
Abstract
Investigation of the potential adverse effects of chemicals and drugs is essential during the drug development process. In vitro cell model systems have been developed over the past years towards such toxicity investigation. 96-well plate is the common platform for screening drug toxicity due to its simplicity. However, this platform only offers 2D cell culture environment and lacks the flow of solutions, which fails to provide the suitable environment for the cells to adequately metabolize the drugs, for the media to replenish, and for the metabolites and wastes to be removed. Microfluidic chips populated with human or animal cells, known as organ-on-a-chip (OOC), can reconcile many issues of in vitro cell models, such as the lack of extracellular matrix and flow as well as the species difference. However, OOC can be complicated to fabricate and operate. To bridge this gap, we utilized paper as a primary substrate for OOC, considering its fibrous structure that can mimic natural extracellular matrix, as well as a syringe pump and filter that are commonly available in most laboratories. Paper microfluidic model was designed and fabricated by wax printing on nitrocellulose paper, seeded and proliferated with liver cells (primary rat hepatocytes and HepG2 cells), and two paper substrates were stacked together to complete the paper model. To this paper-based liver cell model, the following drugs were added: Phenacetin (pain reliever and fever reducer), Bupropion (antidepressant), Dextromethorphan (antidepressant), and phosphate-buffered saline (PBS) as a control, all under a physiologically relevant flow rate. The combination of these drugs with Fluconazole (antifungal drug) was also investigated. Cell count, cell morphology, protein production, and urea secretion after drug treatment confirmed that the model successfully predicted toxicity within 40 minutes. This simple, paper-based liver cell model provided enhanced and faster cell response to drug toxicity and showed comparable or better behavior than the cells cultured in conventional 2D in vitro models.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Toxicity studies of newly discovered drugs are crucial in drug development process. Traditionally, such studies have been performed first on an in vitro platform, where mammalian cells are cultured on a multi-well plate with varying doses of drugs, followed by an animal model to minimize the time, cost, and ethical concerns associated with it1–3. In vitro models populated with liver cells have widely utilized for studying drug absorption, drug metabolism, enzymatic kinetics, and dose-response relationship, in addition to drug toxicity. Primary animal or human hepatocytes, as well as immortalized liver cell lines, have been used extensively for the in vitro liver cell models for evaluating the liver toxicity to drugs. As expected, such in vitro cell models lack blood flow and extracellular matrix (ECM), leading to the loss of viability, decrease in liver-specific functionality and gene expression, etc. 3D culture models of liver cells have been suggested as an alternative. Recently, the active fluid flow has also been incorporated to provide a physiologically relevant environment for the cultured liver cells, including oxygen supply, nutrient diffusion, and waste removal, leading to liver-specific morphology and functionality beyond those provided by 2D liver cell culture4,5. Such models are often referred to as organ-on-a-chip (OOC)5.
As OOCs have continuously been evolving, the fabrication and operation of such OOC devices have tended to become more complicated to fabricate and operate. There certainly exists a gap between those sophisticated OOCs and the in vitro 2D cell culture models. In this work, we propose a simpler and inexpensive solution through utilizing a wax-printed paper microfluidic chip, a syringe pump, and a syringe filter, that are readily available in most laboratories. While paper microfluidic chips have been popularly utilized for biosensor applications, especially point-of-care diagnostics6, their use towards microfluidic cell models (or OOC) has been rarely demonstrated. Paper substrate, consisting of cellulose (or nitrocellulose) fibers, offers a permeable substrate mimicking the natural ECM (in our case sinusoidal endothelium). Such porous and permeable structure can provide more rigorous and flow-resistant cellular adhesion than the smooth and hydrophobic polydimethylsiloxane (PDMS; popularly used for many microfluidic chips and OOCs). Paper microfluidic chips can be fabricated more simply and cost-effectively than the PDMS-based microfluidic chips, requiring only a wax printer and a hot plate. These paper-based chips are seeded and proliferated with liver cells, followed by stacking two paper chips together and encasing them into a syringe filter to construct a liver cell model.
Three commercially available drugs are evaluated for their toxicities in this work: Phenacetin, Bupropion, and Dextromethorphan. Phenacetin was first introduced in 1887 and has been extensively used as analgesic and fever-reducer. The usual prescribed dose was 300 mg 4–6 times a day for adults, not exceeding 2 g daily. However, Phenacetin was removed from the United States market in 1983 and was legally banned in most countries due to its carcinogenic effects in the kidney when used in large amounts7. Bupropion is widely used as antidepressant, which is associated with noradrenergic or dopaminergic mechanisms. This drug was first used in the United States in 1985 to treat moderate and severe depression. Currently, it is widely prescribed for 4 million individuals yearly in the U.S.8 Bupropion is generally prescribed at 75 to 300 mg once or twice a day for adults. Nevertheless, liver abnormalities have been reported for < 1% of patients and acute liver injury for rare cases. While most abnormalities and injuries by Bupropion are self-limited, fatal cases have been nonetheless reported8. Dextromethorphan is commonly used to relieve cough caused by cold and flu by decreasing the brain activity that causes coughing9. Due to its function of decreasing brain activity, it is also widely used as anti-depressant. This drug is generally prescribed at 10 to 20 mg orally every 4 hours as liquid or syrup for adults10. While serious toxicity of Dextromethorphan occurs relatively rarely, there have been reports of major toxicity and death caused by co-ingestion of Dextromethorphan with other drugs11. To study such coingestion-induced toxicity, Fluconazole was additionally evaluated for its toxicity combined with the aforementioned drugs. Fluconazole is widely used to treat systemic and superficial fungal infections and currently prescribed more than 12 million times per year. The recommended dose of Fluconazole ranges from 100 to 400 mg daily for adults. Hepatotoxicity (liver toxicity) by Fluconazole has been reported within the first few weeks of therapy12. While such complications are generally self-limited, fatal cases have nonetheless been reported13.
