
Abstract
In the current study, gene network analysis revealed five novel disease-resistance proteins against bacterial leaf blight (BB) and rice blast (RB) diseases caused by Xanthomonas oryzae pv. oryzae (Xoo) and Magnaporthe oryzae (M. oryzae), respectively. In silico modeling, refinement, and model quality assessment were performed to predict the best structures of these five proteins and submitted to ModelArchive for future use. An in-silico annotation indicated that the five proteins functioned in signal transduction pathways as kinases, phospholipases, transcription factors, and DNA-modifying enzymes. The proteins were localized in the nucleus and plasma membrane. Phylogenetic analysis showed the evolutionary relation of the five proteins with disease-resistance proteins (XA21, OsTRX1, PLD, and HKD-motif-containing proteins). This indicates similar disease-resistant properties between five unknown proteins and their evolutionary-related proteins. Furthermore, gene expression profiling of these proteins using public microarray data showed their differential expression under Xoo and M. oryzae infection. This study provides an insight into developing disease-resistant rice varieties by predicting novel candidate resistance proteins, which will assist rice breeders in improving crop yield to address future food security through molecular breeding and biotechnology.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa sp. japonica) is a monocotyledonous angiosperm whose genome comprises of 12 chromosomes. It is consumed by over 90% of the world's population but produced by only a few countries. The top rice-producing countries, China, India, and Indonesia, produce 30.85%, 20.12%, and 8.21% of total global rice production, respectively (Kumar et al. 2017). By 2050, the demand for food and fiber will rise by up to 70% globally (Singh and Trivedi 2017). This increasing demand for agricultural production must be achieved in existing arable land, under harsher climatic conditions, with deteriorating soil and water quality.
Several pathogenic bacteria and fungi attack the Oryza sativa sp. japonica and massively reduce its production. In recent years, the use of pesticides has decreased bacterial and fungal disease incidence but, at the same time, adversely impacted human health and the entire ecosystem. Alternatively, using disease-resistant varieties of Oryza sativa sp. japonica is considered an effective and sustainable disease control approach. Therefore, extensive studies have been conducted recently on pathogen identification in rice and the signaling mechanism responsible for recognizing innate immunity (Gowda et al. 2015).
The biotic stressors, such as bacteria and fungi, are the primary source of infection in different parts of the rice plants. Xoo is a causative agent in BB disease. It is a major devastating disease in the Oryza sativa sp. japonica, affecting millions of hectares of rice annually, with an essential crop loss of as high as 75% (He et al. 2022). Recently, in vitro studies showed that plant growth-promoting rhizobacteria like Bacillus pumilus SE34 and Bacillus subtilis GBO3 induce systematic resistance against Xoo and improved nutrient uptake and yield (Chithrashree et al. 2011). Though the use of these rhizobacteria might be beneficial, the genetic selection of resistant rice cultivars is the most effective control method for BB disease (Bakade et al. 2021). Further, M. oryzae is another causative agent of a major fungal disease known as RB. This disease causes a severe threat to rice yield across many rice-producing countries all over the world. It has been reported that RB causes 10–30% of the world’s food loss each year (Qi et al. 2023). RB has also hampered cereal crop production worldwide due to its high genetic variability, significantly affecting rice breeders and pathologists (Li et al. 2017). Recent studies have identified resistance genes like Pigm, Ptr, Pi65 (t), and bsr-d1 to show strong resistance against RB disease without reducing the quality or yield of rice (Zhai et al. 2019). Moreover, several broad-spectrum pathogen-resistant varieties of Oryza sativa sp. japonica are now available. For instance, in 2017, some researchers found that the GY129 variety was resistant to all types of M. oryzae isolates and was predicted to carry a novel blast R gene in northern China. In addition, a gene Pi65 (t) in the GY129 variety provides resistance against RB disease in the japonica rice cultivar (Wang et al. 2016).
Plants have a variety of defense mechanisms against biotic stresses, including two types of the immune system. The first type consists of pattern recognition receptors (PRRs) that identify pathogen-associated molecular patterns (PAMPs) leading to PAMP-triggered immunity (PTI). The second type comprises highly polymorphic resistance R proteins against effectors on the pathogen's external surface that initiate effector-triggered immunity (ETI). Several experimental studies have been performed on disease-resistance genes and proteins in Oryza sativa sp. japonica. For instance, disease-resistant rice varieties were developed using molecular marker-assisted selection by transferring broad-spectrum rice blast resistance genes into susceptible genotypes (Mi et al. 2018). In another study, a Quantitative Train Loci (QTL), qBBR11-1, was discovered in a cross of Teqing and Lemont cultivars, showing two years of resistance to BB infection caused by three types of Xoo, i.e., C2, C4, and C510 (Kumar et al. 2018).
Despite the identification and well-documented phenotypes of several R genes in rice (Pradhan et al. 2023), the role of other disease-resistance genes and their mechanisms of action remained unexplored. In this context, a comprehensive analysis of disease-resistance genes at the genome-wide level is lacking for Oryza sativa sp. japonica. In addition, resistance proteins are structurally and functionally diverse, including high copy number variations, sequence diversity, and duplications. Moreover, the identification of resistance gene paralogs that individually contribute to strong resistance phenotypes is challenging. Thus, their identification, characterization, and pyramiding are crucial in preventing and controlling the spread of BB and RB diseases in the face of continually evolving pathogens. Hence, the rice breeders are looking for newer and more effective gene paralogs and alleles for molecular breeding. Consequently, bioinformatics-based approaches augment breeding efforts to introgress stable disease resistance traits in hybrid rice varieties. Thus, the current study will aid rice breeders to utilize in silico-analyzed novel disease-resistance proteins to develop resistant rice cultivars in the long run. Specifically, the current study has following objectives: (1) in silico identification of novel disease-resistance proteins in rice, (2) prediction and analysis of the three-dimensional (3D) structures of the identified proteins, (3) analysis of the evolutionary relationship of the identified proteins with known reference disease-resistance proteins, (4) functional investigation of the proteins through enrichment analysis, (5) identification of the functional interactors of identified proteins with known disease-resistance proteins and, finally (6) in silico gene expression profiling of the corresponding genes using publicly available data.
Our study is highly important for researchers working in the field of agricultural and crop science. Rice scientists can utilize these novel disease-resistant genes in genetic transformation and marker development for better improvement in the rice varieties. The genes identified in the current study can be used to develop an integrated map that may be adopted by rice breeders, specifically against BB and RB. In silico analysis of new gene orthologs is the first step for functional characterization, which will pave the way for further experimentation in planta. Therefore, the current study is a steppingstone toward global efforts to maintain food security and stabilize the production of disease-resistant rice varieties, thus reducing pesticide dependency.
Materials and methods
Data collection
The nucleotide sequences of the 182,437 genes from 12 chromosomes of the Oryza sativa sp. japonica were retrieved from the Ensembl plants database using the Biomart tool (https://plants.ensembl.org/index.html). Of these, 177,317 genes were known, and 5,120 genes were labeled as hypothetically conserved. The SNP-Seek database (https://snp-seek.irri.org/) retrieved previously known disease-resistance genes, including 56 and 85 disease-resistance genes for BB and RB diseases, respectively. Table S1 provides an overview of the different tools and databases used in this study.
Gene network construction and functional analysis
The gene network was constructed for the above-mentioned hypothetically conserved genes and known disease-resistance genes using the Expasy STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) tool (Snel et al. 2000). The STRING is the knowledgebase and software tool for known and predicted protein–protein interactions. Moreover, it includes direct (physical) and indirect (functional) associations derived from various sources, such as genomic context, high-throughput experiments, (conserved) co-expression, and the literature. The Cytoscape (version 3.9.1) tool was used for gene network analysis (Shannon et al. 2003). At first, the gene networks for BB and RB were separately exported from the Expasy STRING tool into the Cytoscape tool. Next, the genes (or nodes) in the network were colored differentially based on their function. The gene function information can be found in the 'function' column inside the node table available in the Cytoscape. Then, the first neighbors of each disease-resistance gene were selected, and the protein identifiers of these first neighbors were extracted from the node table. Only those proteins for which structural and functional information was unavailable in the UniProtKnowledgebase (UniProtKB) database were considered for further functional characterization.
