ME, López-Cortés N, Beloqui A, Ferrer M (2010) Metagenomic era for biocatalyst identification. Curr Opin Biotechnol 21:725–733
Folsom J, Richards L, Pitts B, Roe F, Ehrlich G, Parker A, Mazurie A, Stewart P (2010) Physiology of Pseudomonas aeruginosa in biofilms as revealed by transcriptome analysis. BMC Microbiol 10:294–301
Geng A, Soh A, Lim C, Loke L (2006) Isolation and characterization of phenol degrading bacterium from an industrial active sludge. Appl Microbiol Cell Physiol 71:728–735
Gibson D, Parales R (2000) Aromatic hydrocarbon dioxygenases in environmental biotechnology. Curr Opin Biotechnol 11:236–243
Giegerich R, Meyer F, Schleiermacher C (1996) GeneFisher software support for the detection of postulated genes. Proc Int Conf Intell Syst Mol Biol 4:68–77
Grimm A, Harwood C (1999) NahY a catabolic plasmid encoded receptor required for chemotaxis of Pseudomonas putida to the aromatic hydrocarbon naphthalene. J Bacteriol 181:3310–3316
Handelsman J, Rondon R, Brady F, Clardy J, Goodman R (1998) Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol 5:245–249
Heuer H, Krsek M, Baker P, Smalla K, Wellington M (1997) Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl Environ Microbiol 63:3233–3241
Hirsch R, Mauchline H, Clark M (2010) Culture independent molecular techniques for soil microbial ecology. Soil Biol Biochem 42:878–887
Hosokawa R, Nagai M, Morikawa M, Okuyama H (2009) Autochthonous bioaugmentation and its possible application to oil spills. World J Microbiol Biotechnol 25:1519–1528
Iwai S, Chai B, Sul W, Cole J, Hashsham J, Tiedje J (2010) Gene-targeted-metagenomics reveals extensive diversity of aromatic dioxygenase genes in the environment. ISME J 4:279–285
Iwai S, Chai B, Jesus E, Penton R, Lee T, Cole J, Tiedje J (2011) In: de Bruijn FJ (ed) Handbook of molecular microbial ecology 1: metagenomics and complementary approaches. Wiley Blackwell, Hoboken, pp 235–243
Jiang L, Ruan Q, Li R, Li T (2013) Biodegradation of phenol by using free and immobilized cells of Acinetobacter sp. BS8Y J Basic Microbiol 53(3):224–30
Jorgensen S (2008) Advances in monitoring of catabolic genes during bioremediation. Indian J Microbiol 48:152–155
Kakirde K, Parsley L, Liles M (2010) Size does matter: application-driven approaches for soil metagenomics. Soil Biol Biochem 42:1911–1923
Khara P, Roy M, Chakraborty J, Ghosal D, Dutta K (2014) Functional characterization of diverse ring-hydroxylating oxygenases and induction of complex aromatic catabolic gene clusters in Sphingobium sp. PNB. FEBS Open Biol 4:290–300
Kumar A, Kumar S, Kuma S (2005) Biodegradation kinetics of phenol and catechol using Pseudomonas putida MTCC 1194. Biochem Eng J 22:151–159
Liang R, Wu X, Dai Q, Jin D, Wang Y (2010) Genetic diversity of phthalic acid esters degrading bacteria isolated from different geographical regions of China. Antonie Van Leeuwenhoek 97:79–89
Linhart C, Shamir R (2005) The degenerate primer design problem: theory and applications. Bioinformatics 18:172–180
Lisek A, Sas Paszt L, Trzciński P (2014) Identification and assessment of genetic similarity of soil bacterial isolates of Pseudomonas spp. using molecular techniques. Pol J Microbiol 63(3):291–8
Lorenz P, Eck J (2005) Metagenomics and industrial applications. Nat Rev Microbiol 3:510–516
Louws FJ, Fulbright DW, Stephens CT, de Bruijn FJ (1994) Specific genomic fingerprints of phytopathogenic Xanthomonas and Pseudomonas pathovars and strains generated with repetitive sequences and PCR. Appl Environ Microbiol 60(7):2286–95
Malik S, Beer M, Megharaj M (2008) The use of molecular techniques to characterize the microbial communities in contaminated soil and water. Environ Int 34:265–276
Manickam N, Pathak A, Saini S, Mayilraj S, Shanker R (2010) Metabolic profiles and phylogenetic diversity of microbial communities from chlorinated pesticides contaminated sites of different geographical habitats of India. J Appl Microbiol 109:1458–1468
Mocali S, Bendetti A (2010) Exploring research frontiers in microbiology: the challenge of metagenomics in soil microbiology. Res Microbiol 161:497–505
Morimoto S, Fujii T (2009) A new approach to retrieve full lengths of functional genes from soil by PCR-DGGE and metagenome walking. Appl Microbiol Biotechnol 83:389–396