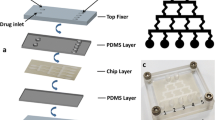
The overarching aim of this study is to design and demonstrate a paper-based liver cell model that is simple, low-cost, and easy-to-use to evaluate drug toxicity, particularly in patients with a pre-existing condition. Figure 1 shows the device setup, requiring only a syringe pump and filter, where the paper-based liver cell model is encased within a filter holder. These small equipment and supplies are either extremely low-cost or readily available in most laboratories, potentially enabling its widespread use.
Results
Selection of Cell Type
To develop the paper-based liver cell model for drug toxicity screening, two types of liver cells were first cultured in order to determine the appropriate cell line to use in the model. Primary rat hepatocytes (PRH) and immortalized human liver carcinoma cells (HepG2) were evaluated on a 96-well tissue culture plate (TCP) pre-treated with type I rat tail collagen. PRH maintained cuboidal morphology while HepG2 exhibited relatively rounded morphology (Figure S1). Recommended minimum daily dosage for Phenacetin, Bupropion, and Dextromethorphan is 1,200 mg (= 300 mg × 4), 75 mg, and 120 mg (= 20 mg × 6), respectively7–10. As the average number of hepatocytes in humans is 1.68 × 1011 and there were 1 × 104 cells in our model, the drug amounts were scaled down to 70 ng Phenacetin, 5 ng Bupropion and 7 ng Dextromethorphan based on the total number of liver cells in our system. Distribution over blood and lymph systems, absorption and metabolism in the other tissues and organs, and excretion were not considered; only the hepatic first pass effect was modeled. The number of PRH and HepG2 cells treated with Phenacetin, Bupropion, and Dextromethorphan significantly decreased in comparison to the control while the Phenacetin showed the highest toxicity represented by the lowest number of cells on the surface after drug treatment (Figure 2). The numbers of HepG2 cells after drug treatment were comparable to those of PRHs, indicating comparable responses to the tested drugs.

Paper-based liver cell model for in vitro drug toxicity test. Paper-based liver cell model consists of two layers of wax-printed NC paper. Top layer design consists of six square inlets void of wax (white) surrounded by hydrophobic wax (black). The bottom layer design consists of six hydrophilic squares aligned to the top layer’s inlets, converging into the central hole via six channels, where HepG2 cells are loaded as shown in top view (left). The top layer was stacked on the bottom layer. The drug solution was delivered to HepG2 cells via a controllable syringe pump and connected tubes with a flow rate of 333 nL/min above the cell culture area. The flow-through solution was collected at the central hole of a bottom layer and subsequently delivered to the collecting tube. Blue arrows represent the direction of liquid flow.

Comparison between PRH vs. HepG2 on TCP for drug toxicity testing under static condition. Cell count, protein concentration, and urea concentration in the supernatant secreted from PRH and HepG2 cells after treated with recommended daily dose/cells of Phenacetin, Bupropion, Dextromethorphan, and a control (PBS). Error bars represent standard errors. * indicates p < 0.05 in comparison to the control.
The concentrations of proteins secreted from both PRH and HepG2 cells treated with all three drugs were significantly lower than the control (Figure 2).
Again, protein productions from PRH and HepG2 cells were also comparable to each other. The concentrations of urea secreted from HepG2 cells were significantly higher in control and Dextromethorphan than with Phenacetin and Bupropion (Figure 2). However, urea productions from PRHs decreased regardless of drug treatments. As represented in Figure 2, HepG2 cells showed similar or better metabolic activities compared to the results from primary cells (PRH). Also considering its superior culturability, HepG2 was chosen for the paper-based liver cell model in this work.
Comparison between Paper and TCP
While paper fibers offer a 3D, ECM-like microenvironment, the adhesion of HepG2 cells on paper fibers is poor. To address this issue, the nitrocellulose (NC) paper substrate was coated by using collagen. This collagen-coated NC paper substrate was further evaluated for HepG2 adhesion and subsequent drug toxicity test in comparison to the collagen-coated tissue culture plate (TCP), which is in a 96-well platform under static condition. The collagen-coated NC paper substrates were cut and fit each well of untreated 96-well TCP, to make HepG2 cells be exposed only to the collagen-coated NC paper. The number of HepG2 cells were evaluated after adding Phenacetin, Bupropion, and Dextromethorphan, in comparison to the control (PBS), using a regular light microscope on TCP and a fluorescence microscope on NC paper. (Paper fibers prevented the cells to be properly imaged on a light microscope, requiring fluorescence microscopy with nuclei and actin staining.) As a result, the number of HepG2 cells significantly decreased with all three drugs in comparison to the control (PBS), on both NC paper and TCP. Phenacetin showed the highest toxicity with the lowest number of HepG2 cells after drug treatment on both NC paper and TCP (Figure 3). The numbers of HepG2 on NC paper were not significantly different from those on TCP, regardless of the added drugs. Protein production was also evaluated, which was significantly lower with all three drugs compared to the control. Again, there was no significant difference between NC paper and TCP (Figure 3). Urea secretion was additionally evaluated. On TCP, urea production decreased the most with Phenacetin and Bupropion, followed by Dextromethorphan. On NC paper, the trend was the same, although at smaller extents especially with Phenacetin, i.e. less compromised urea production upon drug treatments. Overall, NC paper model showed similar or better metabolic activities compared to that of TCP model. Also considering its flexibility and 3D ECM-like microenvironment (shown as mesh-like autofluorescence in Figure S2), NC paper was accordingly chosen as a substrate for the paper-based liver cell model in this work.