Physicochemical analysis of unknown disease-resistance proteins
The amino acid sequences of the above-identified proteins were retrieved, with no structural and functional information, from the UniProtKB database (Bteman et al. 2021). Furthermore, the retrieved amino acid sequences from the UniProtKB database were then subjected to validation for their various stereochemical and physicochemical properties. The physical and chemical properties, such as molecular weight, amino acid composition, theoretical isoelectric point, instability index, extinction coefficient, etc., of the identified unknown disease-resistance proteins were determined using the ExPASy ProtParam tool (Gasteiger et al. 2003).
Structure prediction and stereochemical property analysis of unknown disease-resistance proteins
The secondary structures of the unknown proteins were predicted using their amino acid sequences with the PSI-BLAST Based Secondary Structure Prediction (PSIPRED) tool (McGuffin et al. 2000). This tool employs machine learning techniques to predict secondary structures for the input amino acid sequence. In addition, the 3D structures of these unknown proteins were predicted using SWISS-MODEL, a fully automated protein homology modeling server (Waterhouse et al. 2018). Homology modeling by the SWISS-MODEL describes the stoichiometry and the overall structure of the protein complexes. Its modeling functionality includes modeling homo- and heteromeric complexes with amino acid sequences of the interacting partners as a starting point. In this server, the template sequences for each protein were selected based on their Global Model Quality Estimate (GMQE) score and sequence identity. The GMQE score is a quality estimate that describes the accuracy of the tertiary structure of the resulting model. It combines properties from the target-template alignment and the template structure, which aids in selecting optimal templates for the modeling problem. The GMQE score ranges from 0 to 1, with higher scores indicating more reliability of the template with the target sequence. In addition to the quality score, the sequence identity explains the percentage of identity between target and template sequences during target-template alignment. The identity of the template sequence with the target sequence should be between 30% to 100%. Apart from SWISS-MODEL, there are other methods for protein structure modeling based on threading and deep learning (DL), such as Iterative Threading ASSEmbly Refinement (I-TASSER) and AlphaFold, respectively. I-TASSER estimated low-quality protein models with less confidence score, whereas AlphaFold anticipated a low predicted local distance difference test score. The 3D structures predicted using SWISS-MODEL were further refined with the GalaxyRefine2 server (Lee et al. 2019) and ModRefiner (Xu and Zhang 2011) and later visualized using PyMOL (Schrödinger and DeLano 2020). GalaxyRefine2 server performs short molecular dynamics (MD) relaxations after repeated side chain repacking perturbations. It is used in functional studies that involve protein modeling to improve the quality of the model structures obtained using other prediction methods. Moreover, ModRefiner is a tool for quick and efficient protein structure construction and refinement starting from Cα traces. It is based on two steps, i.e., low-resolution backbone structural construction and high-resolution full-atomic refinements, where a composite physics and knowledge-based force field guides the simulations. Moreover, homology-based model prediction consists of several limitations during modeling. Firstly, it is unlikely to find full-length templates for the larger proteins (> ~ 200 residues). However, the length of the identified proteins in our study was less than 200 residues. Secondly, the alignment between the query and template sequences sometimes produces errors and uncertainties due to the presence of gaps in the sequence alignment. This led to errors in the positioning of the query residues on the template fold and, thus, produced errors in the 3D model of the proteins. Thirdly, even if the sequence alignment and template result in a correct 3D model, the sidechain rotamer positions will be incorrect and produce errors. Although there are several limitations in the homology modeling of the proteins, but the backbone fold of the model is likely to be correct. Thus, this allows us to utilize the homology modeling for the identified disease-resistance proteins. It greatly benefits the wet lab experiments in terms of high coverage, time, and resources. In addition, homology-based modeling suggests information on which residue is on the surface and which is buried. Further, homology-based modelling can study functional sites in the evolutionary conserved residues.
The refined protein structures obtained from the above analysis were then subjected to validation for their various stereochemical properties. The stereochemical properties of the proteins were verified using the Ramachandran plot server (Sheik et al. 2002) and the PROCHECK tool (Laskowski et al. 1993). The Ramachandran plot server includes a graphics package that displays the main chain torsion angles phi, psi (φ, ψ) in a protein of known structure. This server calculates the torsion angles at the central residue in the stretch of three amino acids with specified flanking residue types. In contrast, PROCHECK studies the overall model geometry with the residue-by-residue geometry and provides the stereochemical quality of predicted models. It aims to assess how normal or unusual the geometry of the residues in a given protein structure is, compared with stereochemical parameters derived from well-refined, high-resolution structures. Further, the quality of the protein models was evaluated using Verify3D (Eisenberg et al. 1997) and ERRAT (Colovos and Yeates 1993). The Verify3D tool determines the compatibility of the atomic models with their own amino acid sequences. According to this tool, the compatibility percentage should be greater than 80%. It differentiates between correctly and incorrectly determined regions of protein structures based on characteristic atomic interactions. The ERRAT tool validates crystallography-determined protein structures in which a resolution of 95% or more is acceptable. The error values in the proteins are plotted as a function of the position of a sliding 9-residue window. The error function is based on the statistics of non-bonded atom–atom interactions in the reported structure. Finally, all the predicted and refined structures of the unknown disease-resistance proteins identified in our study are submitted for public access at ModelArchive. The ModelArchive provides a unique, stable accession code for each deposited model. Furthermore, the neural network-based predictor CYSPRED (Fariselli et al. 1999) was used to determine the disulfide bonds between the cysteine residues of these disease-resistance proteins. It provides information regarding the number of cysteine residues, their bonding or non-bonding state, and their reliability score.
Subcellular localization analysis
DeepLoc 1.0 is a Deep Neural Network (DNN) based model that predicts protein subcellular localization relying only on protein sequence information (Almagro Armenteros et al. 2017). In the background, the prediction model uses a recurrent neural network that processes the entire protein sequence by identifying protein regions important for subcellular localization. Moreover, the model was trained and tested on a protein dataset extracted from one of the latest UniProt releases. In addition, the topology of these proteins was predicted using the HMMTOP server (Tusnády and Simon 2001). HMMTOP transmembrane topology prediction server predicts both the localization of helical transmembrane segments and the topology of transmembrane proteins.
Multiple sequence alignment and phylogenetic analysis
The protein family and domain information for the identified unknown disease-resistance proteins were retrieved from the InterProScan (Blum et al. 2021) and Conserved Domain Database (Lu et al. 2020), respectively. Furthermore, the disease-resistant domains were predicted in the identified proteins using the Leucine Rich Repeats (LRR) predictor (Martin et al. 2020) and the Plant Resistance Genes Database (PRGdb) (Calle García et al. 2022). LRR predictor is a web server based on an ensemble of estimators designed to identify LRR motifs better. In the PRGdb, the DRAGO3 tool was used to predict disease-resistant domains and their positions. Additionally, we used ScanProsite (de Castro et al. 2006) and Motif Scan tools (Pagni et al. 2007) to find the consensus sequences and motifs for unknown disease-resistance proteins, respectively. The motifs identified using the Motif Scan tool were used further as input to ScanProsite for identifying the families of respective disease-resistance proteins. The amino acid sequences of the top five proteins from each identified family were retrieved for performing a BLAST search. The BLASTP tool was used to perform a BLAST search, taking each unknown disease-resistance protein and the identified top proteins from each family as input queries against various databases such as UniProtKB, SwissProt, and TrEMBL. We selected the top 12–15 hits from the BLAST search with an alignment score > 100 as a threshold, e-value close to 0, and similarity percentage greater than 60% for performing the local multiple sequence alignments (MSA). The ggmsa package (Zhou et al. 2022) in R software (version 4.2.2) was used to perform the MSA.
Furthermore, the phylogenetic analysis of the MSA results was performed using the neighbour-joining method available in the phylogram package (Wilkinson and Davy 2018) in R software. The phylogenetic tree was constructed through bootstrapping for 100 cycles to get more statistically confident node distances. The phylogenetic trees were plotted using the ggplot package (Wickham 2014) in R software.