Mrozik A, Piotrowska-Seget Z (2010) Bioaugmentation as a strategy for cleaning up of soils contaminated with aromatic compounds. Microbiol Res 165:363–375
Newton CR, Graham A (1996) PCR: introduction to biotechniques. 2nd edn. Bios Scientific Publishers, Milton Park
Parales E, Lee K, Resnick S, Jiang H, Lessner D, Gibson D (2000) Substrate specificity of naphthalene dioxygenase: effect of specific amino acids at the active site of the enzyme. J Bacteriol 182:1641–1649
Prakash T, Taylor T (2012) Functional assignment of metagenomic data: challenges and applications. Brief Bioinform 10:1–17
Rondon M, August P, Bettermann A, Brady S (2000) Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microbiol 6:2541–2547
Sambrook J, Russel D (2001) Molecular cloning. 3rd edn. Cold Spring Harbor Laboratory Press, New York
SantaLucia J (1998) A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci USA 95:1460–1465
Sei K, Asano K, Tateishi N, Mori K, Ike M, Fujita M (1999) Design of PCR primers and gene probes for the general detection of bacterial populations capable of degrading aromatic compounds via catechol cleavage pathways". J Biosci Bioeng 88:542–550
Silva C, Hayden H, Sawbridge T, Mele P, Paula S, Silva L, Vidigal P, Vicentini R, Sousa M, Torres A, Santiago M, Oliveira V (2013) Identification of genes and pathways related to phenol degradation in metagenomic libraries from petroleum refinery wastewater. PLoS One 4:1–11
Steele L, Streit R (2005) Metagenomics: advances in ecology and biotechnology. FEMS Microbiol Lett 247:105–111
Stenuit B, Eyers L, Schuler L, Agathos S, George I (2008) Emerging high-throughput approaches to analyse bioremediation of sites contaminated with hazardous and/or recalcitrant wastes. Biotechnol Adv 26:561–575
Suenaga H, Watanabe T, Sato M, Furukawa K (2002) Alteration of regiospecificity in biphenyl dioxygenase by active-site engineering. J Bacteriol 184:3682–3688
Suenaga H, Mizuta S, Miyazaki K, Yaoi K (2014) Diversity of extradiol dioxygenases in aromatic-degrading microbial community explored using both culture-dependent and culture-independent approaches. FEMS Microbiol Ecol 90(2014):367–379
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, and Kumar S (2011) MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Tancsics A, Szoboszlay S, Kriszt B, Kukolya J, Baka E, Marialigeti K, Revesz S (2008) Applicability of the functional gene catechol 1 2-dioxygenase as a biomarker in the detection of BTEX-degrading Rhodococcus species. J Appl Microbiol 105:1026–1033
Uhlik O, Jecna K, Leigh M, Mackova M, Macek T (2009) DNA based stable isotope probing: a link between community structure and function. Sci Total Environ 407:3611–3619
Vardar G, Wood K (2005) Protein engineering of toluene-o-xylene monooxygenase from Pseudomonas stutzeri OX1 for enhanced chlorinated ethene degradation and o-xylene oxidation. Appl Microbiol Biotechnol 68:510–517
Warnecke F, Hugenholtz P (2007) Building on basic metagenomics with complementary technologies. Genome Biol 8:231
Watanabe K, Teramoto M, Futamata H, Harayama S (1998) Molecular detection isolation and physiological characterization of functionally dominant phenol-degrading bacteria in activated sludge. Appl Environ Microbiol 11:4396–4402
Webster R (2008) Soil sampling and methods of analysis - edited by M.R. Carter & E.G. Gregorich. Eur J Soil Sci 59:1010–1011. doi:10.1111/j.1365-2389.2008.01052_5.x
Witzig R, Junca H, Hecht J, Pieper D (2006) Assessment of toluene/biphenyl dioxygenase gene diversity in benzene-polluted soils: links between benzene biodegradation and genes similar to those encoding isopropylbenzene dioxygenases. Appl Environ Microbiol 72:3504–3514
Acknowledgments
The authors acknowledge VP Research, Alzahra University for funding of this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that no conflict of interest exists.
Rights and permissions
About this article

Cite this article
Nafian, F., Gharavi, S. & Soudi, M.R. Degenerate primers as biomarker for gene-targeted metagenomics of the catechol 1, 2-dioxygenase-encoding gene in microbial populations of petroleum-contaminated environments. Ann Microbiol 66, 1127–1136 (2016). https://doi.org/10.1007/s13213-016-1197-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13213-016-1197-3