Comparison between HepG2 on TCP vs. NC paper under static condition. Cell count, protein concentration, and urea concentration in the supernatant secreted from HepG2 cells on TCP and NC paper after drug treatments. Error bars represent standard errors. * indicates p < 0.05 in comparison to the control.
Paper-based Fluidic Liver Cell Model in Comparison with Static TCP Model
In order to address the effect of the flow, the experiments were conducted for the paper-based liver cell model with and without flow. The liver cell model on dual-layer NC paper was designed and fabricated as shown in Figure 1. Drugs and media were pumped into six inlets of the top layer, mimicking portal veins of a liver lobule. Again, the system is designed to simulate the hepatic first pass effect. The dark region was printed with hydrophobic wax and thus impermeable, while the white areas (inlets) were free of wax and allowed the liquid to flow in and out through paper. This top layer was stacked directly on top of the bottom layer, where six different channels were imprinted mimicking liver sinusoids. HepG2 cells were pre-seeded into these channels before stacking these two NC paper substrates. This stacked system was inserted into a syringe filter, which is connected to two 7-mm plastic tubes. The inlet tube was connected to a 10-mL syringe containing 5 mL of drugs: Phenacetin, Bupropion, Dextromethorphan, and control (PBS). This inlet solution was distributed to six inlets of the top layer. In the bottom layer, all solutions from six channels converged to a central area, which was then connected to an outlet tube. In this work, 1 × 104 HepG2 cells were seeded on each 0.8-mm diameter channel. We used a flow rate of 333 nL/min in each sinusoid-like channel, considering the human liver sinusoid where each unit consisted of 250 tightly packed hepatocytes receiving the flow of approximately 100 pL/s or 6 nL/min14. As there were 1 × 104 cells in each channel, the physiologically relevant volumetric flow rate should be 240 nL/min. However, we used a slightly higher flow rate of 333 nL/min in order to compensate for the hydrodynamic flow resistance build-up from paper fibers, as demonstrated by the previous study on the polystyrene beads’ movements through paper fibers15. The overall flow rate perfused through this system was set at 2 µL/min (= 333 nL/min × 6) as there were six channels in the system. Continuous and steady perfusion was ensured by checking the constant outflow generated at the outlet of a collection tube. This continuous perfusion prevented the toxic metabolites’ build-up and allowed the in-flow of liquid with adequate oxygen supply to the system.

Fluorescence microscopic images (10x) of HepG2 cells on the paper-based fluidic liver cell model (left) and the static 96-well model on NC paper (right). Cells were treated with recommended daily doses of Phenacetin, Bupropion, and Dextromethorphan, as well as a control (PBS). Blue represents the DAPI’s stained nuclei and pink represents the TRITC’s stained phalloidin.

Comparison between paper-based fluidic liver cell model and static 96-well model, both with HepG2 cells and NC paper. Cell count, protein concentration, and urea concentration were evaluated from the supernatant secreted from HepG2 cells after drug treatments. Error bars represent standard errors. * indicates p < 0.05 in comparison to the control.
The number of HepG2 cells on this paper-based liver cells on a paper model was significantly lower than that under the static condition on plain NC paper substrate (Figures 4 and 5), which can be attributed to the removal of weakly adhered cells as the flow washed the detached and dead cells out from the paper substrates that did not interact with the drugs loaded. Specifically, TRITC-phalloidin-stained actin filaments were almost always found where DAPI-stained nuclei existed, while those actin filaments maintained polymerized fibrous structure and nuclei were not fragmented. These results indicate that most of the dead cells were removed and washed away from our model after the drug treatments.
Protein and urea productions under the flow condition were also significantly lower than those under the static condition (Figure 5). The overall trends in drug toxicities (cell counts, protein, and urea production) are similar between static and flow models, in the order of Phenacetin, Bupropion, and Dextromethorphan, indicating the unit cell productions of protein and urea actually improved, which corresponds to the previous studies5,16,17. A continuously perfused system prevents the build-up of toxic metabolites and ensures adequate oxygenation to 3D tissue culture, also contributing to higher unit cell production of protein and urea18. Taken together, the developed paper-based liver cell model provides the microstructure analogous to that of the hepatic sinusoid, generating in vivo-like liver phenotype and metabolic functions19.
The number of cells was also evaluated at four different areas of each channel, as depicted in Figure 6. Again, the system is optimized for evaluating the hepatic first pass effect. A general decreasing trend in cell count, protein production, and urea secretion can easily be identified with a weakly toxic drug of Dextromethorphan along the channel from inlet to outlet. However, no such trend can be identified with control (PBS), showing a consistent number of cells along the channel (Figure 6). It will be discussed further in the Discussion section.

Drug toxicity analysis along the length of the channel (under the flow condition). Top: Graphical illustration of the areas from 1 to 4 along the channel of a bottom layer. Bottom: Number of HepG2 cells for each area after treated with Dextromethorphan and a control (PBS). Error bars represent standard errors. * indicates p < 0.05 in comparison to the control (PBS).