Functional enrichment analysis of disease-resistance proteins
All disease-resistance proteins were functionally annotated using the PANNZER (Protein Annotation with Z-score) web server (Törönen and Holm 2022). It is a high-throughput functional annotation web server that provides gene ontology annotations and free text description predictions. It uses a weighted K-nearest neighbour (KNN) classifier based on sequence similarity and enrichment statistics. It consists of three servers, i.e., the frontend web server containing the interface, the suffix array neighborhood search (SANS) parallel server for fast homology search, and the DictServer for managing associated metadata. The annotation pipeline of the PANNZER includes homology search, gene ontology annotation, and free text description prediction. The functional annotation of disease-resistance proteins by PANNZER was based on a SANS parallel, a protein homology search tool faster than BLAST. Furthermore, the pathway analysis was performed for the identified disease-resistance proteins to identify the presence or absence of a disease-resistance protein against Xoo and M. oryzae. For this purpose, the Assign KO tool from the KEGG Database (Kanehisa and Subramaniam 2002) was used in the interactive mode. This tool is an interface to the BlastKOALA server, which assigns K numbers to the sequences provided by the user as input and performs a BLAST against a nonredundant set of KEGG genes.
Identification of functional interactors in disease-resistance proteins
The proteins and their functional interactors play an essential role in maintaining cellular processes. These interactors must be studied in order to gain a better understanding of various biological phenomena. Thus, to study the interaction of disease-resistance proteins with other similar proteins, the STRING database (Szklarczyk et al. 2019) was used. It is a biological database and web resource that incorporates all known and predicted physical and functional interactions between proteins.
In silico gene expression analysis
In silico expression analysis of genes was performed using EMBL-EBI Expression Atlas (Moreno et al. 2022). It is an open science resource that provides users with an efficient way to locate gene and protein expression information. It uses ArrayExpress, NCBI’s Gene Expression Omnibus (GEO), European Nucleotide Archive (ENA), and Proteomics IDEntification (PRIDE) database as its sources for microarray data. The ArrayExpress is a database for functional genomics data with two parts, i.e., ArrayExpress Repository and Array Express Data Warehouse. These two parts are based on Minimum Information About a Microarray Experiment (MIAME) and gene expression profiles, respectively. The ArrayExpress uses Expression Profiler, an online microarray-data analysis tool, to analyze the data. This tool provides various components for clustering, pattern discovery, statistical analysis, machine-learning algorithms, and visualization. These components can be assessed by the Simple Object Access Protocol (SOAP)-based web services through bioinformatics workflows using Taverna, a tool to build and run workflows of services. Further, GEO is a repository for high-throughput microarray and next-generation sequence functional genomic data. It includes user-specified series and parameters, i.e., P-value and its passage to the back end where the ‘GEOquery’ call loads the corresponding SeriesMatrix file. In addition, the annotation files are then sent via File Transfer Protocol (FTP) and return the ExpressionSet object and contrasts. This will serve as input for the two R scripts, i.e., boxplot and limma, which give a boxplot of expression value distribution and computation of top-ranked genes, respectively. Next, ENA is an archive for publicly available nucleotide sequencing data consisting of Webin command line submission interface (Webin-CLI) validation and submission tool. It includes an ENA back-end infrastructure with cross-reference services, distribution, indexing processes and International Nucleotide Sequence Database Collaboration (INSDC) exchange with NCBI. GEO deploys a workflow that routes high-volume INSDC sequence records in assembly contig sets directly to FTP servers, which are immediately indexed to produce output. Furthermore, PRIDE is a repository for the mass spectrometry-based proteomics data. It includes all types of datasets, i.e., data-dependent acquisition (DDA), Data Independent Acquisition (DIA), MS imaging and top-down proteomics. The analysis performed by the PRIDE on the input data includes resources (PRIDE Archive and Peptidome), tools (PRIDE Inspector and ProteomeXchange submission tool), software libraries such as mzTab, mgf, and pkl, web interface and Application programming interface (API) and external sources where data in PRIDE are disseminated.
In addition, these data sources may have a few limitations that can serve as future directions for the EMBL-EBI Expression Atlas. These limitations include single cell RNA-Seq data visualisation, gene expression across pathways, and proteomics and metabolomics datasets. Further, Single Cell Expression Atlas, including single-cell RNA-Seq data, will include retrieval of not only genes of interest but also for specific conditions such as diseases or cell type. In addition, more datasets of the plant genomics community, such as A. thaliana, are integrated to generate a comprehensive understanding of organism genetics. Furthermore, additional efforts will be put into representing differential datasets in the Expression Atlas interface for the proteomics and metabolomics datasets, thus improving the representation of protein entities. Although there are several limitations in the data sources of the EMBL-EBI Expression Atlas, these data sources enabled us to identify the expression of the studied disease-resistance proteins in tissue and experimental conditions in the rice plant. After accurate curation to represent the experimental design of the identified proteins, the findings were freely available in an easy-to-visualised form. Thus, the gene IDs were retrieved from the UniProtKB database and queried in the Expression Atlas to get the gene expression levels under various biotic stresses. Moreover, the Atlas consists of a log2 (fold-change) scale with a range of −1 to −2.5, which is shown by blue cells indicating the gene is down-regulated. Similarly, red cells depict another range of 1–1.7, indicating the gene is up-regulated. Among these ranges for the current study, we have selected upregulation with log2 (fold-change) > 1.1 and downregulation with log2 (fold-change) < −2.2. In addition, we have selected ‘infect’ as the experimental variable for the study.
Results
Identification of unknown disease-resistance genes in rice through gene network analysis
Gene networks were created by the Expasy STRING tool between hypothetically conserved and disease-resistance genes for BB and RB diseases (Fig. 1). For BB disease, we have found 27 genes to be involved in disease resistance as well as in defense-related mechanisms. Similarly, for RB disease, we identified 26 genes engaged in disease resistance and defense-related mechanisms. The protein identifiers of the first neighbors of the disease-resistance genes were selected from the node table in Cytoscape. There were 125 and 129 first neighbors identified for BB and RB diseases, respectively. These 254 protein identifiers were further analyzed in the UniProtKB database. Of these, we selected 5 proteins marked as “unknown” in the UniProtKB database because only these proteins do not have any structural and functional information available. Noteworthy, these five proteins showed strong interactions with known disease-resistance genes in our gene network analysis. For the purpose of this study, we named these five proteins according to their involvement in the disease. For instance, the two proteins involved in BB disease are Protein BB.1 and Protein BB.2. Similarly, three proteins involved in RB were named Protein RB.1, Protein RB.2, and Protein RB.3.

Gene–gene interaction network of hypothetically conserved and known disease-resistance genes against bacterial blight and rice blast diseases in rice (Oryza sativa). a A gene–gene interaction network of bacterial blight disease-resistance genes in rice is depicted. The network was constructed through the Expasy STRING tool and analyzed in the Cytoscape software (see “Materials and methods” section). b A gene–gene interaction network of rice blast disease-resistance genes in rice is shown. The nodes indicate genes, and branches indicate the interactions in the network. The nodes were colored differentially based on their function. Yellow, black, green, purple, red, and grey colored nodes indicate their involvement in disease resistance, defense mechanism, disease and defense mechanism, stress-related, and less annotation, respectively
Physicochemical characteristics of unknown rice disease-resistance proteins
The physiochemical features of the five disease-resistance proteins were calculated using ExPASy's ProtParam tool. Table 1 shows the physicochemical features, such as atomic composition, molecular weight, theoretical isoelectric point (pI), instability index, and extinction coefficient, for all five proteins. The BB.2 protein has the highest number (1212) of amino acids, whereas Protein RB.3 has the least number (487) of amino acids. Further, BB.2 has a maximum molecular weight of 134.85 kilodaltons (kDa), while Protein BB.3 has a minimum molecular weight of 50.98 kDa. The Protein RB.3 was found to have a maximum pI of 8.05, indicating that it is a positively charged protein, followed by RB.1 (pI = 7.38), RB.2 (pI = 7.29), BB.1 (pI = 6.51), and BB.2 (pI = 6.42). Protein BB.1 has the lowest instability index of 39.24, showing that it is a stable protein. Moreover, it has a high aliphatic index depicting high thermostability over a wide range of temperatures. All five proteins showed low GRAVY values ranging from -0.65 to 0.04. This indicates that these proteins are globular (hydrophilic) rather than membranous (hydrophobic). The extinction coefficients for the proteins range from 141,900 to 168,180 with respect to cysteine (C), tryptophan (W), and tyrosine (Y), showing the presence of high concentrations of C, W, and Y in all the proteins. The presence of disulfide bonds is important in stabilizing protein structures and understanding structural–functional relationships. CYSPRED showed both bonding and non-bonding states of cysteine residues. In addition, CYSPRED predicted the highest reliability score for all five proteins, showing the significance of the prediction of the disulfide bonds.
Three-dimensional structure of unknown rice disease-resistance proteins
PSIPRED tool predicted that all five proteins (i.e., BB.1, BB.2, RB.1, RB.2, and RB.3) had a significant helix and coil content. Of these, BB.2 has the highest helix and coil content, comprising 363 and 758 amino acid residues, respectively (Table S2). We performed homology modeling using SWISS-MODEL to predict the 3D structure of five disease-resistance proteins. We noticed that the template for RB.2 protein has the best QMQE score of 0.63, whereas the templates of RB.1 and RB.3 proteins have the best sequence identity of 84.75%. The GalaxyRefine2 server and ModRefiner refined the models for all five proteins. Among the five proteins, BB.2 has the best model refined by the GalaxyRefine2 server. Moreover, the presence of disulphide bonds is important in stabilizing protein structures and understanding structural–functional relationships. CYSPRED showed both bonding and non-bonding states of cysteine residues. In addition, CYSPRED predicted the highest reliability score for all five proteins, showing the significance of the prediction of the disulphide bonds.
Stereochemical features of unknown rice disease-resistance proteins
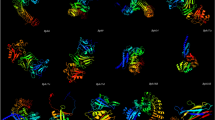
Table S3 shows the results from the analysis performed using the Ramachandran Plot server, PROCHECK, Verify3D, and ERRAT tools. The Ramachandran plot shows the backbone dihedral angles of the five proteins (Figure S1). Furthermore, Table S4 includes a comparison of the stereochemical quality of the 3D models for the disease-resistance proteins predicted by the SWISS-MODEL, I-TASSER, and AlphaFold applications. It shows a comparative analysis of the percentage of the amino acid residues present in the disallowed region of the Ramachandran plots for the 3D models. The lowest percentage of amino acid residues was present in the disallowed region of the Ramachandran plots of the 3D models predicted by the SWISS-MODEL. The percentages were 0.8%, 0%, 0%, 0.3%, and 0% for BB.1, BB.2, RB.1, RB.2, and RB.3, respectively, much lower than the I-TASSER and AlphaFold-predicted models. Thus, we considered SWISS-MODEL as the best-suited program to perform the structure modeling of the disease-resistance proteins. A structural alignment of the 3D models of the disease-resistance proteins predicted by the SWISS-MODEL with those predicted by I-TASSER and AlphaFold is presented in Figure S2. The alignment was performed by the ‘align’ function in PyMOL which superimposes the protein structures based on the homology between their amino acid residues. The analysis showed that the SWISS-MODEL faithfully predicted the functional conformation of the disease-resistance proteins as compared to I-TASSER and AlphaFold, which predicted the misfolded conformation of the proteins. The Ramachandran plot analysis shows that BB.2, RB.1, and RB.3 had the best distribution of φ and ψ angles. In addition, for the same three proteins, more than 90% of amino acids were in the favored regions, and none of the amino acids were in the disallowed regions. The PROCHECK tool checks the stereochemical quality of protein structure by analyzing the residue-by-residue geometry and overall structure geometry. Likewise, in the Ramachandran Plot server, the PROCHECK results also showed that amino acids of BB.2, RB.1, and RB.3 proteins were in the allowed region of the Ramachandran plot. The Verify3D tool determines the compatibility of an atomic model (3D) with its own amino acid sequence (1D). According to Verify3D, the compatibility percentage was found to be 100% for BB.2 (100%), followed by BB.1 (96.58%) and RB.2 (91.11%). The ERRAT tool analyses the statistics of non-bonded interactions between different atom types. It shows that the best score out of 100 was for RB.1, followed by BB.2 (95.58) and BB.1 (94.11). The 3D models of the five proteins were visualized using PyMOL and shown in Fig. 2. In addition, the structural alignment of the predicted and refined models was performed and visualized in PyMOL. Thus, to understand the significant structural changes achieved during the refinement process, it has been found that Protein RB.3 consists of 0.118 as the best Root Mean Square Deviation (RMSD). Furthermore, after obtaining the RMSD value of RB.3 in its structure alignment, proteins BB.1, BB.2, RB.1, and RB.2 consist of 0.279, 0.450, 0.831, and 0.445, respectively, as RMSD values. The RMSTD values of all the identified disease-resistance proteins indicate that there were minor changes in the conformation of the protein structures and, hence, representing that both the models were very similar. The structural alignment of the predicted and refined models of the identified disease-resistance proteins is presented in Figure S3.

A comparative analysis of the 3D models of the disease-resistance proteins against bacterial blight and rice blast diseases predicted by the SWISS-MODEL and refined by the Galaxy Refine2 server and ModRefiner in rice. a–d 3D models of the proteins BB.1 and BB.2 against bacterial blight disease in rice. The models were predicted and refined by the SWISS-MODEL and Galaxy Refine2 server, respectively. The template sequences for each protein were selected based on their Global Model Quality Estimate (GMQE) score and sequence identity (see “Materials and methods” section). e-j 3D models of the proteins RB.1, RB.2, and RB.3 against rice blast disease predicted by the SWISS-MODEL server
Subcellular localization of unknown rice disease-resistance proteins
The subcellular localization of five proteins was carried out using DeepLoc 1.0 to understand their functions and role in regulating the biological processes at the cellular level. The DeepLoc 1.0 result revealed that BB.1 was the only protein found in the cell membrane, with a likelihood score of 0.969. The remaining three proteins (i.e., BB.2, RB.1, RB.2) were localized in the nucleus with likelihood scores of 0.996, 0.882, and 0.51, respectively. The visual findings of the subcellular localization of the identified proteins are depicted in Fig. 3. According to the HMMTOP tool, proteins BB.1 and BB.2 consist of two and one transmembrane helices. In addition, Protein RB.2 was predicted to contain one transmembrane helix. Moreover, we used a neural network-based predictor, CYSPRED, to predict the cysteine residues (Table S5).

Subcellular localization of the disease-resistance proteins in the rice plant cell against bacterial blight and rice blast diseases. a, b Subcellular localization of proteins BB.1 and BB.2 against bacterial blight disease in rice. DeepLoc 1.0, a Deep Neural Network (DNN) based model, was executed to identify the subcellular localization through BB.1 and BB.2 amino acid sequence information. c–e The subcellular localization of the proteins RB.1, RB.2, and RB.3 against rice blast disease, identified through DeepLoc 1.0, are shown. The red line in the prediction of the subcellular localization indicates the existence of RB.1, RB.2, and RB.3 (a, b) at various locations in the rice plant cell, such as cell membrane, mitochondria, nucleus, etc., along with a likelihood score at each location
Phylogeny of unknown rice disease-resistance proteins
InterProScan and CDD tools were used for studying the domains, motifs, families, and superfamilies of the five unknown proteins. Three proteins, BB.1, BB.2, RB.1, RB.2, and RB.3, were found to contain the domains such as LRR, Kinase, Transmembrane (TM), Su(var)3–9, Enhancer-of-zeste and Trithorax (SET), WRKY and Phospholipase D known to be involved in disease resistance. Additionally, DRAGO3 also predicted 5 LRR, 4 Kinase, and 1 TM domains for the Protein BB.1. Similarly, from the LRR predictor, we found that there were in total 22 LRR domains in the Protein BB.1. The presence of these disease-resistant domains indicated that BB.1 might be involved in the disease-resistance mechanism against Xoo and M. oryzae. Table 2 shows information on the conserved domains, motifs, families, and superfamilies of the five unknown proteins.
The ScanProsite was used to identify specific motif consensus sequences of conserved domains from the iterative BLASTP search for the five unknown proteins. Some hundreds of proteins with identical domains and similar motif consensus were retrieved for each query motif. From these BLASTP hits, some 12 to 15 sequences were selected from related organisms either from the Oryza genus or Arabidopsis thaliana (A. thaliana). The above-selected sequences were found to have high similarity and annotation scores in the range of 4–5, as reported in the UniProtKB. On average, we observed 70% similarity between our query proteins (BB.1, BB.2, RB.1, RB.1, and RB.3) and their respective similar proteins. For two proteins, BB.1 and RB.1, the similarity was around 80% with their similar proteins. For each of the five proteins, we performed MSA with their respective similar proteins as described above.