Fluconazole toxicity test using the paper-based liver cell model. Cell count, protein concentration, and urea concentration were evaluated from the supernatant secreted from HepG2 cells after Fluconazole treatment. Error bars represent standard errors. * indicates p < 0.05 in comparison to the control.
Drug Toxicity Test with Pre-existing Drug (Drug Combination) Using the Paper-based Liver Cell Model
Fluconazole, an anti-fungal drug, is prescribed to a patient with a fungal infection, with varied dosage based upon the seriousness of the infection. Generally, 400 mg-dose is prescribed to adults on the first day of treatment and followed by 200 mg dose for at least 10 to 12 weeks for treating many different fungal infections, including Cryptococcal meningitis20. In severe cases, 800 mg can be prescribed on the first day of treatment for adults. While Fluconazole is generally considered safe, a higher dose can pose a threat to infants and elderly patients, especially with pre-existing conditions. Therefore, low, medium, and high concentrations of Fluconazole (200, 400, and 800 mg, respectively) were evaluated in this work prior to the drug combination experiment, again using the paper-based liver cell model. The actual drug amount applied to the paper-based liver cell model was scaled down to 0.8, 1.6, and 3.2 ng, considering the number of hepatocytes in human adults and that of the paper-based liver cell model (refer to the first section of Results). The cell counts were significantly lower with 400 and 800 mg doses, indicating their toxicity. Protein and urea productions were also reduced with an increasing dose of Fluconazole (Figure 7). These results correspond to the known hepatotoxicity of Fluconazole to healthy adults, where 800 mg being toxic, 400 mg being occasionally toxic, and 200 mg being generally safe21.

Drug combination toxicity test using the paper-based liver cell model. Cell count, protein concentration, and urea concentration were evaluated from the supernatant secreted from HepG2 cells after treated with PBS (control), Phenacetin, Bupropion, and Dextromethorphan (gray) and those combined with 200 mg of Fluconazole (black). Error bars represent standard errors. * indicates p < 0.05 between each pair – without vs. with Fluconazole.
Fungal infection treatment usually takes up to 3 months. Therefore, it is quite likely for a patient to take this anti-fungal drug together with other drugs, for example, painkiller or anti-depressant, etc. In this work, we investigated Phenacetin, Bupropion, and Dextromethorphan, along with Fluconazole at 200 mg dose (e.g. a safe dose). The numbers of cells decreased upon the addition of 200 mg dose Fluconazole to all three drugs (Figure 8). Protein and urea productions also decreased upon adding 200 mg dose of Fluconazole to all three drugs (Figure 8).
Discussion
PRHs were freshly isolated from rat liver and transported to the laboratory prior to the same day experiments. These cells were cultured on a collagen-coated 96-well TCP under the static condition and able to maintain their liver-specific cuboidal morphology facilitating their metabolic functions22. While HepG2 cells (non-tumorigenic cells with high proliferation rate) were not able to maintain the characteristic cuboidal morphology, they still maintained epithelial-like morphology (Figure S1) and performed liver-specific functions including albumin and urea production similar to PRH23 as shown in Figure 2. HepG2 cells are known to be an alternative cell line of primary hepatocyte commonly used for in vitro drug metabolism and hepatotoxicity studies, due to its unlimited life span, stable phenotype, high availability, and easy handling24. Both PRH and HepG2 cells were initially seeded into each TCP well and static-incubated with control (PBS), Phenacetin, Bupropion, or Dextromethorphan. After 40 min of incubation, the total number of cells generally decreased with three commercial drugs compared to the control (PBS), due to the necrosis resulting from their toxicity and subsequent detachment from surface5. Both protein production (mostly albumin) and urea secretion from cells were reduced the most with Phenacetin and Bupropion, followed by Dextromethorphan. Under the toxic environment, the drug-induced necrosis leads to a decrease in protein production and urea secretion25. Specifically, reduction in urea production was more apparent with HepG2 cells than with PRHs. This can be explained by the fact that PRHs tend to be more sensitive and susceptible to the change of environment (in these experiments, it was changed from culture media to drug solutions) than HepG2 cells26. Given such limitation, HepG2 cells should be a better choice for building a paper-based liver cell model than PRHs.
Next, HepG2 cells were cultured under the static condition on collagen-coated NC paper and compared with the results on collagen-coated TCP (both in 96-well platforms). While the results on NC paper were generally similar to those on TCP (Figure 3), they outperformed in urea secretion. This can be attributed to the fibrous structure of the collagen-coated NC paper substrate, which better recapitulates the 3D environment of the extracellular matrix (ECM). This structure, in turn, facilitates the maintenance of liver-specific phenotype in vitro27 and tolerance to toxins23.
To better imitate the liver tissue, a two-layer NC paper-based liver cell model was designed and fabricated as depicted in Figure 1. It has been known that in vitro liver cells typically respond to drug toxicity at different concentrations than those in vivo, due to the absence of ECM, flow, and other physiological parameters that strongly affect their metabolic activities. The cell counts with the paper-based liver cell model were significantly lower than those under the static condition (Figure 4). These results are acceptable as the flow washed the detached and dead cells out from the paper substrates that did not interact with the drugs loaded. In fact, protein and urea secretions were not reduced as much as the cell counts, indicating the unit cell productions of protein and urea actually improved, which corresponds to the previous studies5,16,17. A continuously perfused system prevents the build-up of toxic metabolites and ensures adequate oxygenation to 3D tissue culture, also contributing to higher unit cell productions of protein and urea18. Taken together, the developed paper-based liver cell model provides the microstructure analogous to that of the hepatic sinusoid, generating in vivo-like liver phenotype and metabolic functions, which provides a more physiologically relevant flow environment better than TCP model19.