The phylogenetic analysis showed that the protein BB.1 was placed in the monophyletic clade with receptor kinase-like protein XA21, which is annotated with a score of 5 as per the UniProtKB (Fig. 4). The clade bootstrapping confidence is 100, and the subsequent alignment was found to be 84% similar. Similarly, the BB.2 protein was close to the Histone-lysine N-methyltransferase (OsTRX1) protein. We noticed high bootstrap scores of 100 and 98, respectively, for the protein RB.1 and RB.3, which implies that they shared a common ancestry with the conserved WRKY domain with a zinc-finger motif. This domain is commonly found in protein superfamilies involved in various plant physiological regulations, including disease resistance. Finally, the protein RB.2 shared similar phospholipase D (PLD) phosphodiesterase domain activity with its orthologs in A. thaliana. The active site profile in Protein RB.2 was confirmed by the presence of histidine (H), lysine (K), and aspartic acid (D) residues, which refer to the popular HKD motif consensus.

Multiple Sequence Alignment and phylogenetic tree analysis of the disease-resistance proteins against bacterial blight and rice blast diseases. a, b Schematic representation of the phylogenetic trees for the proteins BB.1 and BB.2 against bacterial blight disease. First, the Multiple Sequence Alignment was performed using the ggmsa package of the R software (version 4.2.2). Second, the phylogenetic analysis of the Multiple Sequence Alignment results was performed using the neighbour-joining method available in the phylogram package of the R software. The trees were constructed through bootstrapping for 100 cycles to get more statistically confident node distances (see “Materials and methods” section). c–e Phylogenetic analysis of the proteins RB.1, RB.2, and RB.3 against rice blast disease. The Multiple Sequence Alignment and phylogeny analysis were performed by the packages of the R program (a, b), and the dendrogram of all the disease-resistant proteins in the rice plant was in the form of unrooted trees
Moreover, the phylogenetic analysis of the identified disease-resistance proteins further confirmed their involvement in the disease-resistance mechanism as they are in close evolutionary relationship with the known disease-resistance proteins. Further, the bootstrap scores of the identified proteins show a significant relationship between identified and known disease-resistance proteins. For instance, BB.1, BB.2, RB.1, RB.2, and RB.3 were close to Xa21, OsTRX1, WRKY24, and PLD with their bootstrap scores of 100, 4, 100, 65, and 98, respectively.
Predicted functions of unknown rice disease-resistance proteins
Table 3 shows the functional enrichment analysis results using the PANNZER tool. This tool predicted that Protein BB.1, RB.1, RB.2, and RB.3 have biological functions such as protein phosphorylation, defense response, response to the bacterium, positive regulation of defense response to the bacterium, phospholipase D activity and presence of WRKY domain. This might indicate that BB.1, RB.1, RB.2, and RB.3 are involved in the disease resistance mechanism. Moreover, the information on genes involved in the proteins BB.1, RB.1, RB.2, and RB.3 can be retrieved from publicly available databases such as NCBI. This information can then be utilized to identify disease-resistant Quantitative Trait Loci (QTL) through tools like QTG-Finder (Lin et al. 2020) to develop novel rice varieties. Kumar et al. (2018) analyzed qBBR11-1, a QTL of rice, as a good candidate for defense and resistance analysis towards BB. Further, Wang et al. (2019) identified and analyzed a nucleotide binding site (NBS) LRR, i.e., NLR genes having broad-spectrum resistance against M. oryzae in a rice cultivar. These disease-resistance genes were predicted by a bioinformatics tool, NLR-Parser, and were cloned and tested in susceptible cultivars showing resistance against RB. Thus, the biological functions of the identified disease-resistance proteins can be experimentally validated and utilized for developing resistant varieties against BB and RB. In addition, we can perform molecular marker and pathogenicity assays through the knowledge of functional annotation of the identified disease-resistance proteins. This will also confirm the validity of the information on functional annotation by the PANNZER tool on BB.1, RB.1, RB.2, and RB.3. Biological pathways analysis was performed using the Assign KO tool to find their role in the disease resistance mechanism. This tool predicted that all three proteins against RB were involved in plant-pathogen interaction, Ras, cAMP, and MAPK signaling pathways (Table 4, Table S6). This indicates that these proteins might be involved in the pathways responsible for providing resistance against diseases.
Predicted functional interactors of unknown rice disease-resistance proteins
The protein–protein interactions are of central importance for every process in a living cell. The protein–protein interactions predicted by the Expasy STRING database for the identified disease-resistance proteins are shown in Figure S4. The Expasy STRING database indicated the interactions between transcription initiation factor IIA subunit 2 (Q0DLD3, TFIIAy) and WRKY transcription factor WRKY24 (Q6IEQ7, WRKY24) with the Protein BB.1. This further suggests the involvement of BB.1 in the disease resistance mechanism as WRK24 is a well-known transcription factor for biotic stress signaling. Furthermore, Protein BB.2 has important domains nearby, such as histone H4, lysine-specific demethylase SE14la and RYanodine receptor (SPRY). The histone H4 domain is crucial in transcription regulation, DNA repair, replication, and chromosomal stability. SE14 is a histone demethylase that demethylates ‘Lys-4’ (H3K4me) of histone H3 and controls flowering time in plants. In addition, the SPRY domain is reportedly involved in plant-parasitic interactions, providing innate immunity (Diaz-Granados et al. 2016). Further, Protein RB.1 consists of WRKY24 and Cyclin-D3-1 at the closest distance. This indicates that, like WRKY24, the Protein RB.1 might be involved in the disease resistance mechanism. Cyclin-D3-1 is found to be involved in the control of the cell cycle at the G1/S (start) transition, activating the G1/S phase transition in response to the cytokinin hormone signal. Additionally, Protein RB.2 has Phosphatidylserine decarboxylase proenzyme 1, which has a central role in phospholipid metabolism and the inter-organelle trafficking of phosphatidylserine. RB.2 also interacts with the Phosphoesterase family protein, which has hydrolase activity and is involved in phospholipid catabolic process. Further, we identified the interaction of Protein RB.3 with mitogen-activated protein kinase 1 (Q84UI5, MPK1), which is found to be involved in disease resistance and stress tolerance signaling pathways (Sharma et al. 2020). In addition, Protein RB.3 contains WRKY24 as its interactor, indicating the involvement of RB.3 in the disease resistance mechanism.
Gene expression under biotic stress of unknown rice disease-resistance proteins
Public microarray data indicated differential expression of the genes encoding the five proteins under study, during infection by different strains of rice pathogens Xoo and M. oryzae, suggesting their clear involvement in the biotic stress responses of rice. Eight experiments in Gene Expression Atlas indicated a downregulation of Os11g0559200 encoding BB.1 with log2 (fold-change) in the range of −2.2 to −1.6, having the same values for Xoo pv. oryzicola showed upregulation of Os12g0613200 encoding BB.2 with the value of 1.1 in one experimental condition. Other experiments indicated the downregulation of Os08g0499300, encoding RB.1 with −1.1 as log2 (fold-change), and upregulation of Os10g0524400 and Os05g0343400, encoding RB.2 and RB.3 with highest values of 3.9 and 3.2, respectively. The relative expression levels of these genes under biotic stress are presented in Table 5.
Discussion
Food security is a global concern, especially for staple crops such as rice, which prompts a greater focus on developing and improving crop protection methods. The regular increase in the application of chemical fertilizers and pesticides in rice has made the pathogens more resistant, and hence, there is a global effort for pest biocontrol, including the development of disease-resistant varieties for future global food security. This work aimed to identify unknown resistance proteins against major bacterial and fungal diseases in Oryza sativa sp. japonica and their in silico characterization using state-of-the-art bioinformatics tools. We focused on the disease-resistance proteins for BB and RB diseases following a genome-wide study. We identified five unknown disease-resistance proteins, two for BB and three for RB, and characterized them according to their functions, domains, protein–protein interactions, and involvement in biological pathways.
Gene co-expression and protein–protein interaction network analyses are powerful in silico tools to identify unknown candidate genes (Klasberg et al. 2016). First, we collected data for known and disease-resistance genes from Ensembl plants and SNP-Seek databases. The Ensembl plants database provides vast information on the genome level, thus providing free access to the complete nucleotide sequences of the rice plant. In addition, the SNP-Seek database consists of information on the known disease-resistance genes with a user-friendly web interface. Next, we performed a network-based analysis to identify unknown disease-resistance proteins in rice. Thus, for BB disease, network analysis using Cytoscape revealed seventeen disease-resistance genes, five defense-related genes, and five genes involved in disease resistance and defense mechanisms. Similarly, for RB disease, we identified eleven genes for disease resistance, eight genes as defense-related, and seven genes involved in both disease resistance and defense mechanisms. After this, we performed various in silico analyses of these five identified proteins.