The number of cells along the length of the channel was investigated in the paper-based liver cell model. While the cell count was consistent along the channel with non-toxic PBS (control), it increased along the channel from the inlet to the outlet with Dextromethorphan (Figure 6). As the flow rate was very low and the steady outflow was observed, this gradient phenomenon is not a drug concentration gradient caused by unsteady accumulation but rather a cellular response gradient28. HepG2 cells at the inlet were exposed first to the toxic drugs, causing a higher necrosis rate. These drugs were metabolized by HepG2 cells at the inlet, resulting in lower drug concentration at the next position, thus creating the gradient of drug concentration along the channel towards the outlet. Our observation was, however, contradicted to the in vivo toxicity gradient, mainly because of the in vivo oxygen gradient. Low toxicity is presented at the sinusoidal inlet where the drug carried by the portal vein is mixed with oxygen-rich blood from the hepatic artery. As mixed blood flow along sinusoid, hepatocytes utilize oxygen to metabolize the drugs while excreting waste products into the blood. These resulted in high toxicity to the cells close to a central vein due to the secreted waste and oxygen depletion29. Such an oxygen gradient was not presented in our system because of the unidirectional flow from the syringe to the collecting tube and no media circulation in our system that provided sufficient oxygen supply to the cells.
An anti-fungal drug, Fluconazole, exhibited a similar trend to other commercial drugs (Figure 7). Increasing Fluconazole dosage resulted in lower cell count and reduced protein and urea production, thus increase in hepatotoxicity, as high dosage of Fluconazole has been reported to cause acute liver failure13. Nevertheless, HepG2 cells incubated with 800 mg Fluconazole produced higher protein than those incubated with the lower dosage because hepatocytes tend to secrete and accumulate more proteins, especially albumin, to stabilize the superoxide and free radicals in order to protect themselves from damage induced by drug toxicity30.
Finally, 200 mg dose of Fluconazole (considered as a safe dose) was mixed with Phenacetin, Bupropion, or Dextromethorphan. The toxicity of commercial drugs plus Fluconazole was higher than those without, as observed with cell count, protein production, and urea secretion. Interaction of two different drugs often inhibits the metabolism of each drug, leading to toxic effects31. Interaction of Fluconazole and Phenacetin towards decreased Phenacetin metabolism has been reported32, as well as Fluconazole/Bupropion towards decreased Bupropion metabolism and Fluconazole/Dextromethorphan towards decreased Dextromethorphan metabolism33. Such compromised metabolism resulted in the accumulation of drug, which consequently increased the toxicity to the cells.
Conclusion
The developed paper-based fluidic liver cell model shows an improved version of the traditional in vitro drug toxicity screening with the addition of flow to enhance cellular response to drug toxicity, represented by its overall results that can outperform the traditional in vitro model. The developed paper-based fluidic liver cell model consists of materials that can be easily found in most laboratories: 1) liver sinusoids were constructed on nitrocellulose paper using a commercial wax printer; 2) a syringe filter was used as a chamber for holding liver cells on paper; 3) a syringe pump was used to generate physiologically relevant flow to deliver drugs19. Each compartment was simply connected via plastic tubes. Three different commercial drugs, Phenacetin, Bupropion, and Dextromethorphan were successfully tested on the developed system with HepG2 cells for their toxicity. In addition, anti-fungal drug Fluconazole, which can be frequently used over a long period of time, was also evaluated for its hepatotoxicity by itself alone and combined with the above three commercial drugs (drug combination test). This simple paper-based liver cell model showed comparable or better behavior than the cells cultured in traditional TCP, while generating results as early as 40 min compared to other methods that may require greater than 48 hours34. Due to its simplicity, this device can be easily adapted for personalized testing using the patient’s own cells in a physician’s office, towards personalized medicine. Future work includes the development of a handheld imaging device compatible with this model for real-time quantification of drug toxicity level and software to allow the automated and user-friendly data analysis.
Materials and Methods
Selection of Cell Type
Primary rat hepatocytes (PRH; Lonza, Alpharetta, GA, USA) were freshly isolated and received from the vendor. PRHs were then centrifuged at 1,500 rpm for 5 min. The cell pellet was resuspended at a final concentration of 1 × 106 cells/mL with William’s E medium (Thermo-Fisher, Waltham, MA, USA) supplemented with 10% v/v fetal bovine serum (FBS; Fisher Scientific, Pittsburgh, PA, USA), 0.2% v/v of 250 µg/mL Amphotericin B (GE Healthcare, Maidstone, Kent, UK), and 0.1% v/v of 50 mg/mL Gentamycin sulfate. HepG2 (ATCC, Manassas, VA, USA) were maintained in Eagle’s Minimum Essential Medium (Corning Inc., Corning, NY, USA) supplemented with 10% v/v FBS, 0.2% v/v amphotericin B, and 0.1% v/v Gentamycin sulfate on T-75 cell culture flasks. Cells were cultured under static conditions at 37°C in 5% CO2 until 90% confluency. Cells were passaged using standard procedures then resuspended at a final concentration of 1 × 106 cells/mL.