MSA and phylogeny analyses were performed, which showed the evolutionary relationship of the predicted proteins with reported disease-resistance proteins. In addition, secondary and tertiary structures were predicted and validated along with the stereochemical and physicochemical properties. The subcellular localization revealed their presence in the nucleus and plasma membrane, whereas the topology study indicated the presence of transmembrane helices in them. Furthermore, the biological sequence analysis, including disulfide bonds, conserved domains, motifs, folds, families, and superfamilies, was performed, revealing structural features of plant disease-resistance proteins.
We found that BB.1, BB.2, RB.1, RB.2, and RB.3 carry LRR, Kinase, TM, SET, WRKY, and Phospholipase D domains known to be involved in disease resistance against Xoo and M. oryzae. Our results indicated a high similarity of Protein BB.1 with Rice Receptor Kinase XA21 (RRK XA21), indicating a similar immune response action against BB disease. Park and Ronald (2012) have found that the RRK XA21 is responsible for broad-spectrum innate immunity against the bacterial pathogen Xoo. During bacterial infection, RRK XA21 recognizes the Xoo Ax21 protein and binds to its conserved sulfated peptide region to finally release a kinase domain upon cleavage, and this kinase domain ultimately translocates into the rice protoplast nucleus. Following the nuclear localization, the XA21 kinase domain binds with transcriptional regulation factors to trigger the immune response. Furthermore, Choi et al. (2014) experimentally validated the function of OsTRX1, which contains a SET domain similar to our Protein BB.2. The SET domain is essential for the methylation of a lysine residue of the N-terminus of Histone H3 (Rea et al. 2000). In rice, this methylation is crucial for the OsTRX1-mediated negative regulation of transcriptional activation, which is part of a broader defense mechanism against BB disease. Protein BB.2 and OsTRX1 share a matched lysine residue in their respective SET domains, followed by a high cystine signature in the post-SET region towards the C-termini. Thus, the prediction of methyltransferase activity in Protein BB.2 is supported by its similarity with high cystine and lysine-rich consensus in OsTRX1. Moreover, proteins RB.1 and 3 RB.3 shared a common ancestry with the conserved WRKY domain with a zinc-finger motif, which is commonly found in protein superfamilies involved in various plant physiological regulations, including disease resistance. The high confidence score of proteins in subsequent phylogenetic analysis indicates a strong probability of their functioning as defense-related transcriptional regulators. In addition, RB.2 shared a similar phospholipase D (PLD) phosphodiesterase domain with its orthologs in A. thaliana. The active site profile in RB.2 was confirmed by the presence of histidine (H), lysine (K), and aspartic acid (D) residues, which constitute the well-known HKD motif consensus. Our phylogenetic study illustrated the common ancestry of HKD motifs presented in various copies of phospholipase phosphodiesterase proteins of A. thaliana. These paralogs are known candidates in the resistance mechanism of A. thaliana against RB disease (Bargmann and Munnik 2006). Therefore, from a strong homology perspective, RB.2 is predicted to possess similar hydrolyzing activity of glycerol phospholipids during the resistance against the RB disease in rice.
Functional enrichment analysis of the predicted proteins showed their involvement in the disease resistance mechanism. BB.1, RB.1, RB.2, and RB.3 were found to have roles in biological functions such as protein phosphorylation and phospholipase D activity. In addition, BB.2 was predicted to be involved in Histone H3-K4 methylation, RNA binding, and DNA-binding transcription factor activity. The pathway analysis of BB.1 and BB.2 has also indicated their roles in transcription under control conditions as well as bacterial infection. Moreover, RB.1 was involved in secondary metabolite synthesis and lipid metabolism, whereas RB.2 and RB.3 were involved in Ras, phospholipase D, cAMP, and MAPK signaling pathways, indicating their possible functional mechanisms in the resistance mechanism against Xoo and M. oryzae. Furthermore, the functional interactors of our identified proteins were also found to be involved in disease resistance. For example, the interactors of BB.1 and RB.3, viz. TFIIAy and WRKY24, respectively, are known players of bacterial disease resistance in rice and are highly expressed in disease-resistant rice varieties (Kumar et al. 2022; Zhang et al. 2017). Moreover, gene expression profiling showed upregulation of four of five predicted disease-resistance proteins under Xoo and M. oryzae infection. This strongly suggested that our identified proteins are likely to be involved in rice disease resistance against RB and BB. As an exception, Os11g0559200 encoding BB.1, an LRR kinase family protein, was downregulated under infection by several strains of Xoo (Table 5). Previous reports suggest that some LRR kinases are upregulated under infection by some pathogenic strains while downregulated under others, indicating negative regulation of biotic stress signaling (Shumayla et al. 2016; Zhang et al. 2004). Interestingly, in our protein–protein interaction analysis, BB.1 was found to physically interact with WRKY24, a known negative regulator of abscisic acid (ABA) and gibberellic acid (GA) signaling, which cross-talks with biotic stress signaling (Zhang et al. 2004; Singh et al. 2022). WRKY transcription factors (TFs) are the largest families of transcriptional regulators and have diverse roles in disease resistance mechanisms. These TFs have components of signaling that regulate responses to biotic stresses. They can act as activators or repressors through TF net that participates in many cytoplasmic and nuclear processes. These processes include signaling events from organelles and the cytoplasm to the nucleus. In addition, WRKYs work along with TFs in clusters to mediate various responses in stress tolerance. The hormones, including salicylic acid and jasmonic acid/ethylene, are primarily involved in this response. Further, the salicylic acid-mediated signaling pathway is associated with resistance to biotrophic (Xoo) pathogens. Moreover, OsWRKY24 negatively regulates GA and ABA signaling through two WRKY DNA binding domains. However, further study will be necessary to dissect this negative regulation of stress signaling through BB.1 under Xoo infection in rice.
In summary, the current study predicted five unknown disease-resistance proteins in rice against Xoo and M. oryzae. It has been found that long-lasting disease resistance is a crucial trait for rice breeding programs. However, achieving durable resistance is challenging because Xoo and M. oryzae continuously evolve to overcome the rice defense system. Its population changes through mutations in the pathogen effector genes targeted by rice resistance genes or proteins. Thus, this study sheds light on identifying broad-spectrum disease-resistance proteins that can provide resistance to wide strains of the Xoo and M. oryzae. Therefore, the identified disease-resistance proteins in the present study can be utilized to provide broad-spectrum resistance in rice. Further, through in silico studies, we can identify nearby QTLs responsible for disease resistance mechanisms against BB and RB diseases. These QTLs can then be anticipated to develop and improve disease-resistant cultivars in rice. Hence, future experimental validation of the predicted roles of these proteins will provide new candidates for disease resistance in rice. Furthermore, detailed research in the future can be performed on the identified disease-resistance proteins, including (1) genome-wide analysis of these proteins in the japonica sp., such as the study on cis-regulatory elements, synteny with other similar proteins, etc., (2) transcriptome analysis to gain in-depth knowledge of molecular mechanisms of these proteins and to apply this in genetic engineering (3) involvement of these proteins in the signaling pathways of hormones responsible in the disease resistance mechanism. Next, to develop novel disease-resistant varieties through the identified disease-resistance proteins, a few investigating steps are needed to be followed in the future. These steps include gene sequence retrieval responsible for these proteins, their isolation, amplification, and cloning, molecular marker and pathogenicity assays to confirm resistance further, and finally, gene pyramiding into pure rice lines. These lines will first be tested in the lab environment and then, under field conditions, to enhance durability and spectrum of resistance. However, after developing disease-resistant rice varieties, it must pass through several hurdles. This includes approval of regulatory authorities considering ethical concerns of disease-resistant varieties that will be grown or imported into a country. The requirements of these authorities in different countries are not standardized, making the approval much more complex. In addition, there can be various ethical considerations in the identified disease-resistance proteins, such as the effects of non-target crops, disease susceptibility, suspected allergy, potential for gene flow, changes in plant metabolism, consumer risk, etc. Thus, addressing ethical concerns in the rice breeding experiments is essential, including reducing fertilizer use and food losses and increasing sustainability. Therefore, interdisciplinary approaches that include social scientists, scientists, and policymakers are required to achieve the aim of successful rice breeding in a short duration. Additionally, it is up to scientists, seed companies, international agricultural organizations, and legislators to utilize genetic disease solutions responsibly, aiding in rice improvement. This will help rice breeders and biotechnologists for developing resistant rice varieties against BB and RB diseases. This information will also be used to develop molecular markers that can be used to screen varieties for resistance to both BB and RB diseases, as well as for marker-assisted selection.