The 10 µL of cell suspension was seeded onto a 96-well tissue culture plate (TCP; Corning Inc.) pretreated with type I rat tail collagen (50 µg/mL; BD, Franklin Lakes, NJ, USA). Cell suspension was left for 15 min to allow complete attachment and 90 µL of warm media was added. The cells seeded on TCP were incubated for 4 h (PRH) and overnight (HepG2) in a CO2 incubator prior to drug toxicity experiment.
Comparison between Paper and TCP
Nitrocellulose (NC) paper substrates were hole-punched into 5-mm diameter circles, individualized into a 96-well plate, and UV-sterilized prior to experiments. For comparison purposes, 96-well TCP was also used. Both NC paper and TCP surfaces were incubated with type I rat tail collagen for 1 h, then washed twice with phosphate buffer saline (PBS; Sigma-Aldrich, St. Louis, MO, USA). HepG2 (1 × 104 cells) was seeded onto each substrate following the aforementioned-protocol and incubated overnight in a CO2 incubator to allow cell attachment. After incubation, the media was removed, and cells were washed twice with DPBS. Cells were then incubated with 100 µL of three different drugs: Phenacetin (pain reliever and fever reducer), Bupropion and Dextromethorphan (both are antidepressant) (Sigma-Aldrich) were tested at low doses (5–70 ng, much lower than daily recommended 300–1200 mg doses) and phosphate-buffered saline (PBS) was also tested as a control. All drugs were added to the 96-well plate for 40 min under static condition. Following drug treatments, supernatants were transferred to a new TCP and stored at -20°C if cell metabolic activity is not measured immediately. Cells on the substrate were fixed with a 4% solution of paraformaldehyde (Affymetrix, Santa Clara, CA, USA) and washed twice with PBS before fluorescent staining and imaging.
Drug Toxicity Test on the Liver Cell Model
The paper-based microfluidic chip was used as a flexible substrate for the liver cell model. Chip layout was designed using SolidWorks software (Solid-Works Corp., Waltham, MA, USA) and wax-printed (ColorQube, Xerox, Norwalk, CT, USA) on NC paper. The paper microfluidic chip consists of two layers (Figure 1). Top layer design consists of six 0.5 mm × 1 mm squares void of wax (white), surrounded by hydrophobic wax (black), all printed on NC chromatography paper (GE Healthcare). They serve as inlets to the paper-based liver cell model and mimic the portal veins in the liver. The bottom layer consisted of six hydrophilic squares with 0.5 mm × 1 mm inlets, perfectly aligned with the top layer’s inlets. Each square is connected to a 0.8 mm × 5 mm channel, mimicking liver sinusoid. All six channels converge to a 1-mm diameter circle at the center, which was hole-punched using a tissue biopsy puncher with 1-mm diameter. This serves as an outlet of the liver cell model on paper and mimics the central vein in the liver.
To seed HepG2 cells on the NC paper, its bottom layer was initially placed into a 35 mm × 15 mm petri dish (Sigma-Aldrich). 1 × 104 of HepG2 cells were seeded on this bottom layer paper chip and submerged under 3 mL of media, followed by incubation overnight under static culture in a CO2 incubator. After incubation, the media were removed, and the paper chip was washed twice with DPBS. The bottom layer paper chip was then transferred and stacked with the top layer in a syringe filter (Fisher Scientific, Pittsburgh, PA, USA), connected to two 7-mm plastic tubes (Fisher Scientific). The inlet tube was connected to a 10-mL syringe (Fisher Scientific) containing 5 mL of drugs as listed in the previous section and placed on a syringe pump (New Era Pump Systems Inc., Farmingdale, NY, USA). The outlet tube was connected to a 15-mL centrifuge tube (VWR, Radnor, PA, USA) (Figure 1). The syringe pump was set at 2 µL/min (333 nL/min for each channel of the liver cell model). The drug in the syringe was pumped and flew through the cells in a syringe filter for 40 min. After drug treatments, supernatants were collected in 15-mL centrifuge tubes and kept at -20°C if cell metabolic activity was not measured immediately. Cells on the paper substrate were fixed with a 4% solution of paraformaldehyde (Affymetrix, Santa Clara, CA, USA) and washed twice with PBS before the fluorescence staining and imaging.
Drug Combination Toxicity Test on the Liver Cell Model
In addition to three drugs described in the above, antifungal drug Fluconazole were also evaluated at 200, 400, and 800 mg dosages. The actual Fluconazole concentration was scaled down to 0.8, 1.6, and 3.2 ng considering the number of liver cells in the paper model. After this initial testing, Fluconazole at 200 mg dosage was additionally added to the other three commercial drugs, Phenacetin, Bupropion, Dextromethorphan, as well as control (PBS), to evaluate drug combination toxicity.
Fluorescence Imaging
Waste media was removed from the collagen-coated NC paper models, which were then fixed with a 4% solution of paraformaldehyde for 15 min. Paraformaldehyde was removed and paper models were rinsed twice with washing buffer solution (1X PBS with 0.05% Tween-20). Cell membranes were perforated with 0.1% Triton-X-100 (Fisher Scientific) for 5 min. Triton-X was removed and paper models were rinsed twice with washing buffer solution. Paper models were treated with blocking buffer solution (1% bovine serum albumin - BSA in 1X PBS) and then were incubated for 30 min. The cells’ nuclei were stained with DAPI (4’,6-diamidino-2-phenylindole; EMD Millipore, Burlington, MA, USA) and actin filaments with TRITC (tetramethylrhodamine B isothiocyanate) conjugated phalloidin (EMD Millipore, Burlington, MA, USA). Images were collected using the NIS element software on a personal computer connected to a benchtop fluorescence microscope (Nikon Eclipse TS100, Minato, Tokyo, Japan) with UV and TRITC filter attachments (A.G. Heinze, Lake Forest, CA, USA). Greyscale images were taken from each filter cube, transferred to ImageJ software, and pseudocolors were added: blue to DAPI and red to Phalloidin-TRITC. These two images were stacked onto each other to represent the entire cell images.
Cell Counting
Number of cells after drug treatment was quantified by taking light microscopic images (on TCP) or fluorescence microscopic images (on NC paper), and further counting the number of cells using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA).
Evaluation of Cells’ Metabolic Activities
Besides the cell morphology and counting, drug toxicity can be also evaluated by cells’ metabolic activities. The protein and urea secretions from liver cells can be determined by Bradford assay and Bromocresol purple assay, respectively. Total protein concentration was determined by mixing the supernatant from a well plate or a liver cell model after drug treatment with Bradford’s reagent (at 1:1 ratio) and incubating at room temperature for 10 min prior to measurements. Total urea concentration was determined by mixing the same supernatant with Bromocresol purple solution (at 4:1 ratio) and incubating at room temperature for 20 min prior to measurements. The resulting absorbance is measured using the Ocean Optics miniature spectrometer (Largo, FL, USA) and its OceanView software version 1.6.7 at 595 nm (for protein quantification) and 588 nm (for urea quantification). The measured absorbance values were calculated to the concentration using the standard curve constructed with a known concentration of protein and urea. Finally, total protein and urea concentrations from each treatment were normalized by the initial number of seeded cells.
Statistical Analysis
All data were derived from at least three replicates, each using a different paper model. Statistical analyses were performed using analyses of variance (ANOVA). Differences at p < 0.05 were considered statistically significant.
References
Zhang, J., Zhao, X., Liang, L., Li, J., Demirci, U. & Wang, SQ. A decade of progress in liver regenerative medicine. Biomaterials157, 161–176 (2018).
LeCluyse, E.L., Bullock, P.L. & Parkinson, A. Strategies for restoration and maintenance of normal hepatic structure and function in long-term cultures of rat hepatocytes. Adv. Drug Delivery Rev.22, 133–186 (1996).
DelRaso, N.J. In vitro methodologies for enhanced toxicity testing. Toxicol. Lett.68, 91–99 (1993).
Soldatow, V.Y., LeCluyse, E.L., Griffith, L.G. & Rusyn, I. In vitro models for liver toxicity testing. Toxicol. Res. (Cambridge, U. K.)2, 23–39 (2013).
Kang, I.-K., Kim, G.J., Kwon, O.H. & Ito, Y. Coculture of hepatocytes and fibroblasts by micropatterned immobilization of β-galactose derivatives. Biomaterials25, 4225–4232 (2004).
Renault, C., Li, X., Fosdick, S.E. & Crooks, R.M. Hollow-channel paper analytical devices. Anal. Chem.85, 7976–7979 (2013).
Phenacetin. Available at: https://www.ncbi.nlm.nih.gov/books/NBK304337/?report=classic. (Accessed: 21st March 2020).
Bupropion. Available at: https://livertox.nlm.nih.gov/Bupropion.htm. (Accessed: 21st March 2020)
Dextromethorphan: MedlinePlus Drug Information. Available at: https://medlineplus.gov/druginfo/meds/a682492.html. (Accessed: 21st March 2020)
Dextromethorphan: Side Effects, Dosages, Treatment, Interactions, Warnings. Available at: https://www.rxlist.com/consumer_dextromethorphan/drugs-condition.htm. (Accessed: 21st March 2020)
Anderson, I.B. Chapter 60. Dextromethorphan. in Poisoning & Drug Overdose (ed. Olson, K.R.) (McGraw-Hill: New York, 2012).
Fluconazole. Available at: https://livertox.nlm.nih.gov/Fluconazole.htm. (Accessed: 21st March 2020)
Jacobson, M.A., Hanks, D.K. & Ferrell, L.D. Fatal acute hepatic necrosis due to fluconazole. Am. J. Med.96, 188–190 (1994).
Puhl, G., Schaser, K.D., Vollmar, B., Menger, M.D. & Settmacher, U. Noninvasive in vivo analysis of the human hepatic microcirculation using orthogonal polarization spectral imaging. Transplantation75, 756–761 (2003).
Chumo, B., Muluneh, M. & Issadore, D. Laser micromachined hybrid open/paper microfluidic chips. Biomicrofluidics7, 64109 (2013).
Prodanov, L., Jindal, R., Bale, S.S., Hegde, M., McCarty, W.J., Golberg, I., Bhushan, A., Yarmush, M.L. & Usta, O.B. Long-term maintenance of a microfluidic 3D human liver sinusoid: maintenance of a microfluidic 3D human liver sinusoid. Biotechnol. Bioeng.113, 241–246 (2016).
Wang, Y., Wang, H., Deng, P., Chen, W., Guo, Y., Tao, T. & Qin, J. In situ differentiation and generation of functional liver organoids from human iPSCs in a 3D perfusable chip system. Lab Chip18, 3606–3616 (2018).
Du, C., Narayanan, K., Leong, M.F. & Wan, A.C.A. Induced pluripotent stem cell-derived hepatocytes and endothelial cells in multi-component hydrogel fibers for liver tissue engineering. Biomaterials35, 6006–6014 (2014).