Data availability
The datasets generated during the current study are available in the ModelArchive repository with IDs ma-u68j8, ma-yfta8, ma-xdomw, ma-n3mnn, and ma-y1dh3.
References
Almagro Armenteros JJ, Sønderby CK, Sønderby SK, Nielsen H, Winther O (2017) DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics (Oxf Engl) 33(21):3387–3395. https://doi.org/10.1093/bioinformatics/btx431
Bakade R, Ingole KD, Deshpande S, Pal G, Patil SS, Bhattacharjee S, Prasannakumar MK, Ramu VS (2021) Comparative transcriptome analysis of rice resistant and susceptible genotypes to Xanthomonas oryzae pv. oryzae identifies novel genes to control bacterial leaf blight. Mol Biotechnol 63(8):719–731. https://doi.org/10.1007/S12033-021-00338-3/FIGURES/6
Bargmann BOR, Munnik T (2006) The role of phospholipase D in plant stress responses. Curr Opin Plant Biol 9(5):515–522. https://doi.org/10.1016/J.PBI.2006.07.011
Blum M, Chang HY, Chuguransky S, Grego T, Kandasaamy S, Mitchell A, Nuka G, Paysan-Lafosse T, Qureshi M, Raj S, Richardson L, Salazar GA, Williams L, Bork P, Bridge A, Gough J, Haft DH, Letunic I, Marchler-Bauer A et al (2021) The InterPro protein families and domains database: 20 years on. Nucleic Acids Res 49(D1):D344–D354. https://doi.org/10.1093/NAR/GKAA977
Bteman A, Martin MJ, Orchard S, Magrane M, Agivetova R, Ahmad S, Alpi E, Bowler-Barnett EH, Britto R, Bursteinas B, Bye-A-Jee H, Coetzee R, Cukura A, da Silva A, Denny P, Dogan T, Ebenezer TG, Fan J, Castro LG et al (2021) UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49(D1):D480–D489. https://doi.org/10.1093/NAR/GKAA1100
Calle García J, Guadagno A, Paytuvi-Gallart A, Saera-Vila A, Amoroso CG, D’esposito D, Andolfo G, AieseCigliano R, Sanseverino W, Ercolano MR, Sanseverino W (2022) PRGdb 4.0: an updated database dedicated to genes involved in plant disease resistance process. Nucleic Acids Res 50(D1):D1483–D1490. https://doi.org/10.1093/NAR/GKAB1087
Chithrashree, Udayashankar AC, Chandra Nayaka S, Reddy MS, Srinivas C (2011) Plant growth-promoting rhizobacteria mediate induced systemic resistance in rice against bacterial leaf blight caused by Xanthomonas oryzae pv. oryzae. Biol Control 59(2):114–122. https://doi.org/10.1016/J.BIOCONTROL.2011.06.010
Choi SC, Lee S, Kim SR, Lee YS, Liu C, Cao X, An G (2014) Trithorax group protein Oryza sativa trithorax1 controls flowering time in rice via interaction with early heading date3. Plant Physiol 164(3):1326–1337. https://doi.org/10.1104/PP.113.228049
Colovos C, Yeates TO (1993) Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci Publ Protein Soc 2(9):1511–1519. https://doi.org/10.1002/PRO.5560020916
de Castro E, Sigrist CJA, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N (2006) ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res 34(suppl_2):W362–W365. https://doi.org/10.1093/NAR/GKL124
Diaz-Granados A, Petrescu AJ, Goverse A, Smant G (2016) SPRYSEC effectors: a versatile protein-binding platform to disrupt plant innate immunity. Front Plant Sci. https://doi.org/10.3389/fpls.2016.01575
Eisenberg D, Lüthy R, Bowie JU (1997) VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol 277:396–404. https://doi.org/10.1016/S0076-6879(97)77022-8
Fariselli P, Riccobelli P, Casadio R (1999) Role of evolutionary information in predicting the disulfide-bonding state of cysteine in proteins. Proteins. https://doi.org/10.1002/(SICI)1097-0134(19990815)36:3
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A (2003) ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31(13):3784–3788. https://doi.org/10.1093/NAR/GKG563
Gowda M, Shirke MD, Mahesh HB, Chandarana P, Rajamani A, Chattoo BB (2015) Genome analysis of rice-blast fungus Magnaporthe oryzae field isolates from southern India. Genomics Data 5:284–291. https://doi.org/10.1016/J.GDATA.2015.06.018
He Z, Xin Y, Wang C, Yang H, Xu Z, Cheng J, Li Z, Ye C, Yin H, Xie Z, Jiang N, Huang J, Xiao J, Tian B, Liang Y, Zhao K, Peng J (2022) Genomics-assisted improvement of super high-yield hybrid rice variety “Super 1000” for resistance to bacterial blight and blast diseases. Front Plant Sci 13:1430. https://doi.org/10.3389/FPLS.2022.881244/BIBTEX
Kanehisa M, Subramaniam (2002) The KEGG database. Novartis Found Symp 247:91–103. https://doi.org/10.1002/0470857897.CH8
Klasberg S, Bitard-Feildel T, Mallet L (2016) Computational identification of novel genes: current and future perspectives. Bioinform Biol Insights 10:121–131. https://doi.org/10.4137/BBI.S39950
Kumar S, Meshram S, Sinha A (2017).Bacterial diseases in rice and their eco-friendly management. www.tjprc.org
Kumar S, Zaharin N, Nadarajah K (2018) In silico identification of resistance and defense related genes for bacterial leaf blight (BLB) in rice. J Pure Appl Microbiol 12(4):1867–1876. https://doi.org/10.22207/JPAM.12.4.22
Kumar A, Kumar R, Shukla P, Singh S, Alam M, Singh DK (2022) Update on cloning and molecular characterization of bacterial blight resistance genes in rice. In: Shukla P, Kumar A, Kumar R, Pandey MK (eds) Molecular response and genetic engineering for stress in plants, vol 2. Biotic stress. IOP Publishing, UK, pp 71–715
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26(2):283–291. https://doi.org/10.1107/S0021889892009944
Lee GR, Won J, Heo L, Seok C (2019) GalaxyRefine2: simultaneous refinement of inaccurate local regions and overall protein structure. Nucleic Acids Res 47(W1):W451–W455. https://doi.org/10.1093/NAR/GKZ288
Li W, Zhu Z, Chern M, Yin J, Yang C, Ran L, Cheng M, He M, Wang K, Wang J, Zhou X, Zhu X, Chen Z, Wang J, Zhao W, Ma B, Qin P, Chen W, Wang Y et al (2017) A natural allele of a transcription factor in rice confers broad-spectrum blast resistance. Cell 170(1):114-126.e15. https://doi.org/10.1016/J.CELL.2017.06.008
Lin F, Lazarus EZ, Rhee SY (2020) QTG-Finder2: a generalized machine-learning algorithm for prioritizing QTL causal genes in plants. G3 Genes Genomes Genet 10(7):2411–2421. https://doi.org/10.1534/g3.120.401122
Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Marchler GH, Song JS, Thanki N, Yamashita RA, Yang M, Zhang D, Zheng C, Lanczycki CJ, Marchler-Bauer A (2020) CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res 48(D1):D265–D268. https://doi.org/10.1093/NAR/GKZ991
Martin EC, Sukarta OCA, Spiridon L, Grigore LG, Constantinescu V, Tacutu R, Goverse A, Petrescu AJ (2020) LRRpredictor—a new LRR motif detection method for irregular motifs of plant NLR proteins using an ensemble of classifiers. Genes 11(3):286. https://doi.org/10.3390/GENES11030286
McGuffin LJ, Bryson K, Jones DT (2000) The PSIPRED protein structure prediction server. Bioinformatics 16(4):404–405. https://doi.org/10.1093/BIOINFORMATICS/16.4.404
Mi J, Yang D, Chen Y, Jiang J, Mou H, Huang J, Ouyang Y, Mou T (2018) Accelerated molecular breeding of a novel P/TGMS line with broad-spectrum resistance to rice blast and bacterial blight in two-line hybrid rice. Rice 11(1):1–12. https://doi.org/10.1186/S12284-018-0203-8/TABLES/
Moreno P, Fexova S, George N, Manning JR, Miao Z, Mohammed S, Muñoz-Pomer A, Fullgrabe A, Bi Y, Bush N, Iqbal H, Kumbham U, Solovyev A, Zhao L, Prakash A, García-Seisdedos D, Kundu DJ, Wang S, Walzer M et al (2022) Expression atlas update: gene and protein expression in multiple species. Nucleic Acids Res 50(D1):D129–D140. https://doi.org/10.1093/nar/gkab1030
Pagni M, Ioannidis V, Cerutti L, Zahn-Zabal M, Jongeneel CV, Hau J, Martin O, Kuznetsov D, Falquet L (2007) MyHits: improvements to an interactive resource for analyzing protein sequences. Nucleic Acids Res 35(suppl_2):W433–W437. https://doi.org/10.1093/NAR/GKM352
Park CJ, Ronald PC (2012) Cleavage and nuclear localization of the rice XA21 immune receptor. Nat Commun 3(1):1–6. https://doi.org/10.1038/ncomms1932
Pradhan M, Bastia D, Samal KC, Dash M, Sahoo JP (2023) Pyramiding resistance genes for bacterial leaf blight (Xanthomonas oryzae pv. Oryzae) into the popular rice variety, Pratikshya through marker assisted backcrossing. Mol Biol Rep. https://doi.org/10.1007/S11033-023-08805-7/TABLES/6
Qi Z, Du Y, Yu J, Zhang R, Yu M, Cao H, Song T, Pan X, Liang D, Liu Y (2023) molecular detection and analysis of blast resistance genes in rice main varieties in Jiangsu Province, China. Agronomy 13(1):157. https://doi.org/10.3390/AGRONOMY13010157/S1
Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtier K, Ponting CP, Allis CD, Jenuwein T (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406(6796):593–599. https://doi.org/10.1038/35020506
Schrödinger L, DeLano W (2020) PyMOL. Source: http://www.pymol.org/pymol.. Accessed 2 Jun 2022
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–2504. https://doi.org/10.1101/GR.1239303
Sharma D, Verma N, Pandey C, Verma D, Bhagat PK, Noryang S, Singh K, Tayyeba S, Banerjee G, Sinha AK (2020) MAP kinase as regulators for stress responses in plants. In: Protein kinases and stress signaling in plants. https://doi.org/10.1002/9781119541578.ch15
Sheik SS, Sundararajan P, Hussain ASZ, Sekar K (2002) Ramachandran plot on the web. Bioinformatics 18(11):1548–1549. https://doi.org/10.1093/BIOINFORMATICS/18.11.1548
Shumayla S, Sharma S, Kumar R, Mendu V, Singh K, Upadhyay SK (2016) Genomic dissection and expression profiling revealed functional divergence in Triticum aestivum leucine rich repeat receptor like kinases (TaLRRKs). Front Plant Sci. https://doi.org/10.3389/fpls.2016.01374
Singh BK, Trivedi P (2017) Microbiome and the future for food and nutrient security. Microb Biotechnol 10(1):50. https://doi.org/10.1111/1751-7915.12592
Singh D, Dhiman VK, Pandey H, Dhiman VK, Pandey D (2022) Crosstalk between salicylic acid and auxins, cytokinins, and gibberellins under biotic. Stress. https://doi.org/10.1007/978-3-031-05427-3_11
Snel B, Lehmann G, Bork P, Huynen MA (2000) STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res 28(18):3442–3444. https://doi.org/10.1093/NAR/28.18.3442
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Von Mering C (2019) STRING v11: protein–protein association networks with increased coverage, supporting functional discovery ingenome-wide experimental datasets. Nucleic Acids Res 47(D1):D607–D613. https://doi.org/10.1093/NAR/GKY1131
Törönen P, Holm L (2022) PANNZER-A practical tool for protein function prediction. Protein Sci Publ Protein Soc 31(1):118–128. https://doi.org/10.1002/PRO.4193
Tusnády GE, Simon I (2001) The HMMTOP transmembrane topology prediction server. Bioinformatics 17(9):849–850. https://doi.org/10.1093/BIOINFORMATICS/17.9.849
Wang Y, Zhao JM, Zhang LX, Wang P, Wang SW, Wang H, Wang XX, Liu Z, Zheng WJ (2016) Analysis of the diversity and function of the alleles of the rice blast resistance genes Piz-t, Pita and Pik in 24 rice cultivars. J Integr Agric 15(7):1423–1431. https://doi.org/10.1016/S2095-3119(15)61207-2
Wang L, Zhao L, Zhang X, Zhang Q, Jia Y, Wang G, Li S, Tian D, Li WH, Yang S (2019) Large-scale identification and functional analysis of NLR genes in blast resistance in the Tetep rice genome sequence. Proc Natl Acad Sci U S A 116(37):18479–18487. https://doi.org/10.1073/pnas.1910229116
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, De Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46(W1):W296–W303. https://doi.org/10.1093/NAR/GKY427
Wilkinson L (2011) ggplot2: elegant graphics for data analysis by WICKHAM, H. Biometrics, 67(2). https://doi.org/10.1111/j.1541-0420.2011.01616.x
Wilkinson SP, Davy SK (2018) phylogram: an R package for phylogenetic analysis with nested lists. J Open Source Softw 3:790. https://doi.org/10.21105/joss.00790
Xu D, Zhang Y (2011) Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J 101(10):2525–2534. https://doi.org/10.1016/J.BPJ.2011.10.024
Zhai K, Deng Y, Liang D, Tang J, Liu J, Yan B, Yin X, Lin H, Chen F, Yang D, Xie Z, Liu JY, Li Q, Zhang L, He Z (2019) RRM transcription factors interact with NLRs and regulate broad-spectrum blast resistance in rice. Mol Cell 74(5):996-1009.e7. https://doi.org/10.1016/J.MOLCEL.2019.03.013
Zhang ZL, Xie Z, Zou X, Casaretto J, Ho THD, Shen QJ (2004) A rice WRKY gene encodes a transcriptional repressor of the gibberellin signaling pathway in aleurone cells 1. Plant Physiol 134(4):1500–1513. https://doi.org/10.1104/pp.103.034967
Zhang J, Chen L, Fu C, Wang L, Liu H, Cheng Y, Li S, Deng Q, Wang S, Zhu J, Liang Y, Li P, Zheng A (2017) Comparative transcriptome analyses of gene expression changes triggered by Rhizoctonia solani AG1 IA infection in resistant and susceptible rice varieties. Front Plant Sci 8:1422. https://doi.org/10.3389/FPLS.2017.01422/BIBTEX
Zhou L, Feng T, Xu S, Gao F, Lam TT, Wang Q, Wu T, Huang H, Zhan L, Li L, Guan Y, Dai Z, Yu G (2022) ggmsa: a visual exploration tool for multiple sequence alignment and associated data. Brief Bioinf 23(4):1–12. https://doi.org/10.1093/BIB/BBAC222
Acknowledgements
PY acknowledges the support from seed grant (project number I/SEED/PY/20200037) funded by the Indian Institute of Technology, Jodhpur, India; VD is thankful for the financial support from the Ministry of Education (MoE), India; and RSS (file number: 09/1125(0019)/2021-EMR-I) is financially supported by the CSIR-NET fellowship.
Funding
VD collected study data, performed primary analysis, and designed the working strategy; SB carried out basic data analysis and contributed to manuscript writing; RSS performed functional enrichment; PY conceptualized and supervised the study; AS edited the manuscript and provided inputs; all authors contributed to writing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare that there are no competing financial interests.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dhiman, V., Biswas, S., Shekhawat, R.S. et al. In silico characterization of five novel disease-resistance proteins in Oryza sativa sp. japonica against bacterial leaf blight and rice blast diseases. 3 Biotech 14, 48 (2024). https://doi.org/10.1007/s13205-023-03893-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-023-03893-5