Dash, A., Simmers, M.B., Deering, T.G., Berry, D.J., Feaver, R.E., Hastings, N.E., Pruett, T.L., LeCluyse, E.L., Blackman, B.R. & Wamhoff, B.R. Hemodynamic flow improves rat hepatocyte morphology, function, and metabolic activity in vitro. Am. J. Physiol.: Cell Physiol.304, C1053–C1063 (2013).
Fluconazole (Oral Route) Proper Use - Mayo Clinic. Available at: https://www.mayoclinic.org/drugs-supplements/fluconazole-oral-route/proper-use/drg-20071428. (Accessed: 21st March 2020)
Fluconazole Dosage Guide with Precautions. Available at: https://www.drugs.com/dosage/fluconazole.html. (Accessed: 21st March 2020)
Shulman, M. & Nahmias, Y. Long-term culture and coculture of primary rat and human hepatocytes. Methods Mol. Biol.945, 287–302 (2013).
Bokhari, M., Carnachan, R.J., Cameron, N.R. & Przyborski, S.A. Culture of HepG2 liver cells on three-dimensional polystyrene scaffolds enhances cell structure and function during toxicological challenge. J. Anat.211, 567–576 (2007).
Donato, M.T., Tolosa, L. & Gómez-Lechón, M.J. Culture and functional characterization of human hepatoma HepG2 cells. in Protocols in In Vitro Hepatocyte Research (eds. Vinken, M. & Rogiers, V.) 77–93 (Springer New York, 2015).
Bhise, N.S., Manoharan, V., Massa, S., Tamayol, A., Ghaderi, M., Miscuglio, M., Lang, Q., Zhang, Y.S., Shin, S.R., Calzone, G., Annabi, N., Shupe, T.D., Bishop, C.E., Atala, A., Dokmeci, M.R. & Khademhosseini, A. A liver-on-a-chip platform with bioprinted hepatic spheroids. Biofabrication8, 014101 (2016).
Sison-Young, R.L., Lauschke, V.M., Johann, E., Alexandre, E., Antherieu, S., Aerts, H., Gerets, H.H.J., Labbe, G., Hoët, D., Dorau, M., Schofield, C.A., Lovatt, C.A., Holder, J.C., Stahl, S.H., Richert, L., Kitteringham, N.R., Jones, R.P., Elmasry, M., Weaver, R.J., Hewitt, P.G., Ingelman-Sundberg, M., Goldring, C.E. & Park, B.K. A multicenter assessment of single-cell models aligned to standard measures of cell health for prediction of acute hepatotoxicity. Arch. Toxicol.91, 1385–1400 (2017).
Wang, Y., Kim, M.H., Shirahama, H., Lee, J.H., Ng, S.S., Glenn, J.S. & Cho, N.-J. ECM proteins in a microporous scaffold influence hepatocyte morphology, function, and gene expression. Sci. Rep.6, 37427 (2016).
Hung, P.J., Lee, P.J., Sabounchi, P., Lin, R. & Lee, L.P. Continuous perfusion microfluidic cell culture array for high-throughput cell-based assays. Biotechnol. Bioeng.89, 1–8 (2005).
Trefts, E., Gannon, M. & Wasserman, D.H. The liver. Curr. Biol.27, R1147–R1151 (2017).
Pyzik, M., Rath, T., Kuo, T.T., Win, S., Baker, K., Hubbard, J.J., Grenha, R., Gandhi, A., Krämer, T.D., Mezo, A.R., Taylor, Z.S., McDonnell, K., Nienaber, V., Andersen, J.T., Mizoguchi, A., Blumberg, L., Purohit, S., Jones, S.D., Christianson, G., Lencer, W.I., Sandlie, I., Kaplowitz, N., Roopenian, D.C. & Blumberg, R.S. Hepatic FcRn regulates albumin homeostasis and susceptibility to liver injury. Proc. Natl. Acad. Sci. U. S. A.114, E2862–E2871 (2017).
Teo, Y.L., Ho, H.K. & Chan, A. Metabolism-related pharmacokinetic drug-drug interactions with tyrosine kinase inhibitors: current understanding, challenges and recommendations. Br. J. Clin. Pharmacol.79, 241–253 (2015).
Phenacetin. Available at: https://www.drugbank.ca/drugs/DB03783. (Accessed: 21st March 2020)
Fluconazole. Available at: https://www.drugbank.ca/drugs/DB00196. (Accessed: 21st March 2020)
Beckwitt, C.H., Clark, A.M., Wheeler, S., Taylor, D.L., Stolz, D.B., Griffith, L. & Wells, A. Liver ‘organ on a chip’. Exp. Cell Res.363, 15–25 (2018).
Acknowledgements
This work was supported by the pilot interdisciplinary grant from the BIO5 Institute at the University of Arizona and cardiovascular biomedical engineering training grant from U.S. National Institutes of Health, grant number T32HL00 7955. K.K. acknowledges the scholarship from the Development and Promotion of Science and Technology Talents Project (DPST) of Thailand. P.A. acknowledges the scholarship from One District One Scholarship (ODOS) of Thailand.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interests The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Kaarj, K., Ngo, J., Loera, C. et al. Simple Paper-based Liver Cell Model for Drug Screening. BioChip J 14, 218–229 (2020). https://doi.org/10.1007/s13206-020-4211-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13206-020-4211-6